")
Back to Journals » Clinical Ophthalmology » Volume 13
Safety of cetirizine ophthalmic solution 0.24% for the treatment of allergic conjunctivitis in adult and pediatric subjects
Authors Malhotra RP, Meier E , Torkildsen G , Gomes PJ, Jasek MC
Received 1 September 2018
Accepted for publication 12 December 2018
Published 19 February 2019 Volume 2019:13 Pages 403—413
DOI https://doi.org/10.2147/OPTH.S186092
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Ranjan P Malhotra,1 Edward Meier,2 Gail Torkildsen,3 Paul J Gomes,4 Mark C Jasek5
1Ophthalmology Associates, St Louis, MO, USA; 2Apex Eye, Mason, OH, USA; 3Andover Eye, Andover, MA, USA; 4Ora, Inc., Andover, MA, USA; 5Eyevance Pharmaceuticals, Fort Worth, TX, USA
Purpose: The studies reported here aimed to assess the safety and tolerability of cetirizine ophthalmic solution 0.24%, a new topical ophthalmic medication approved by the US Food and Drug Administration for the treatment of ocular itching associated with allergic conjunctivitis.
Patients and methods: Three clinical studies evaluated cetirizine ophthalmic solution 0.24% administration: a Phase I prospective, single-center, open-label, pharmacokinetic (PK) study (N=11) evaluating single-dose administration and twice-daily (BID) administration for 1 week in healthy adults, and two Phase III, multi-center, randomized, double-masked, vehicle-controlled, parallel-group studies evaluating the safety and tolerability in adult and pediatric populations (2–18 years of age) for up to 6 consecutive weeks. The first safety and tolerability study evaluated cetirizine BID (study 1, N=512), while the second study examined cetirizine three times daily (TID) (study 2, N=516). Each study assessed best corrected visual acuity, slit-lamp biomicroscopy, IOP, dilated ophthalmoscopy, treatment-emergent adverse events, vital signs, urine pregnancy test, and physical examination (general health, head, eyes, ears, nose, and throat). The PK study also measured hematology, blood chemistry, and urinalysis, while the two Phase III studies additionally assessed corneal endothelial cell counts (ECC) and ECC density in a subset of subjects (via specular microscopy), and drug administration tolerability.
Results: Bilateral administration of cetirizine ophthalmic solution 0.24% resulted in low systemic exposure in the PK study and was associated with a low incidence of mild adverse events. There were no drug-related severe or serious adverse events. The tolerability scores between the active and vehicle groups were comparable, demonstrating high comfort in the administration of cetirizine ophthalmic solution 0.24%.
Conclusion: Cetirizine ophthalmic solution 0.24% dosed BID or TID demonstrated an acceptable safety profile and was well-tolerated when administered to subjects aged ≥2 years.
Keywords: ocular allergy, antihistamine, ocular itching, tolerability, topical administration
Introduction
Allergies affect ~30% of populations in industrialized countries.1 Seasonal allergic conjunctivitis is the most prevalent allergic condition and the cause of approximately one-third of all allergic disorders. The key ocular symptom is ocular itching. Other troublesome ocular sequelae include conjunctival, episcleral and ciliary hyperemia, eyelid swelling, chemosis, mucous discharge, and tearing; the set of accompanying sequelae varying among the afflicted.2,3 The causative agent of allergies include perennial allergens (such as cat dander, cockroach, dog dander, and dust mite) and seasonal allergens (such as Kentucky bluegrass, maple, meadow fescue, oak, ragweed, rye grass, timothy grass, and white birch). In susceptible individuals, these common allergens, and other environmental insults, trigger a well-characterized allergic response: the multifactorial type 1 hypersensitivity reaction characterized by an early, acute phase activated by mast cell degranulation and release of histamine, and a late phase induced by pro-inflammatory mediators (ie, prostaglandins, leukotrienes, cytokines, and ILs).4
Given that allergic reactions are driven by histamine, the therapeutic market has yielded several efficacious systemic antihistamine products. Cetirizine hydrochloride is an antihistamine with a well-characterized systemic safety and efficacy profile due to its use for over 20 years. The anti-allergic efficacy of cetirizine has been documented in various animal and human models.5,6 In vitro receptor binding studies have shown no measurable affinity for histamine receptors other than H1 and negligible induction of anticholinergic and anti-serotonergic activity, supporting an on-target product with minimal adaptive immune response. As a result, cetirizine has been approved for use in the United States since 1995 and has since been adapted to various formulations.
Individual patient response to antihistamines can be highly variable,7 and the efficacy for which systemic antihistamines are valued, including the relief of ocular allergy, is frequently accompanied by ocular adverse events (AEs) such as pupillary changes, blurred vision, and dry eye syndrome.8,9 For example, oral cetirizine in patients with normal ocular health has been shown to induce signs and symptoms associated with ocular dryness, including corneal and conjunctival staining, decreased tear film break-up time, and increased ocular discomfort when exposed to a controlled adverse environment (CAE®).9 Cetirizine ophthalmic solution 0.24% was developed to minimize systemic effects by using a concentration of cetirizine hydrochloride specifically designed for targeted and comfortable ocular administration.
Here we report the results from one pharmacokinetic (PK) and two safety and tolerability studies conducted as part of the drug development program of cetirizine ophthalmic solution 0.24% (Zerviate®). Cetirizine ophthalmic solution 0.24% used in these studies was a sterile, buffered, clear, colorless aqueous solution containing cetirizine 0.24% (equivalent to cetirizine hydrochloride 0.29%) and 0.010% benzalkonium chloride (preservative).10 The vehicle used in these studies was identical to cetirizine ophthalmic solution 0.24% except it did not contain the active drug. Cetirizine ophthalmic solution 0.24% demonstrated broad safety and tolerability along with compelling efficacy (Meier et al, 2018, co-submitted) in the relief of ocular itching associated with allergic conjunctivitis in pediatric (2–18 years of age) and adult subjects.
Patients and methods
Study design
PK study
The PK study was a prospective, single-center, open-label study designed to characterize the plasma PK and safety profile of cetirizine ophthalmic solution 0.24% after a single bilateral dose (ie, one drop in each eye) and after twice-a-day (BID) bilateral dosing for 1 week in healthy, adult subjects. The study comprised five study visits over a period of ~3 weeks. After screening, subjects received one dose of cetirizine ophthalmic solution 0.24% or vehicle and were queried for safety and tolerability. At visit 3, subjects (or their caregiver) instilled one dose under the observation of a trained staff technician. Thereafter, subjects (or their caregiver) administered cetirizine BID for up to 6 days; instillation of the second daily dose was to be administered ~8 hours but no earlier than 7 hours after the first dose of the day. The final dose at visit 4 was instilled by a trained staff technician.
Phase III safety evaluations
The two Phase III, multi-center, randomized, double-masked, vehicle-controlled, parallel-group long-term safety and tolerability studies evaluated the safety and tolerability of BID (study 1) or three times daily (TID, study 2) dosing in adult and pediatric (≥2 to ≤18 years of age) populations for up to 6 weeks. Subjects were randomized (2:1) to receive one bilateral dose of either cetirizine or vehicle, respectively. Each study comprised four study visits over a period of ~6 weeks. A subset of subjects underwent specular microscopy to determine central corneal endothelial cell count (ECC) at the fifth study visit, extending the study length to 12 weeks.
At visit 1, one dose of cetirizine or vehicle was instilled. Thereafter, instillation was performed BID or TID for 6 weeks (with doses instilled by a trained staff technician at visits 2 and 3). In the BID study, the second daily dose was to be instilled ~8 hours, but no earlier than 7 hours, after the first dose of the day. In the TID study, doses were to be instilled approximately every 6 hours within 1 calendar day, but no earlier than 5 hours after the previous dose. The BID and TID studies were conducted at four and six investigative sites in the United States, respectively. All three studies complied with the Declaration of Helsinki 2013 and Good Clinical Practice E6 (R1) guidelines; all three studies were reviewed and approved by an external institutional review board (IRB; Alpha IRB, San Clemente, CA, USA).
Subjects
All three studies were performed in compliance with the Declaration of Helsinki, International Conference of Harmonization, Good Clinical Practice Guidance, and all applicable local, state, and federal requirements. Each subject or their legally acceptable representative provided informed written informed consent/assent (for subjects under the age of 18 years) prior to any study-related procedures. The study protocols, informed consent forms, HIPPAA forms, and related documents were approved by the IRB (Alpha IRB).
The PK study (N=11) enrolled healthy adult subjects of either sex ≥18 and ≤55 years of age with ocular health within normal limits and a best corrected visual acuity (BCVA) of 0.6 logarithm of minimum angle of resolution (logMAR) or better in each eye as measured using an Early Treatment Diabetic Retinopathy Study (ETDRS) chart. No subjects had any active systemic or ocular disorder other than refractive disorder; any abnormality of the lids, ocular surface, or lacrimal duct system that, in the opinion of the investigator, could have affected ophthalmic drop absorption.
The Phase III studies enrolled healthy adult and pediatric subjects (≥2 to ≤18 years of age) with a history or family history of atopic disease, including allergic conjunctivitis. In both the studies, the primary inclusion criteria were as follows: the ability to self-administer eye drops or have a caregiver, legal guardian, or school healthcare provider (eg, nurse) do so, and a calculated visual acuity of 0.3 logMAR or better in each eye as measured using an ETDRS chart. In subjects aged <10 years who were developmentally unable to use the ETDRS chart, a best attempt at visual acuity was made using the LEA symbols or visual behavior. Subject participating in the ECC evaluation was required to have a baseline corneal ECC ≥2,200/cells/mm2.
Both the studies excluded females who were currently pregnant, currently breast feeding or planning to breast feed during the study period; subjects with ongoing active ocular infection; current or historical ocular morbidity, recent ocular surgery; and subjects with any ocular condition that, in the opinion of the investigator, could have affected the subjects’ safety or trial parameters. Subjects were disallowed any topical ophthalmic agents or contact lens use for 5 days prior to visit 1, and any systemic corticosteroids, cancer chemotherapy, or systemic medications that the investigator felt may have confounded study data or interfered with the subjects’ participation within 14 days prior to visit 1.
Assessments
The PK study evaluated dilated ophthalmoscopy, IOP, physical examination, hematology, blood chemistry, and urinalysis at visit 1 (post single exposure) and visit 5 (post 1 week continuous BID exposure). BCVA, slit lamp biomicroscopy (SLE), and vital signs were evaluated at visit 1 (screening), and pre- and post-instillation at visits 2, 4, and 5. AEs were recorded at visits 2 through 5. Blood samples for PK measurements were collected 1 hour prior to study drug instillation, and post-instillation of a single dose at 15±3 minutes, 30±5 minutes, 1 hour ± 10 minutes, 1.5 hours ± 10 minutes, 2 hours ± 10 minutes, 3 hours ± 15 minutes, 4 hours ± 20 minutes, 6 hours ± 20 minutes, 8 hours ± 20 minutes, 12 hours ± 60 minutes, 18 hours ± 60 minutes, and 24 hours ± 60 minutes. PK parameters included maximum observed plasma concentration (Cmax), measured in ng/mL, area under the plasma concentration–time curve (AUC), time to peak plasma concentration (Tmax), and elimination half-life (T1/2), the latter two measured in hours.
In the Phase III studies, the safety parameters (IOP, dilated ophthalmoscopy, ECC evaluations among participating subjects, physical examination, and vital signs) were evaluated at visits 1, 4, and 5. BCVA, SLE, and AE queries were performed at every study visit. The Phase III studies also evaluated tolerability (ie, drop comfort) at visits 1, 2, and 3. Drop comfort was self-assessed by subjects immediately upon instillation, 30 seconds post-dose, and 1 minute post-dose by using a numerical scale ranging from 0 to 10, where 0= very comfortable and 10= very uncomfortable. For each subject, the average score between eyes at each time point was used for analysis.11
Statistical analyses
In all the three studies, analyses were conducted with all randomized subjects who received at least one dose of medication; analyzed as treated with no data excluded for any reason. Safety was assessed by change from baseline (the last assessment prior to the first instillation). PK, comfort, and quantitative safety analyses were summarized using descriptive statistics with the data for BCVA, SLE, dilated ophthalmoscopy, IOP, ECC, ECC density, and drop comfort, stratified by age and treatment group. BCVA and IOP were summarized by categories quantified by degree of change from baseline. Physical examination and pregnancy test results were summarized descriptively. The qualitative safety variables were summarized using counts and percentages. Drop comfort was analyzed using Fisher’s exact test or chi-squared test as appropriate. P-values for safety, drop comfort, and age group comparisons were calculated using a two-sample t-test comparing changes from baseline in the cetirizine group to the vehicle group with an α equal to 0.05. For IOP, a chi-squared test was utilized to compare the active treatment group with the vehicle treatment group for subjects categorized as having changes from baseline pressures ≥10 mmHg. Hypothesis testing was not performed in the long-term safety studies.
Results
Subject disposition and demographics
Subject disposition
In the PK study, eleven subjects were enrolled and ten subjects (90.9%) completed the study. One subject (9.1%) was discontinued from the study prior to completion because of a lack of venous access. In the BID study (study 1), the study population comprised 512 subjects (123 pediatric): 341 subjects received cetirizine and 171 subjects received vehicle. Of the 512 randomized subjects, 488 subjects (95.3%) completed the study and 24 subjects (4.7%) were discontinued from the study prior to completion. Two discontinuations in the cetirizine group were due to AEs. In the TID study, the study population comprised 516 subjects (128 pediatric): 343 subjects received cetirizine and 173 subjects received vehicle. Of the 516 subjects enrolled in the study, 493 subjects (95.5%) completed the study and 23 subjects (4.5%) were discontinued from the study prior to completion. Four discontinuations were due to AEs in the cetirizine treatment group.
Subject demographics
In the PK study, the study population had a mean age of 25.3 (±6.62) years with a range from 19 to 38 years, slightly more female (54.5%) than male subjects, and a majority of subjects who were ethnically not Hispanic or Latino (81.8%). Racially, white subjects predominated (72.7%) with two subjects classified as other (18.2%), and one subject as Black or African American (9.1%).
Demographics in the BID (study 1) and TID (study 2) were comparable across treatment groups. In the BID study, the mean age was 35.4 (±19.42) years with more females (60.4%) than males. Ethnically, the majority of subjects were non-Hispanic/Latino (93.9%). The majority of subjects were white (87.3%), followed by Black or African American (7.2%) and other (3.5%), with Asian and American Indian or Alaskan Native accounting for 1% each. In the TID study, the study population had a mean age of 32.2 (±17.31) years and also contained more females (57.9%) than males. Ethnically, the majority of the subjects were non-Hispanic/Latino (81.0%). The majority of the subjects were white (76.7%) and Black or African American (16.5%), with the races of unknown, Asian, Multiracial, Native Hawaiian or other Pacific Islander, and American Indian or Alaskan Native accounting for the remaining 6.8% of the population.
Pharmacokinetics
All subjects had detectable systemic cetirizine throughout the 24-hour period following single and multiple dosing (Figure 1). The mean extent of exposure was 7.4 (±2.11) days of BID dosing. After a single bilateral dose of cetirizine ophthalmic solution 0.24%, the mean peak plasma concentration (Cmax) was 1.7 ng/mL and the mean time to maximum concentration (Tmax) was 2.2 hours. After a week of bilateral BID dosing, Cmax of cetirizine increased to 3.1 ng/mL, and Tmax dropped to 1.6 hours. Therefore, a week of BID dosing yielded a Cmax ~1.8 times greater than that of a single dose. The mean AUC(0–24) was proportionally consistent with the Cmax when comparing a single to 1 week dosing period. After a single dose the mean AUC(0–24) was 18.972 (±10.4329) ng·h/mL which increased to 31.688 (±18.3170) ng·h/mL after 1 week of exposure. Total drug exposure over time (AUC(0–∞)) was 22.453 (±12.5063) ng·h/mL which similarly increased to 36.486 (±20.3550) ng·h/mL after 1 week of exposure. The terminal half-life (T1/2) of cetirizine was short. T1/2 after a single dose was 8.6 and 8.2 hours after a week of BID dosing. Overall, these data demonstrate low systemic exposure following bilateral BID dosing of cetirizine 0.24% ophthalmic solution.
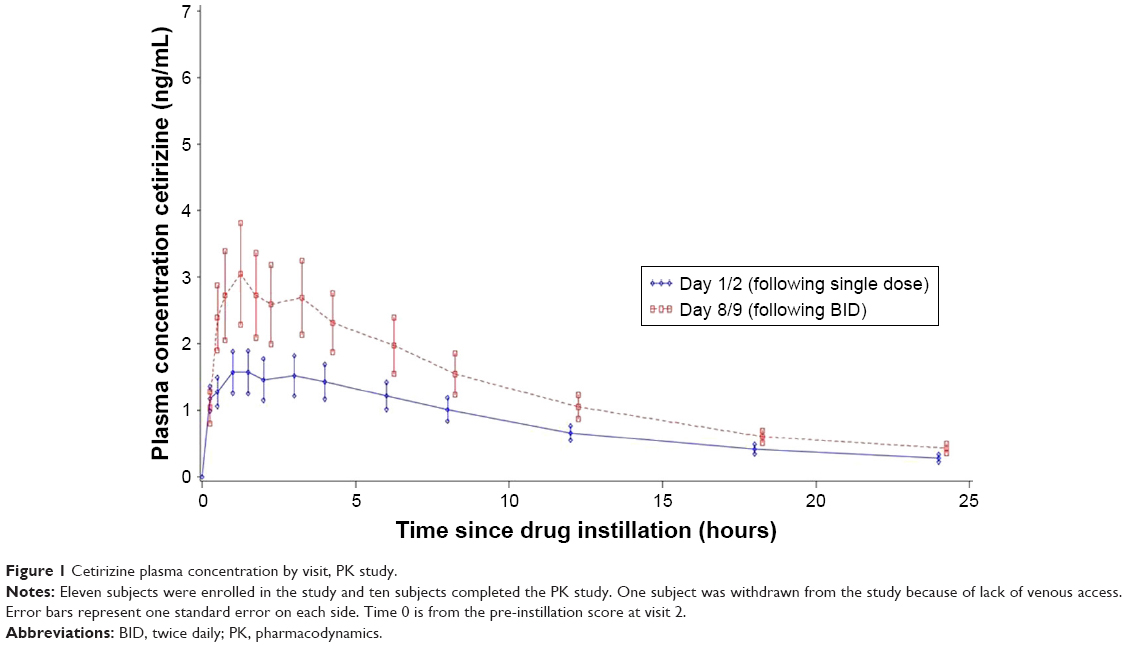 | Figure 1 Cetirizine plasma concentration by visit, PK study. |
Safety
PK Study
No ocular treatment-emergent AEs (TEAEs) occurred during the PK study. Three non-ocular TEAEs were reported by three subjects; none of the non-ocular TEAEs were suspected of being related to cetirizine or indicated any potential safety concern. The non-ocular events were catheter site pain, presyncope (both moderate in intensity and both resolved by study termination), and abnormal liver function test. A shift in abnormal liver function tests in alanine transaminase and aspartate transaminase levels to ~1.5 times the normal upper limit at visit 5 was mild in severity. All other findings in the BCVA, SLE, dilated ophthalmoscopy, IOP, physical examination, and vital signs examinations were normal or not clinically significant, and no clinically relevant changes from baseline were reported in hematology, urinalysis, and the remaining panel of blood chemistry measures (albumin, ALP, bicarbonate, blood urea nitrogen, calcium, chloride, cholesterol, creatinine enzymatic, gamma glutamyl transferase, globulin, glucose, phosphorous, potassium, sodium, bilirubin, protein, triglycerides, and uric acid).
BID study (study 1)
BID dosing study, similar to the PK study, supported an acceptable safety profile for cetirizine ophthalmic solution 0.24%. The mean extent of exposure was 41 days in both the cetirizine and the vehicle groups. No clinically or statistically significant differences in safety parameters were observed between the cetirizine and vehicle groups, with the exception of AEs. AEs were primarily mild. Severe AEs were unrelated to study drug or were resolved at follow-up.
Ocular TEAEs were reported by 121 subjects (23.6%) with 141 ocular TEAE incidences. Events occurring at ≥1% in any treatment group are summarized in Table 1. The prevalence of ocular TEAEs among the cetirizine group (22.9%) was lower than that of vehicle group (25.1%). Of the 91 ocular TEAEs reported by 78 subjects in the cetirizine group, 88 were classified as mild and three moderate (Table 2). The most common ocular TEAEs (≥1%) in the cetirizine group were conjunctival hyperemia, instillation site pain, and ocular hyperemia, collectively accounting for 61 of 91 ocular disorders in the cetirizine group (Table 1). Similarly, TEAEs in the vehicle group ≥1% also included conjunctival hyperemia, instillation site pain, and ocular hyperemia. Blurred vision, dry eye, reduced visual acuity, and eye discharge occurred in up to five subjects from both treatment groups. Although conjunctival hyperemia was the most prevalent TEAE, its prevalence in the cetirizine group was lower than that in the vehicle group (7.9% vs 11.1%, respectively). No incidents of reduced visual acuity were suspected to be related to cetirizine. All other ocular TEAEs occurred in <1% of subjects in both the treatment groups.
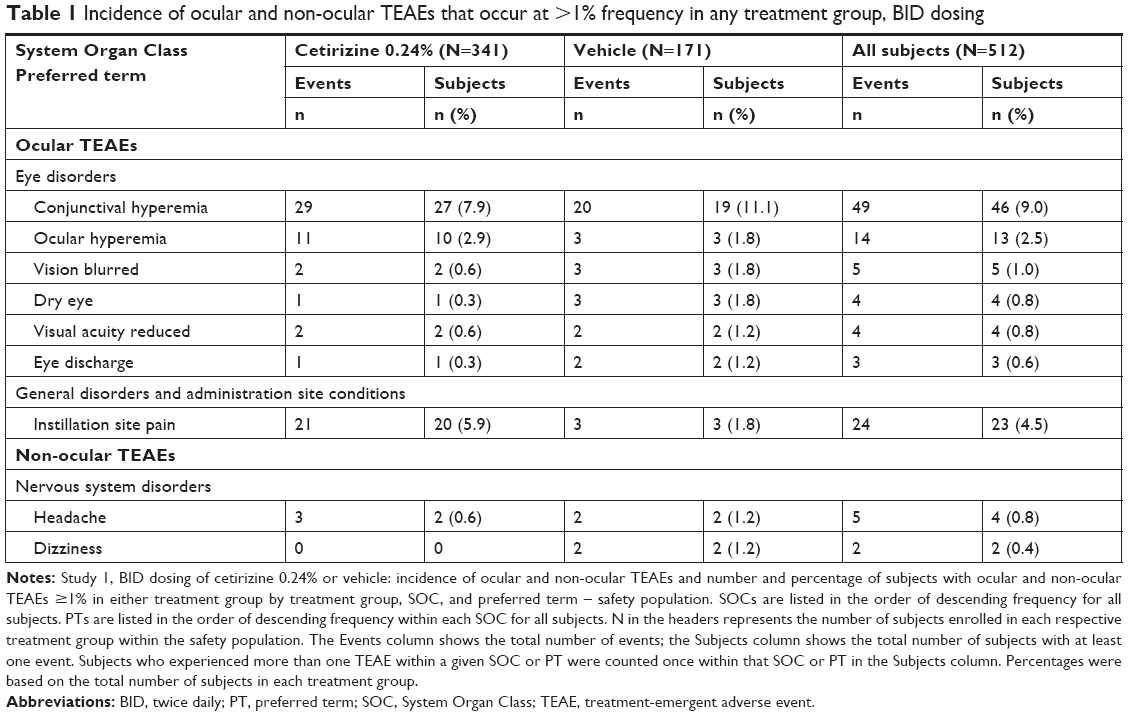 | Table 1 Incidence of ocular and non-ocular TEAEs that occur at >1% frequency in any treatment group, BID dosing |
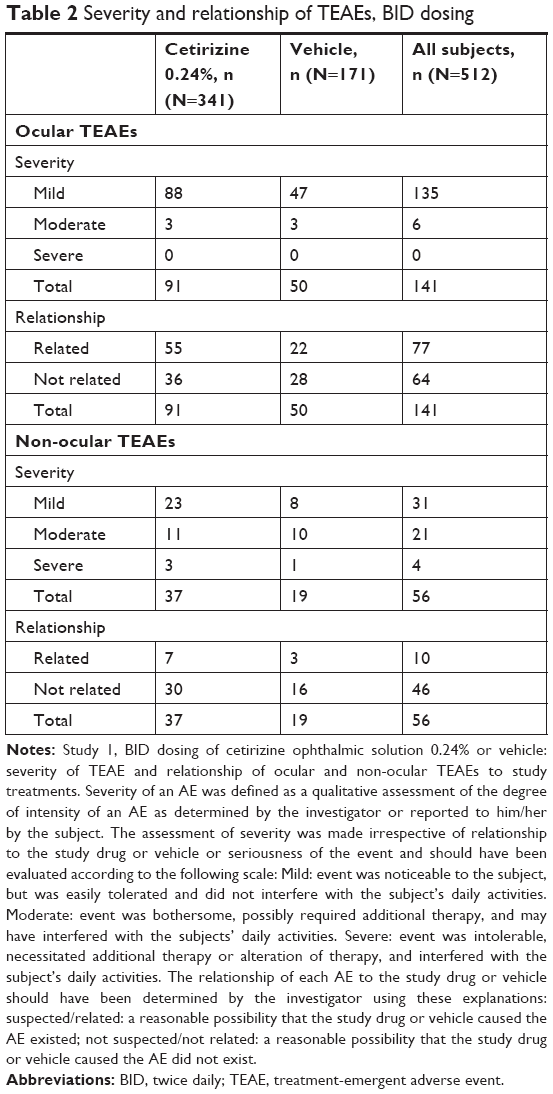 | Table 2 Severity and relationship of TEAEs, BID dosing |
Non-ocular TEAEs that were related to the study drug occurred in <3% of the study population. Overall, 44 subjects (8.6%) reported 56 non-ocular TEAEs. The percentage of subjects with non-ocular TEAEs was comparable between treatment groups (8.5% cetirizine; 8.8% vehicle). Only two TEAEs occurred at a frequency ≥1%: headache and dizziness (Table 1). Of the 37 non-ocular TEAEs reported by 29 subjects in the cetirizine group, 23 non-ocular TEAEs were classified as mild, eleven as moderate, and three as severe (Table 2). These three severe non-ocular TEAEs were trigeminal neuralgia, headache, and herpes zoster. Of severe TEAEs, only headache was suspected to be of related to cetirizine administration.
TID study (study 2)
TID dosing further supported a safe profile for cetirizine ophthalmic solution 0.24%. In the TID study, the extent of exposure was a mean of 41 days in both the groups. As with the BID study, no significant differences in safety measures were observed between the cetirizine and the vehicle groups, except AEs. Overall, 37 subjects (7.2%) reported 45 ocular TEAEs. As with the BID study, the prevalence of ocular TEAEs among subjects in the cetirizine group (6.7%) was lower than in the vehicle group (8.1%). In both the groups, the majority of the TEAEs were classified as mild. No ocular TEAEs of severe intensity or of serious designation were reported. In the cetirizine group, the ocular TEAEs occurring in ≥1% of subjects were reduced visual acuity, conjunctival hyperemia, and instillation site pain (Table 3). In the vehicle group, reduced visual acuity, conjunctival hyperemia, punctate keratitis, and instillation site erythema were all above a frequency of 1% (Table 3). Notably, among the most common ocular TEAEs, only instillation site pain (all classified as mild) had a higher prevalence with cetirizine than with vehicle. All other ocular TEAEs occurred in <1% of subjects in each treatment group.
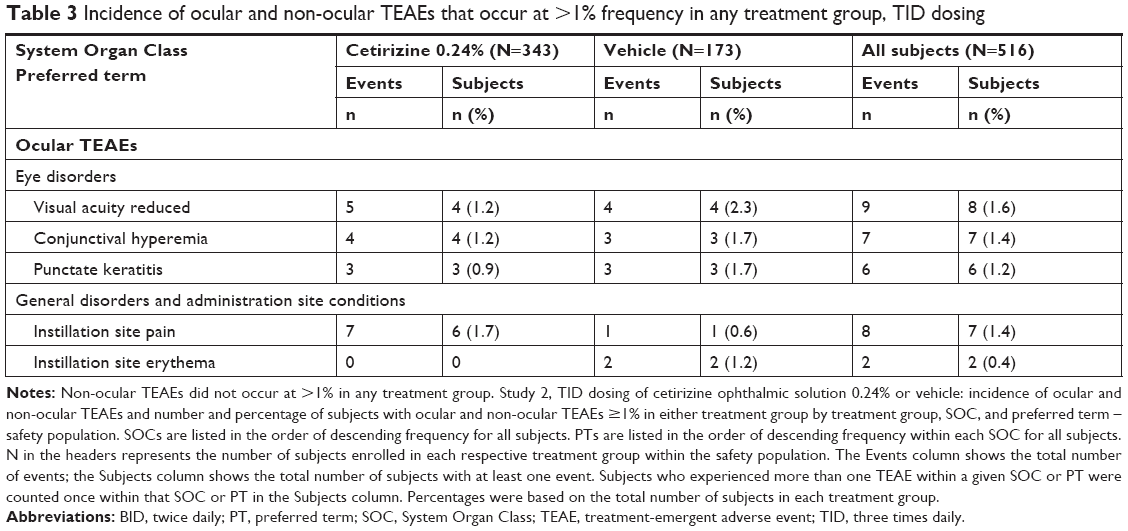 | Table 3 Incidence of ocular and non-ocular TEAEs that occur at >1% frequency in any treatment group, TID dosing |
Related ocular TEAEs were primarily mild. Forty-five ocular TEAEs were reported while only 25 were suspected to be related to treatment: 14 of 28 in the cetirizine group and 11 of 17 in the vehicle group. Overall, the most frequent ocular TEAE was reduced visual acuity, with four cases in each treatment group (1.2% cetirizine; 2.3% vehicle). No incidents of reduced visual acuity were related to study treatment. In both the treatment groups, all incidents of instillation site pain were related to study drug (six cetirizine, 1.7% vs one vehicle, 0.6%). Six incidents of punctate keratitis, three in each treatment group (0.9% cetirizine; 1.7% vehicle), were suspected to be related to treatment. Seven incidents of conjunctival hyperemia were reported, but only one incident in the cetirizine group vs two incidents in the vehicle group were suspected to be related to study drug (0.3% vs 1.2%, respectively). The remaining TEAE that occurred in ≥1% of either population was instillation site erythema: two cases were reported in the vehicle group (1.2%) and both were deemed suspected to be related to the study treatment. All ocular TEAEs resolved during the study period, supporting the safety of cetirizine ophthalmic solution 0.24%.
Twenty-eight incidences of non-ocular TEAEs were reported by 20 subjects. Fifteen subjects in the cetirizine group reported 22 non-ocular TEAEs whereby 19 were of mild and three were of moderate severity. Events of increased severity (ie, migraine) occurred in the vehicle group and were unrelated to the study drug. Six non-ocular TEAEs were related to cetirizine: two incidents each of oropharyngeal pain and headache plus one incident each of pharyngeal hypoesthesia and dry mouth. The non-ocular TEAEs related to cetirizine administration were all mild in intensity. Non-ocular TEAEs did not occur at ≥1% in any treatment group.
Withdrawals due to TEAEs
Six subjects withdrew from the BID study due to TEAEs. Four of these subjects withdrew because of non-ocular TEAEs (pregnancy, anaphylactic shock, gastroenteritis viral, and headache) and one subject withdrew because of an ocular TEAE (viral conjunctivitis of moderate severity). None of these TEAEs were related to cetirizine or vehicle treatment. The remaining subject was in the vehicle group and withdrew due to a combination of three related TEAEs: eye discharge, ocular hyperemia, instillation site pain; and one unrelated TEAE: pain in jaw.
In the TID study, five subjects withdrew from the study due to TEAEs. Two of these TEAEs were ocular. One subject in the cetirizine group withdrew due to an unrelated incidence of allergic conjunctivitis. A second subject, in the vehicle group, withdrew due to instillation site pain of moderate intensity. The three non-ocular TEAEs responsible for subject withdrawal occurred in the cetirizine group and were a mild study drug-related headache, an unrelated mild incidence of throat irritation, and an unrelated incidence of moderately intense substance abuse (an SAE).
Pediatric TEAEs
There were no safety concerns in the pediatric population dosed BID with cetirizine or vehicle. In the pediatric population of the BID study, TEAEs were reported by only one subject aged 2–6 years, eight subjects aged 7–12 years, and one subject aged 13–17 years (Table 4). The majority of incidences were mild in intensity and largely unrelated to study drug. One incidence of conjunctival hyperemia occurred with moderate intensity in the vehicle group.
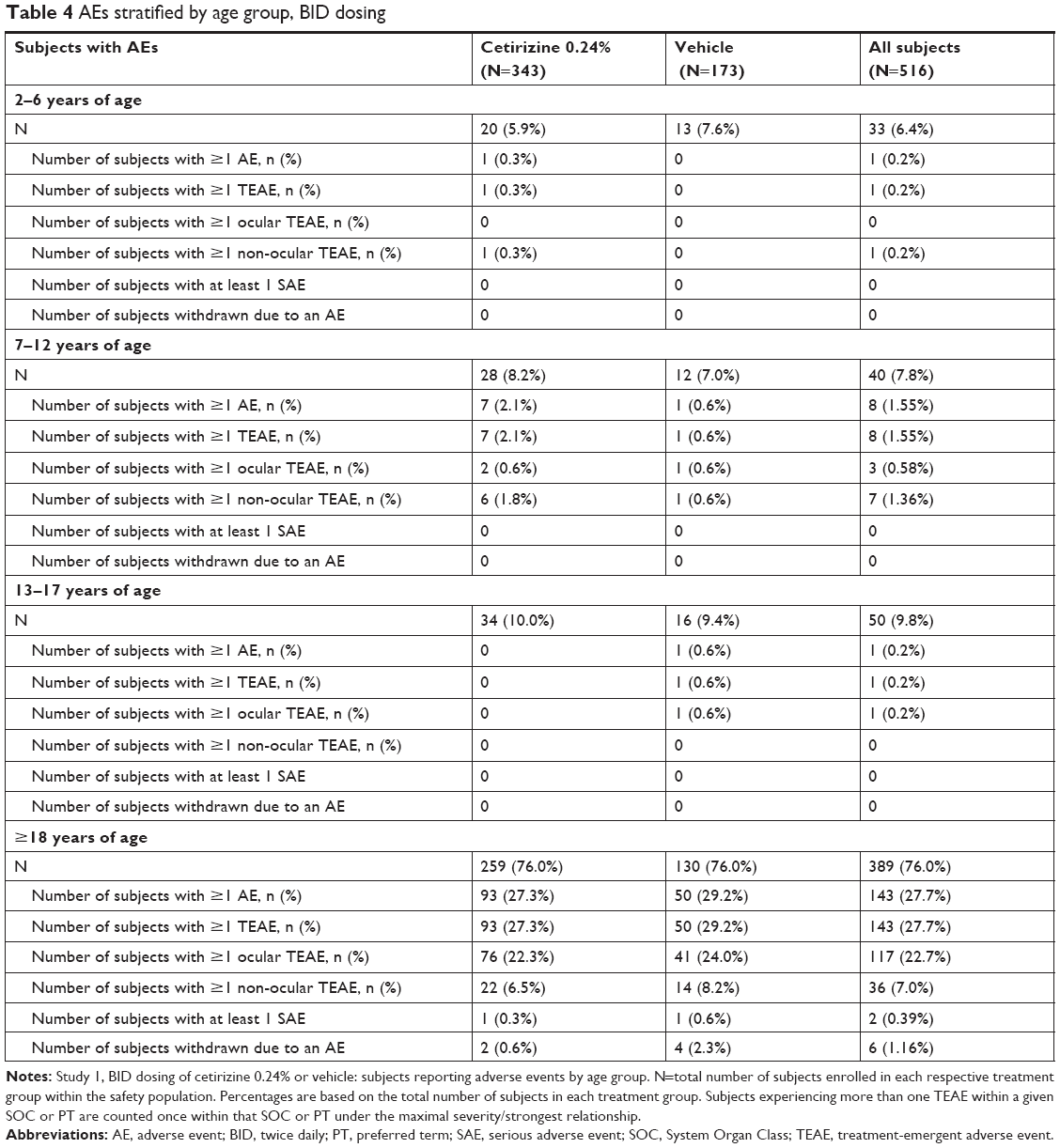 | Table 4 AEs stratified by age group, BID dosing |
The TID study comprised a larger pediatric population (128 subjects) and confirmed the safety of cetirizine ophthalmic solution 0.24% in a pediatric population dosed TID with cetirizine or its vehicle. No ocular or non-ocular events occurred in subjects aged 2–6 years and only one event occurred in subjects aged 7–12 years (Table 5). The non-ocular TEAE, which occurred in subjects aged 7–12 years, was streptococcal pharyngitis in the cetirizine group. In subjects aged 13–17 years, one ocular TEAE of meibomian gland dysfunction occurred in one subject and three non-ocular TEAEs of atrial tachycardia, sinusitis, and dermatitis contact occurred in three subjects (Table 5). None of these events were suspected of being related to study treatment. Thus, both BID or TID dosing demonstrated an acceptable safety profile in patients aged 2–18 years.
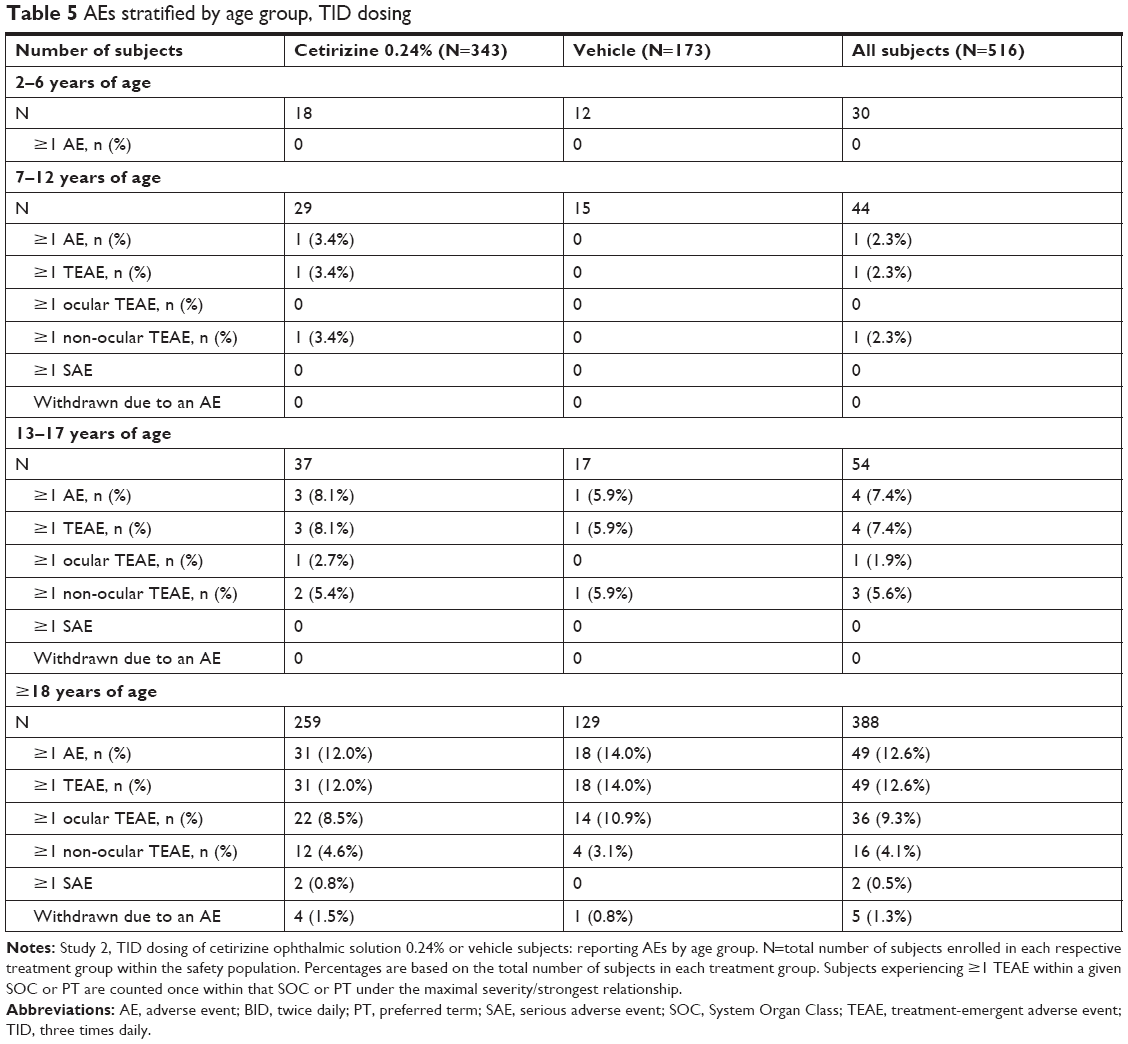 | Table 5 AEs stratified by age group, TID dosing |
Tolerability
Drop comfort (tolerability) was graded using an 11-point scale wherein 0= very comfortable and 10= very uncomfortable. Higher scores indicated less comfort. The difference in comfort scores between treatment groups was not clinically significant (ie, <1 unit change). In the BID study, mean scores across subgroups and whole groups ranged from 0.3 to 0.9 in the cetirizine group vs 0.1–0.5 in the vehicle group. Both cetirizine and vehicle groups were comfortable upon administration (Figure 2). The TID study showed similar results with mean scores across subgroups and whole groups of 0.6–0.9 in the cetirizine group compared to 0.2–0.5 in the vehicle group (Figure 2). Thus, cetirizine ophthalmic solution 0.24% instillation was comfortable relative to vehicle and well tolerated.
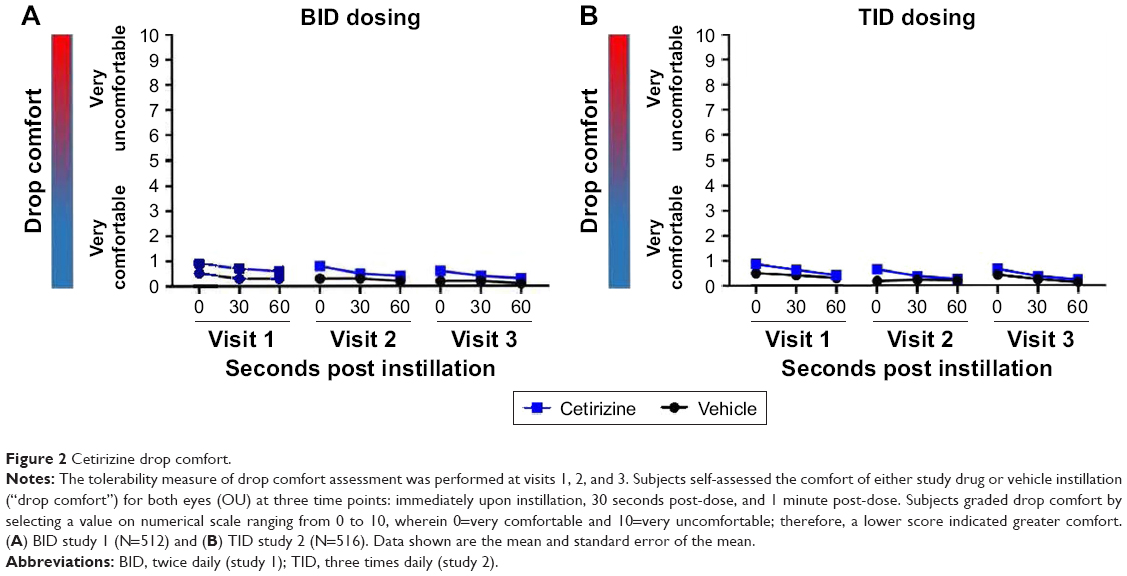 | Figure 2 Cetirizine drop comfort. |
Discussion
Cetirizine ophthalmic solution 0.24% is the first topical ophthalmic formulation of cetirizine hydrochloride approved by the Food and Drug Administration (FDA) for the indication of ocular itching associated with allergic conjunctivitis. The findings reported here demonstrate a favorable safety profile of cetirizine as an ophthalmic solution. Coupled with its strong efficacy in the relief of ocular itching, eyelid swelling, and nasal symptoms (Meier et al 2018, co-submitted), cetirizine ophthalmic solution 0.24% is a robust addition to the alternatives that eye care providers have for allergic conjunctivitis.
Cetirizine ophthalmic solution 0.24% showed a suitable safety profile when administered BID to TID for up to 12 weeks in patients ≥2 years of age. The PK study demonstrated low systemic exposure of cetirizine, 100-fold less than oral administration, when administered as an ophthalmic solution in support of the minimal systemic AEs that were reported in the BID and TID studies. There were no clinically relevant changes in BCVA, SLE, dilated ophthalmoscopy, IOP, physical examination, or vital signs in any study. The most common AEs in BID dosing were mild to moderate cases of conjunctival hyperemia, instillation site pain (all mild), and ocular hyperemia. These AEs were also reported in the TID study at similar severity and also resolved within the study period. An even lower incidence of AEs was reported in pediatric patients aged 2–18 years. Together these data provide the safety basis for FDA approval of cetirizine ophthalmic solution 0.24%.
Cetirizine has been approved as a tablet since 1995 with a long-standing systemic safety profile.12 As a tablet or liquid formulation, cetirizine may cause drowsiness and, in more rare instances, undesired cardiac, gastrointestinal, and renal disorders. Furthermore, oral cetirizine can lead to ocular accommodation disorder, blurred vision, and oculogyration.13 The ophthalmic solution of cetirizine 0.24% reported here caused no undesirable systemic AEs nor did it cause any of the ocular disorders above. As an ophthalmic solution, cetirizine offers a unique advantage over the tablet formulation by limiting systemic exposure and AEs. Further studies comparing or combining oral and ophthalmic cetirizine may prove useful to patients with unmet ocular symptoms caused by allergic conjunctivitis or those that would benefit from a reduced systemic exposure.
Cetirizine ophthalmic solution 0.24% provides an alternative to other ophthalmic solutions currently available for the indication of ocular itch associated with allergic conjunctivitis. Allergic conjunctivitis commonly results in divergent and patient-dependent symptoms; thus, the ideal therapeutic for one patient may not ease the symptoms of all patients,14 supporting the need for a broad anti-allergics market. Cetirizine ophthalmic solution 0.24% comes with the added benefit of patient and physician familiarity, above and beyond its demonstrated efficacy and safety. The high comfort upon instillation, low incidence of AEs, minimal systemic exposure, and lack of clinically relevant changes in safety measures suggest that cetirizine ophthalmic solution 0.24% will add to and improve the currently marketed therapeutics for allergic conjunctivitis.
Acknowledgment
Medical writing assistance was provided by Ora, Inc.
Disclosure
Paul J Gomes is an employee of Ora, Inc. Mark C Jasek is an employee of Eyevance Pharmaceuticals. Zerviate is a proprietary trademark of Nicox SA and is licensed by Eyevance Pharmaceuticals. Controlled adverse environment (CAE®) and Conjunctival Allergen Challenge (Ora-CAC®) are registered trademarks of Ora, Inc.
References
Bielory L. Allergic and immunologic disorders of the eye. Part II: ocular allergy. J Allergy Clin Immunol. 2000;106(6):1019–1032. | ||
Ciprandi G, Buscaglia S, Pesce GP, Bagnasco M, Canonica GW. Ocular challenge and hyperresponsiveness to histamine in patients with allergic conjunctivitis. J Allergy Clin Immunol. 1993;91(6):1227–1230. | ||
Bacon AS, Ahluwalia P, Irani AM, et al. Tear and conjunctival changes during the allergen-induced early- and late-phase responses. J Allergy Clin Immunol. 2000;106(5):948–954. | ||
Abelson MB, Loeffler O. Conjunctival allergen challenge: models in the investigation of ocular allergy. Curr Allergy Asthma Rep. 2003;3(4):363–368. | ||
Charlesworth EN, Kagey-Sobotka A, Norman PS, Lichtenstein LM. Effect of cetirizine on mast cell-mediator release and cellular traffic during the cutaneous late-phase reaction. J Allergy Clin Immunol. 1989;83(5):905–912. | ||
Levi-Schaffer F, Eliashar R. Mast cell stabilizing properties of antihistamines. J Invest Dermatol. 2009;129(11):2549–2551. | ||
Fukushima A, Ebihara N. Efficacy of olopatadine versus epinastine for treating allergic conjunctivitis caused by Japanese cedar pollen: a double-blind randomized controlled trial. Adv Ther. 2014;31(10):1045–1058. | ||
Fraunfelder FW, Fraunfelder FT. Oculogyric crisis in patients taking cetirizine. Am J Ophthalmol. 2004;137(2):355–357. | ||
Ousler GW, Wilcox KA, Gupta G, Abelson MB, Fink K. An evaluation of the ocular drying effects of 2 systemic antihistamines: loratadine and cetirizine hydrochloride. Ann Allergy Asthma Immunol. 2004;93(5):460–464. | ||
FDA. Zerviate™ [prescribing information]. Lake Forest, IL: Akorn, Inc.; 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208694s000lbl.pdf. Accessed January 25, 2018. | ||
Ousler G, Devries DK, Karpecki PM, Ciolino JB. An evaluation of Retaine™ ophthalmic emulsion in the management of tear film stability and ocular surface staining in patients diagnosed with dry eye. Clin Ophthalmol. 2015;9:235–243. | ||
Johnson & Johnson Consumer Companies, Inc. Zyrtec® The #1 Allergist Recommended Brand*; 2011. Available from: http://www.multivu.com/assets/52078/documents/Zyrtec-fact-sheet-99-original.pdf. Accessed June 7, 2018. | ||
FDA. Package insert template for cetirizine tablet/oral drops/ oral solution; 2013. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2004/19835slr016,21150slr005,30346slr011_zyrtec_lbl.pdf. Accessed February 12, 2019. | ||
Ehlers WH, Donshik PC. Allergic ocular disorders: a spectrum of diseases. Clao J. 1992;18(2):117–124. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.