")
Back to Journals » Journal of Asthma and Allergy » Volume 14
Safety of Once-Daily Single-Inhaler Triple Therapy with Fluticasone Furoate/Umeclidinium/Vilanterol in Japanese Patients with Asthma: A Long-Term (52-Week) Phase III Open-Label Study
Authors Hozawa S, Ohbayashi H , Tsuchiya M, Hara Y, Lee LA, Nakayama T, Tamaoki J, Fowler A, Nishi T
Received 9 February 2021
Accepted for publication 3 June 2021
Published 6 July 2021 Volume 2021:14 Pages 809—819
DOI https://doi.org/10.2147/JAA.S305918
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Amrita Dosanjh
Soichiro Hozawa,1 Hiroyuki Ohbayashi,2 Michiko Tsuchiya,3 Yu Hara,4 Laurie A Lee,5 Takashi Nakayama,6 Jun Tamaoki,6 Andrew Fowler,7 Takanobu Nishi6
1Hiroshima Allergy and Respiratory Clinic, Hiroshima, Japan; 2Tohno Chuo Clinic, Gifu, Japan; 3Department of Respiratory Medicine, Rakuwakai Otowa Hospital, Kyoto, Japan; 4Department of Pulmonology, Yokohama City University Graduate School of Medicine, Kanagawa, Japan; 5GSK, Collegeville, PA, USA; 6GSK, Tokyo, Japan; 7GSK, Brentford, Middlesex, UK
Correspondence: Takanobu Nishi
GlaxoSmithKline K.K., 1-8-1 Akasaka, Minato-ku, Tokyo, 107-0052, Japan
Tel +81-70-4135-5303
Fax +81-3-4231-5974
Email [email protected]
Purpose: The pivotal CAPTAIN study reported a favorable safety profile with once-daily inhaled corticosteroid/long-acting muscarinic antagonist/long-acting β2-agonist (ICS/LAMA/LABA) triple combination of fluticasone furoate/umeclidinium/vilanterol (FF/UMEC/VI) in patients with inadequately controlled asthma, some of whom were Japanese. Here, we evaluate the long-term (52 weeks) safety of FF/UMEC/VI in Japanese patients with asthma.
Patients and Methods: This was a Phase III, 52-week, multicenter, non-comparator, non-randomized, open-label study (NCT03184987) in Japanese adults receiving maintenance therapy with ICS/LABA, with or without LAMA. At enrollment, patients were allocated to either FF/UMEC/VI 100/62.5/25mcg (Group 1) or 200/62.5/25mcg (Group 2). Patients in Group 1 could have their treatment stepped up to 200/62.5/25mcg at Week 24 if their Asthma Control Questionnaire (ACQ)-7 score was > 0.75. The primary endpoint was the incidence of adverse events (AEs) and serious AEs (SAEs). Secondary endpoints included vital signs, electrocardiogram measurements, and clinical laboratory tests (biochemistry, hematology, urinalysis). Efficacy was assessed as “other” endpoints.
Results: A total of 111 Japanese patients were included in the intention-to-treat (ITT) population. Overall, 77 (69%) patients reported ≥ 1 AE (Group 1: n=30 [64%]; step-up group: n=7 [78%]; Group 2: n=40 [73%]). SAEs were reported for 1 (2.1%) and 2 (3.6%) patients in Groups 1 and 2, respectively. All SAEs were considered unrelated to study treatment. One AE and one SAE led to study withdrawal: oropharyngeal discomfort (Group 1); eosinophilic granulomatosis with polyangiitis (Group 2). No new safety concerns were identified throughout the 52-week treatment period.
Conclusion: In this uncontrolled open-label study, no new safety concerns were observed with long-term (52 weeks) treatment with once-daily FF/UMEC/VI among 111 Japanese patients with asthma.
Keywords: asthma, inhalers, Japan, safety, treatment outcome
Introduction
It is estimated that up to 18% of the global population and 6–10% of the Japanese population are affected by asthma,1,2 a chronic respiratory airway disease characterized by symptoms such as wheeze, shortness of breath, chest tightness, and cough.1 Around 30–50% of patients with moderate/severe asthma who are prescribed and are adherent to inhaled corticosteroids/long-acting β2-agonist (ICS/LABA) therapy remain symptomatic.3–6 A recent real-world study of patients with asthma in Japan showed that 50% of the overall population had partially controlled asthma with 15% having uncontrolled asthma.7 Furthermore, a greater proportion of patients with uncontrolled severe asthma in Japan require oral corticosteroids (OCS), have higher rates of hospitalization and a greater medical and economic burden compared with patients with controlled severe asthma or mild-to-moderate asthma, indicating a clear need for more effective management.8
For patients with uncontrolled asthma despite ICS/LABA therapy, treatment options include increasing ICS dose and/or adding another bronchodilator, such as a long-acting muscarinic antagonist (LAMA).1 The addition of the LAMA tiotropium is recommended by both the Global Initiative for Asthma (GINA) and the Japanese Society of Allergology for patients with uncontrolled asthma on ICS/LABA therapy,1,2 and has been shown to improve lung function, symptom control, and the rate of asthma exacerbations.9,10 Previous studies have demonstrated favorable safety for ICS, LABA, or LAMA in patients with asthma. An integrated analysis demonstrated that fluticasone furoate (FF), a once-daily ICS, has a favorable safety profile, with the most frequent drug-related adverse events (AEs) being headache, dysphonia, and oral/oropharyngeal candidiasis.11 Similarly, treatment with vilanterol (VI), a LABA, has been reported to be associated with low incidences of AEs, the most common reported being tremor and palpitations.12 Glucose effects (eg, impaired glucose tolerance; raised blood glucose) had a reported incidence of 0–1% among patients treated with VI in one study.12 Safety results of dual combination therapy with FF/VI have also shown a favorable risk profile, with headache, nervous system disorders, and nasopharyngitis reported as the most frequently occurring on-treatment AEs.4,13 Treatment with umeclidinium (UMEC), a LAMA, was also not associated with significant safety concerns; the most frequently reported on-treatment AEs included headache, nasopharyngitis, abnormal product taste, and pharyngitis.14,15
Single-inhaler triple therapy with the combination of FF/UMEC/VI is widely approved as a once-daily treatment for chronic obstructive pulmonary disease (COPD).16 Single-inhaler FF/UMEC/VI triple therapy has also recently been approved in the United States and Japan as a once-daily maintenance treatment for adult patients with asthma, based on positive findings from the recently published Phase IIIA Clinical Study in Asthma Patients Receiving Triple Therapy in a Single Inhaler (CAPTAIN) study.17 CAPTAIN compared the efficacy and safety of once-daily FF/UMEC/VI with FF/VI over 24–52 weeks in patients with asthma inadequately controlled on ICS/LABA. Results showed improved lung function, reduced symptoms and improved asthma control, and numerical reductions in the annualized rate of moderate/severe exacerbations with FF/UMEC/VI versus FF/VI. The safety profile of both treatments was similar, with no new or unexpected safety findings. The intention-to-treat (ITT) population of CAPTAIN included 229 (9.4%) patients across 63 centers in Japan, all of whom were of Japanese ancestry, for whom efficacy and safety outcomes were consistent with the overall study population.18
The aim of this study was to evaluate the long-term (52 weeks) safety of fixed-dose FF/UMEC/VI in Japanese patients with asthma. Treatment efficacy was also assessed as an “other” endpoint in an exploratory manner. This study was used as part of the submission of a regulatory application to the Japanese Ministry of Health, Labour and Welfare for once-daily FF/UMEC/VI for the treatment of adults with asthma.
Patients and Methods
Study Design and Study Population
This was a Phase III, 52-week, multicenter, non-comparator, non-randomized, open-label study in Japanese patients aged 18 years or older, who had received an asthma diagnosis at least a year prior to providing informed consent and were receiving maintenance therapy with ICS/LABA, with or without LAMA, for at least 4 weeks pre-screening (GSK 207236, NCT03184987). Patients were excluded if an asthma exacerbation occurred that required a change in maintenance therapy in the 6 weeks prior to screening, although patients who required a temporary change in asthma therapy (eg, increased dose of ICS or use of OCS) were not necessarily excluded provided the patient’s condition had stabilized upon resuming pre-exacerbation maintenance therapy. Additionally, patients were excluded if they had a diagnosis of other concurrent respiratory disorders, including a diagnosis of COPD.19 Current smokers (within 12 months of screening) or former smokers with a smoking history of ≥10 pack-years were also excluded. The inclusion and exclusion criteria are listed in Supplementary Materials.
The study design is shown in Figure 1. Briefly, eligible patients entered into a 2-week run-in period, during which they continued to receive their pre-screening asthma maintenance therapy. At enrollment, patients were allocated to either FF/UMEC/VI 100/62.5/25mcg (Group 1) or FF/UMEC/VI 200/62.5/25mcg (Group 2) depending on pre-screening therapy (ICS dose/prior use of LAMA) and control status (Asthma Control Questionnaire [ACQ]-7 score ≤0.75 [controlled] or >0.75 [not well controlled]) (Table 1). Patients receiving FF/UMEC/VI 100/62.5/25mcg could have their treatment stepped up to 200/62.5/25mcg at Week 24 if their ACQ-7 score was >0.75; however, this step-up was at the investigator’s discretion. All treatments were administered via the ELLIPTA dry-powder inhaler (Glaxo Operations UK, Hertfordshire, UK) once daily in the morning.
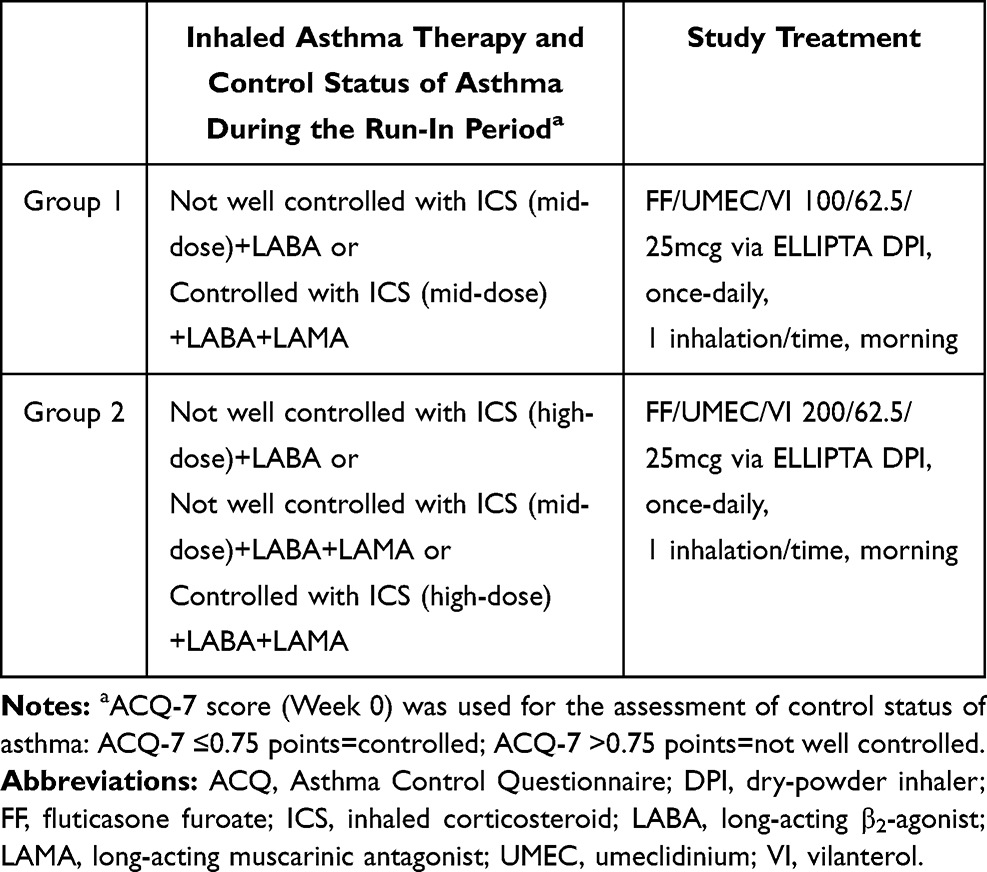 |
Table 1 Study Treatment Assignment |
 |
Figure 1 Study design: 52-week, open-label Phase III safety study. aPatients allocated to receive FF/UMEC/VI 100/62.5/25mcg (selection of FF dose ([100 or 200mcg] depended on patients’ pre-screening therapy [ICS dose/prior use of LAMA] and control status [ACQ-7 total score ≤0.75 or >0.75] [Table 1]); bPatients switching medication from FF/UMEC/VI 100/62.5/25mcg to 200/62.5/25mcg at Week 24 if their ACQ-7 score was >0.75; this step up was at the investigator’s discretion; cPatients allocated to receive FF/UMEC/VI 200/62.5/25mcg (selection of FF dose [100 or 200mcg] depended on patients’ pre-screening therapy [ICS dose/prior use of LAMA] and control status [ACQ-7 total score ≤0.75 or >0.75] (Table 1]). Abbreviations: ACQ, Asthma Control Questionnaire; FF, fluticasone furoate; ICS, inhaled corticosteroid; ITT, intention-to-treat; LABA, long-acting β2-agonist; LAMA, long-acting muscarinic antagonist; UMEC, umeclidinium; VI, vilanterol. |
This study was performed in accordance with the Declaration of Helsinki, International Conference on Harmonisation Good Clinical Practice, and applicable country-specific regulatory requirements. The protocol was reviewed and approved by an internal GSK review board and was approved by applicable central or local institutional review boards or independent ethics committees. All patients enrolled provided written informed consent and patient anonymity was preserved using methods approved by the Ethics Committee.
Safety Endpoints and Assessments
The primary endpoint was the incidence and type of AEs and serious AEs (SAEs) in all dose groups, including the step-up group, and the total study population. AEs of special interest (AESIs) were defined as AEs that were associated with known pharmacological effects of ICS, LAMA, and LABA. For secondary endpoints, raw value and change from baseline were summarized by dose group and in the total population for each visit, and included vital signs (blood pressure, pulse rate) measured at every clinical visit; electrocardiogram (ECG) measurements (QTcF, heart rate, PR interval) at screening and Weeks 4, 24, and 52, or the Early Withdrawal Visit (if applicable); clinical laboratory tests (biochemistry, hematology, urinalysis) at screening and Weeks 12, 24, and 52, or the Early Withdrawal Visit (if applicable).
For clinical laboratory tests, data were summarized by shifts from baseline relative to the normal range by dose group and in the total study population at each visit.
“Other” Endpoints
The efficacy of FF/UMEC/VI combination therapy was pre-defined as “other” endpoints and assessed in an exploratory manner. Summary statistics were provided for change from baseline at Weeks 24 and 52 for the following outcomes, and are reported in the total study population combining all three treatment groups only: trough forced expiratory volume in 1 second (FEV1); ACQ-7 total score; St George’s Respiratory Questionnaire (SGRQ) total score; Asthma Quality of Life Questionnaire (AQLQ) total score; and annualized rate of moderate/severe and severe asthma exacerbations.
Statistical Analyses
Summaries of safety data, patient demographics, and other (efficacy) data were conducted on the ITT population according to the treatment they received. All AEs were classified using Medical Dictionary for Regulatory Activities (MedDRA) version 22.0 and grouped by system organ class and preferred term, unless otherwise stated. As the study was an open-label safety study, and efficacy assessments were specified as “other” endpoints, no statistical analyses were planned, and thus only summary tables for efficacy parameters are provided. Programming of summaries and figures was performed using SAS System version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Patient Demographics and Clinical Characteristics
The study was conducted in 14 centers across Japan from June 22, 2017 (first patient enrolled) to June 25, 2019 (last patient last visit). A total of 111 patients were included in the ITT population: Group 1, n=47; step-up group, n=9; Group 2, n=55. Of these, 46, 8, and 51 patients in the respective dose groups completed 52 weeks of treatment. Six patients withdrew from the study (Group 1: n=1, AE; step-up group: n=1, withdrawal by patient; Group 2: n=4, withdrawal by patient, n=2, AE, n=1, physician decision) (Figure 2).
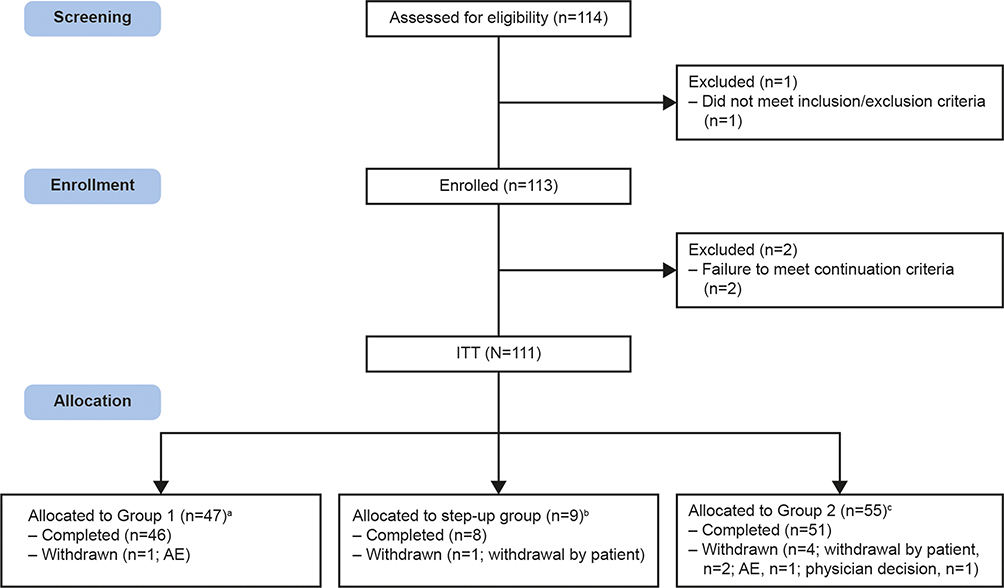 |
Figure 2 Study disposition. aPatients allocated to receive FF/UMEC/VI 100/62.5/25mcg; bPatients switching medication from FF/UMEC/VI 100/62.5/25mcg to 200/62.5/25mcg at Week 24; cPatients allocated to receive FF/UMEC/VI 200/62.5/25mcg. Abbreviations: AE, adverse event; ITT, intention-to-treat. |
Baseline demographics and clinical characteristics are presented in Table 2. Overall, patients had a mean (standard deviation [SD]) age of 50.9 (13.41) years, with slightly more females (n=64 [58%]). Patients had a predicted FEV1 mean (SD) of 86.4% (16.58) and asthma duration of 18.5 years (14.37).
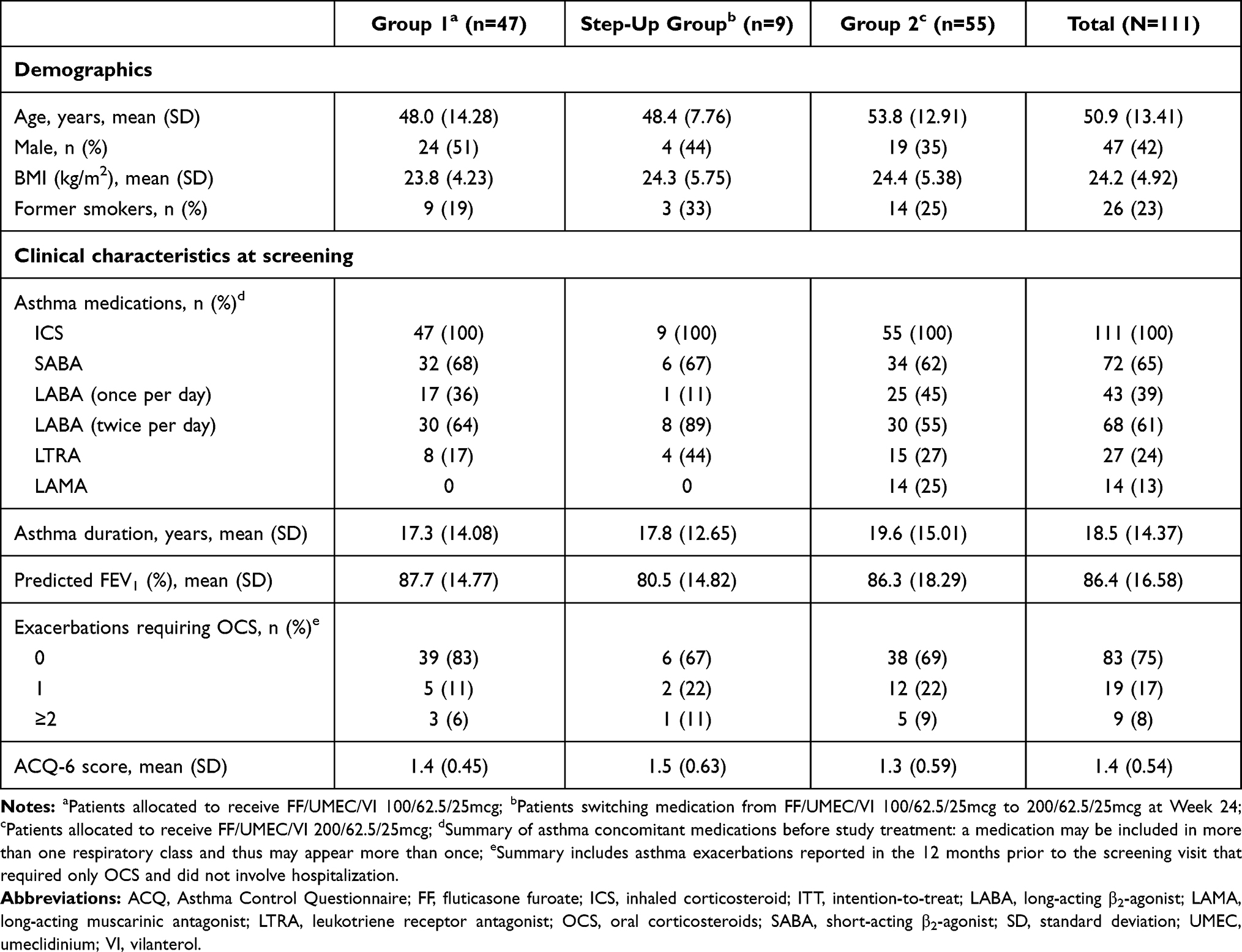 |
Table 2 Baseline Demographics and Clinical Characteristics (ITT Population) |
Safety
Overall, 77 (69.4%) patients reported at least one AE (Group 1: 30 [63.8%]; step-up group: 7 [77.8%]; Group 2: 40 [72.7%] Table 3). The most frequent AEs reported were nasopharyngitis (n=33 [29.7%]), dysgeusia (n=11 [9.9%]), and pharyngitis (n=11 [9.9%]) (Table 3). The severities of almost all AEs were classified as mild or moderate, with only 4 (3.6%) events reported as severe across a diverse range of AEs. Two events led to withdrawal from the study: oropharyngeal discomfort (Group 1) (Table 3) and eosinophilic granulomatosis with polyangiitis (Group 2) (Table 4). The AE of oropharyngeal discomfort was considered related to study treatment, but was mild in severity and non-serious. No fatal AEs were reported.
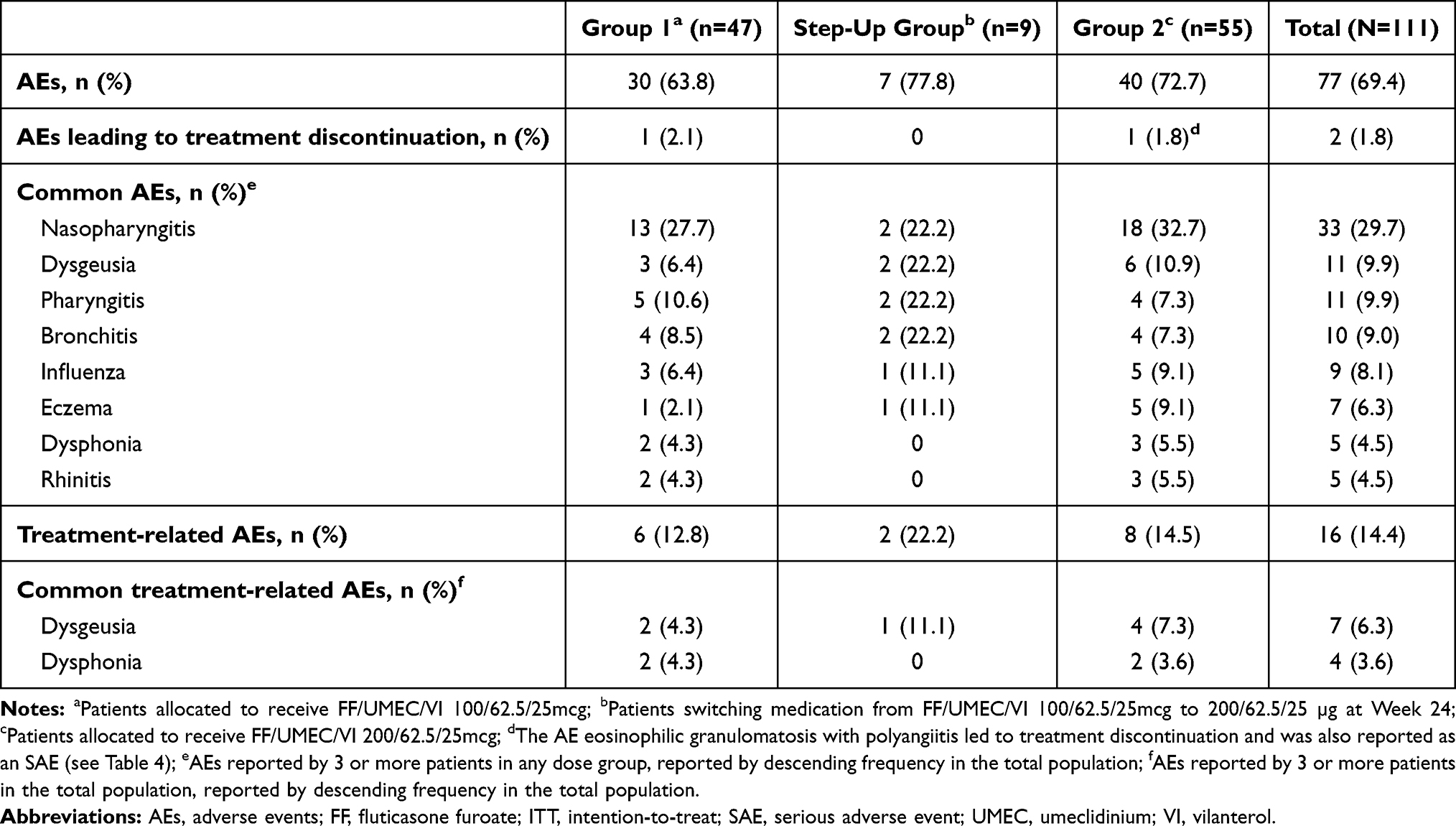 |
Table 3 Summary of On-Treatment AEs (ITT Population) |
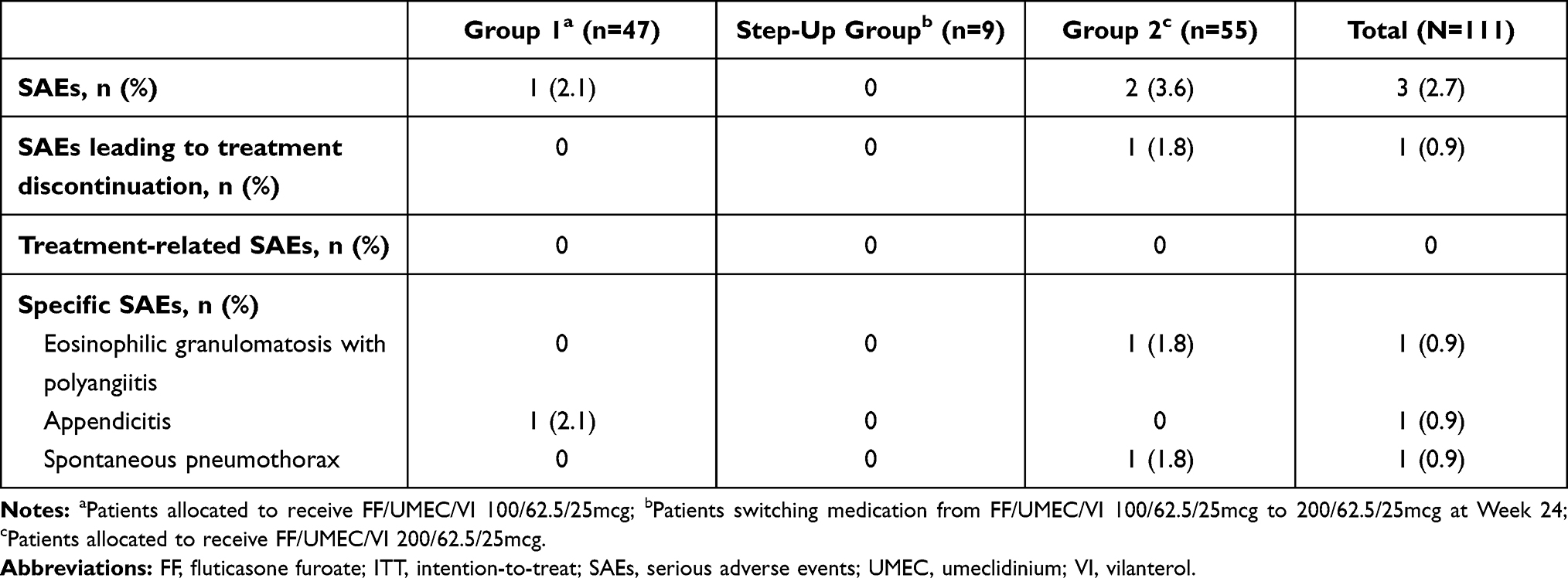 |
Table 4 Summary of On-Treatment SAEs (ITT Population) |
SAEs were reported for 1 patient in Group 1 (appendicitis) and 2 patients in Group 2 (spontaneous pneumothorax, eosinophilic granulomatosis with polyangiitis) (Table 4). None of the SAEs reported were considered by the study investigators to be related to study treatment. The patient reporting eosinophilic granulomatosis with polyangiitis withdrew from the study (see above); however, the SAE did not resolve. The other 2 SAEs did not lead to treatment discontinuation, and both events resolved.
The frequency of AESIs was low overall (Table 5). Dry mouth/drying of the airway secretions (broad focus) was the most frequently reported AESI (n=53 [47.7%]), due to the number of patients reporting nasopharyngitis (n=33 [29.7%]) (Table 5). No participants reported an AESI of dry mouth/drying of the airway secretions (narrow focus [defined using seven pre-specified preferred terms, excluding nasopharyngitis]). No major adverse cardiac events occurred and very few participants (n=5 [4.5%]) experienced AESIs of cardiovascular effects. Only one pneumonia event (non-serious) was reported. The proportion of patients with hypersensitivity in the total study population was 16.2% (n=18). However, individual preferred terms within the overall category of hypersensitivity did not exceed 1 or 2 events, with the exception for eczema and allergic rhinitis, which were reported in 7 (6.3) and 5 (4.5) patients, respectively, in the total study population. None of these events were reported by the study investigators to be related to study treatment.
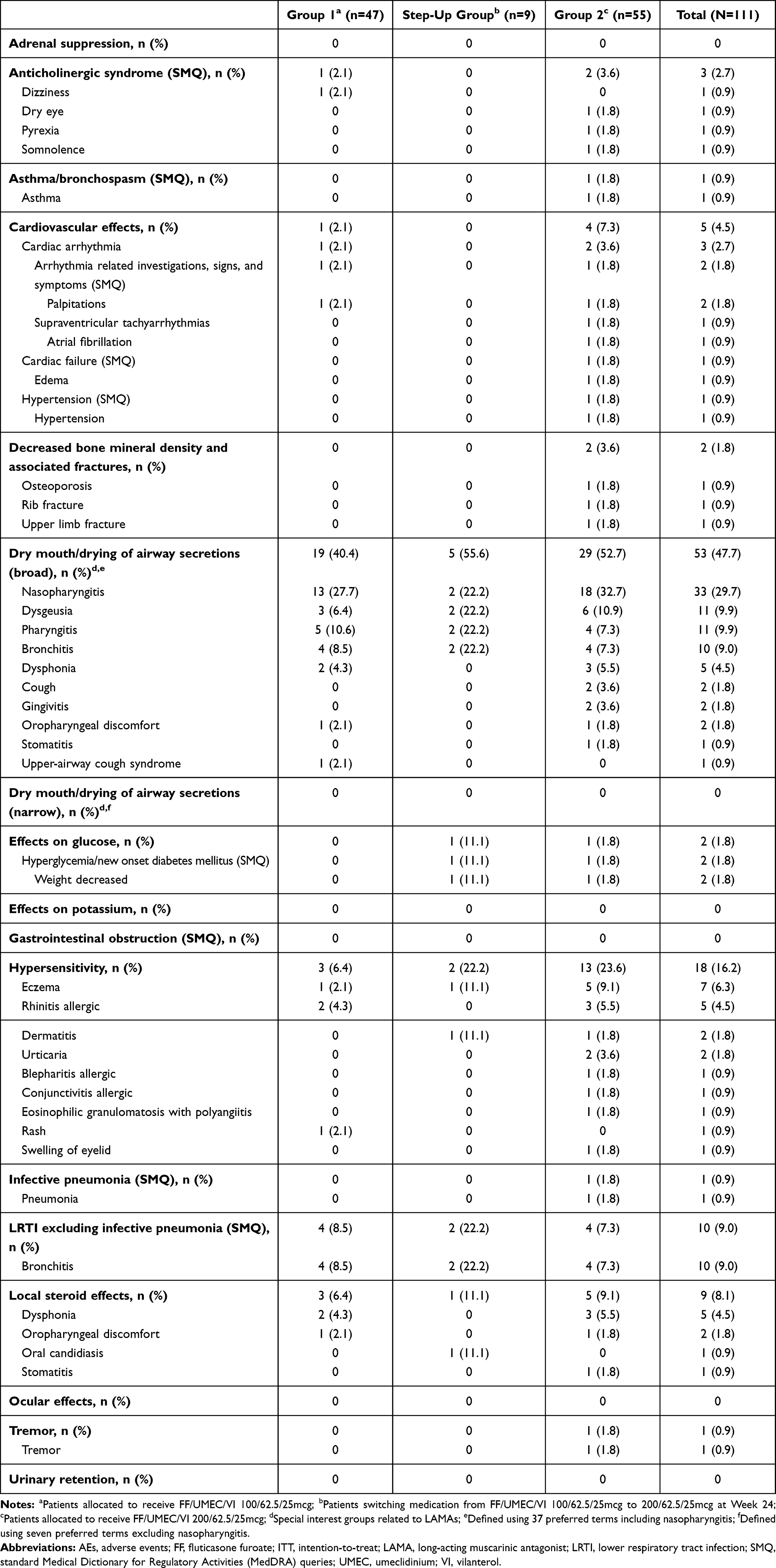 |
Table 5 Summary of On-Treatment AEs of Special Interest (ITT Population) |
No new safety concerns were identified from additional safety evaluations throughout the 52-week treatment period in any of the dose groups. Mean changes from baseline in vital signs were minimal across the total study population (systolic blood pressure: +2.0 mmHg; diastolic blood pressure +2.3 mmHg; pulse rate +0.6 beats/min). There were no clinically significant abnormal findings on the ECG. Few patients had a shift from baseline to high or low value relative to the normal range for each parameter of hematology and clinical chemistry assessed (Supplementary Table 1). The most frequently reported abnormal clinical chemistry parameter was glucose, with a shift from baseline to high in 25% of patients in the total study population (Supplementary Table 1).
“Other” Endpoints
Exploratory efficacy data revealed mean (SD) changes from baseline across the total study population in trough FEV1 (Week 24: 166mL [303]; Week 52: 165mL [294]); ACQ-7 total score (Week 24: −0.51 [0.72]; Week 52: −0.63 [0.68]; minimum clinically important difference [MCID]: −0.5); SGRQ total score (Week 24: −8.27 [11.74]; Week 52: −10.07 [11.73]; MCID: −4.0); AQLQ total score at (Week 24: 0.42 [0.69]; Week 52: 0.55 [0.67]; MCID: 0.5) (Supplementary Table 2). Overall, few patients experienced moderate/severe or severe asthma exacerbations (n=19 [17%] and n=14 [13%], respectively), and corresponding annualized rates were also low (0.41 and 0.30, respectively).
Discussion
This Phase III trial was the first study to evaluate the use of UMEC exclusively in a Japanese asthma population receiving maintenance therapy with ICS/LABA, with or without LAMA. The recently published pivotal Phase IIIA CAPTAIN study17 compared the efficacy and safety of once-daily FF/UMEC/VI with FF/VI in patients with asthma inadequately controlled on ICS/LABA, with approximately 10% of the patient population being of Japanese origin.18 However, compared with Japanese patients in the CAPTAIN study,18 the patients in our study had better disease control, as shown by lower pre-study ACQ-6 mean (SD) scores (1.36 [0.54] vs 1.61 [0.68]) and better lung function, as shown by higher mean (SD) percent predicted pre-bronchodilator FEV1, mean (SD) (86.4 [16.6] vs 73.4 [14.3]). These findings may be related to differences in eligibility criteria between the two studies.
No new safety concerns were observed with long-term (52 weeks) treatment with once-daily FF/UMEC/VI 100/62.5/25mcg or 200/62.5/25mcg, nor among the small group of patients who stepped up treatment at Week 24. Almost all AEs were reported as mild or moderate in their maximum severity, and only 3 SAEs were reported, all of which were not drug related according to the study investigators. No clinically significant findings or abnormalities in clinical laboratory, vital signs, or ECG assessments were observed. AEs leading to treatment discontinuation and SAEs were rare across all groups. The incidence of nasopharyngitis, the most common AE, was also similar across groups. It is important to note that this study was not powered to detect differences in AE rates between treatment groups. Furthermore, the lack of a control group means that it is not possible to conclude as to whether AE rates were higher in patients in this study, who were all treated with FF/UMEC/VI, compared with those on other treatments. The incidence and type of on-treatment AEs or treatment-related AEs were similar to observations from previous studies with FF, VI, UMEC, or their combination treatments in patients with asthma or COPD.
The frequency of dysgeusia (and drug-related events of dysgeusia) reported in this study (n=11 [9.9%]) was higher than reported in CAPTAIN (n=3 [0.1%]).17 Importantly, all cases of dysgeusia were mild and all events were recovered/resolved at the end of follow-up, with no treatment discontinuation. It should be noted that the open-label design of this study may have contributed to increased reporting of some AEs in patients receiving FF/UMEC/VI, and furthermore some events were reported at only a few study sites. Infective pneumonia was reported for 1 patient in this study (0.9%; N=111) and 36 patients (1.5%; N=2436) in the CAPTAIN study. These very low numbers of pneumonia events and the open-label design of this study mean it is difficult to draw comparisons with the CAPTAIN study.
There were no clinically significant changes from baseline in any parameters of clinical laboratory evaluations in this study. As seen in CAPTAIN,17 the most frequently reported clinical chemistry parameter with a shift from baseline to outside the normal range post baseline (worst case post baseline) was glucose in the total study population. However, this may be explained by blood sampling under non-fasting conditions, as permitted in the study. No AEs relevant to glucose increase were reported.
The safety profile of FF/UMEC/VI in Japanese patients with asthma in this study appears to be in line with previous clinical studies of FF/UMEC/VI, VI-containing dual therapies (FF/VI or UMEC/VI), or UMEC monotherapy in Japanese patients with COPD.20,21 Furthermore, the safety profile of FF/UMEC/VI in this study was similar to the safety profile reported for the overall CAPTAIN study population17 and the Japanese cohort.18
This study was neither powered nor designed to assess efficacy endpoints including pulmonary function. However, exploratory assessments revealed improvements in lung function and patient reported outcomes over 52 weeks versus baseline. A MCID for FEV1 has not been formally defined in asthma; however, a previous Phase III study of patients with persistent asthma showed that compared with placebo, FF 100mcg and FF/VI 100/25mcg improved trough FEV1 by 136mL and 172mL, respectively.22 In addition, changes in patient-reported outcome measures exceeded the respective MCIDs at Week 52. Whilst we were unable to compare against a control in our study, these data support efficacy observations seen in the Japanese cohort of the CAPTAIN study.18 Few patients experienced moderate or moderate/severe exacerbations and mean annualized rate of moderate/severe exacerbations were low, in line with observations in the Japanese subpopulation of CAPTAIN.18
The strengths of the study include its long duration, which allows assessment of the safety of FF/UMEC/VI over a 52-week period. Whilst the study focuses on the Japanese population, the eligibility criteria were as permissive as possible to more broadly reflect the asthma population seen in real-world clinical practice compared with randomized controlled trials that have more stringent eligibility criteria. Limitations include its open-label and non-randomized design with no control group and the relatively small population size (N=111), and thus the study was not designed to assess efficacy.
Conclusion
In conclusion, no new safety concerns were associated with long-term (52 weeks) treatment with once-daily FF/UMEC/VI 100/62.5/25mcg or 200/62.5/25mcg, nor among patients stepping up treatment in Japanese patients with asthma. These findings support the positive risk/benefit profile of FF/UMEC/VI in Japanese patients as observed in the pivotal CAPTAIN study.17
Abbreviations
ACQ, Asthma Control Questionnaire; AE, adverse event; AESI, adverse event of special interest; AQLQ, Asthma Quality of Life Questionnaire; CAPTAIN, Clinical Study in Asthma Patients Receiving Triple Therapy in a Single Inhaler; COPD, chronic obstructive pulmonary disease; DPI, dry-powder inhaler; ECG, electrocardiogram; FEV1, forced expiratory volume in 1 second; FF, fluticasone furoate; GINA, Global Initiative for Asthma; ICS, inhaled corticosteroid; ITT, intention-to-treat; LABA, long-acting β2-agonist; LAMA, long-acting muscarinic antagonist; LRTI, lower respiratory tract infection; LTRA, leukotriene receptor antagonist; LS, least squares; MCID, minimum clinically important difference; MedDRA, Medical Dictionary for Regulatory Activities; OCS, oral corticosteroids; SABA, short-acting β2-agonist; SAE, serious adverse event; SD, standard deviation; SMQ, standard Medical Dictionary for Regulatory Activities queries; SGRQ, St George’s Respiratory Questionnaire; UMEC, umeclidinium; VI, vilanterol.
Data Sharing Statement
Anonymized individual participant data and study documents can be requested for further research from www.clinicalstudydatarequest.com.
Acknowledgments
GSK, Collegeville, PA, USA was Laurie A Lee's affiliation at time of study. This study was funded by GlaxoSmithKline (GSK 207236, NCT03184987). GSK was involved at all stages of the study, including study design, data collection, analysis and interpretation, and preparation of the report. Editorial support in the form of preparation of the first draft based on input from all authors, and collation and incorporation of author feedback to develop subsequent drafts was provided by Anne Errichelli, DPhil, and Alexandra Berry, MSc, of Fishawack Indicia Ltd., UK, part of Fishawack Health and was funded by GSK. ELLIPTA is owned by or licensed to the GSK group of companies.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis or interpretation; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
SH has received honoraria from Astellas Pharma, GSK, Novartis Pharma, AstraZeneca, Kyorin Pharmaceutical, and Sanofi. HO has received honoraria from Kyorin Pharmaceutical, and Mylan EPD GK. MT has no conflicts of interest to disclose. YH has received honoraria from GSK, Novartis Pharma, and AstraZeneca. LAL was an employee of GSK at the time of the study and holds stocks or shares in GSK. TNa and AF are employees of GSK and hold stocks or shares in GSK. TNi and JT are employees of GSK. The authors report no other conflicts of interest in this work.
References
1. Global Initiative for Asthma (GINA). Global strategy for asthma management and prevention; 2020. Available from: https://ginasthma.org/wp-content/uploads/2020/06/GINA-2020-report_20_06_04-1-wms.pdf.
2. Nakamura Y, Tamaoki J, Nagase H, et al. Japanese guidelines for adult asthma 2020. Allergol Int. 2020;69(4):519–548. doi:10.1016/j.alit.2020.08.001
3. Sulaiman I, Greene G, MacHale E, et al. A randomised clinical trial of feedback on inhaler adherence and technique in patients with severe uncontrolled asthma. Eur Respir J. 2018;51(1):1701126. doi:10.1183/13993003.01126-2017
4. Bernstein DI, Bateman ED, Woodcock A, et al. Fluticasone furoate (FF)/vilanterol (100/25 mcg or 200/25 mcg) or FF (100 mcg) in persistent asthma. J Asthma. 2015;52(10):1073–1083. doi:10.3109/02770903.2015.1056350
5. Davis J, Trudo F, Siddall J, Small M. Burden of asthma among patients adherent to ICS/LABA: a real-world study. J Asthma. 2019;56(3):332–340. doi:10.1080/02770903.2018.1455858
6. Lee LK, Obi E, Paknis B, Kavati A, Chipps B. Asthma control and disease burden in patients with asthma and allergic comorbidities. J Asthma. 2018;55(2):208–219. doi:10.1080/02770903.2017.1316394
7. Adachi M, Hozawa S, Nishikawa M, Yoshida A, Jinnai T, Tamura G. Asthma control and quality of life in a real-life setting: a cross-sectional study of adult asthma patients in Japan (ACQUIRE-2). J Asthma. 2019;56(9):1016–1025. doi:10.1080/02770903.2018.1514628
8. Nagase H, Adachi M, Matsunaga K, et al. Prevalence, disease burden, and treatment reality of patients with severe, uncontrolled asthma in Japan. Allergol Int. 2020;69(1):53–60. doi:10.1016/j.alit.2019.06.003
9. Kerstjens HA, Engel M, Dahl R, et al. Tiotropium in asthma poorly controlled with standard combination therapy. N Engl J Med. 2012;367(13):1198–1207. doi:10.1056/NEJMoa1208606
10. Kew KM, Dahri K. Long-acting muscarinic antagonists (LAMA) added to combination long-acting beta2-agonists and inhaled corticosteroids (LABA/ICS) versus LABA/ICS for adults with asthma. Cochrane Database Syst Rev. 2016;1:CD011721. doi:10.1002/14651858.CD011721.pub2
11. O’Byrne PM, Jacques L, Goldfrad C, et al. Integrated safety and efficacy analysis of once-daily fluticasone furoate for the treatment of asthma. Respir Res. 2016;17(1):157. doi:10.1186/s12931-016-0473-x
12. Lötvall J, Bateman ED, Bleecker ER, et al. 24-h duration of the novel LABA vilanterol trifenatate in asthma patients treated with inhaled corticosteroids. Eur Respir J. 2012;40(3):570–579. doi:10.1183/09031936.00121411
13. Devillier P, Humbert M, Boye A, et al. Efficacy and safety of once-daily fluticasone furoate/vilanterol (FF/VI) versus twice-daily inhaled corticosteroids/long-acting beta2-agonists (ICS/LABA) in patients with uncontrolled asthma: an open-label, randomized, controlled trial. Respir Med. 2018;141:111–120. doi:10.1016/j.rmed.2018.06.009
14. Lee LA, Briggs A, Edwards LD, Yang S, Pascoe S. A randomized, three-period crossover study of umeclidinium as monotherapy in adult patients with asthma. Respir Med. 2015;109(1):63–73. doi:10.1016/j.rmed.2014.10.009
15. Kerwin E, Pascoe S, Bailes Z, et al. A phase IIb, randomised, parallel-group study: the efficacy, safety and tolerability of once-daily umeclidinium in patients with asthma receiving inhaled corticosteroids. Respir Res. 2020;21(1):148. doi:10.1186/s12931-020-01400-5
16. Food and Drug Administration (FDA). TRELEGY ELLIPTA prescribing information; 2020. Available from: https://www.gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Trelegy/pdf/TRELEGY-ELLIPTA-PI-PIL-IFU.PDF.
17. Lee LA, Bailes Z, Barnes N, et al. Efficacy and safety of once-daily single-inhaler triple therapy (FF/UMEC/VI) versus FF/VI in patients with inadequately controlled asthma (CAPTAIN): a double-blind, randomised, phase 3A trial. Lancet Respir Med. 2021;9(1):69–84. doi:10.1016/S2213-2600(20)30389-1
18. Nakamura N, Hozawa S, Sagara H, et al. Efficacy and safety of once-daily, single-inhaler fluticasone furoate/umeclidinium/vilanterol (FF/UMEC/VI) versus FF/VI in Japanese patients with inadequately controlled asthma: the CAPTAIN study.
19. Global Initiative for Chronic Obstructive Lung Disease (GOLD COPD). Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease; 2020. Available from: https://goldcopd.org/wp-content/uploads/2019/12/GOLD-2020-FINAL-ver1.2-03Dec19_WMV.pdf.
20. Yamagata E, Soutome T, Hashimoto K, Mihara K, Tohda Y. Long-term (52 weeks) safety and tolerability of umeclidinium in Japanese patients with chronic obstructive pulmonary disease. Curr Med Res Opin. 2016;32(5):967–973. doi:10.1185/03007995.2016.1140029
21. Kato M, Tomii K, Hashimoto K, et al. The IMPACT Study - single inhaler triple therapy (FF/UMEC/VI) versus FF/VI and UMEC/VI in patients with COPD: efficacy and safety in a Japanese Population. Int J Chron Obstruct Pulmon Dis. 2019;14:2849–2861. doi:10.2147/COPD.S226601
22. Bleecker ER, Lötvall J, O’Byrne PM, et al. Fluticasone furoate-vilanterol 100-25 mcg compared with fluticasone furoate 100 mcg in asthma: a randomized trial. J Allergy Clin Immunol Pract. 2014;2(5):553–561. doi:10.1016/j.jaip.2014.02.010
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.