")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Safety, efficacy, and clinical utility of macitentan in the treatment of pulmonary arterial hypertension
Received 11 December 2015
Accepted for publication 3 March 2016
Published 18 May 2016 Volume 2016:10 Pages 1675—1682
DOI https://doi.org/10.2147/DDDT.S88612
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Thomas J Monaco,1 Carlos D Davila2
1Division of Pulmonary and Critical Care Medicine, Baylor College of Medicine, Houston, TX, 2Department of Medicine, Einstein Medical Center, Philadelphia, PA, USA
Abstract: Pulmonary arterial hypertension is a progressive, debilitating disease caused by a dysregulation of the pulmonary vascular tone that inevitably leads to right heart failure and death without treatment. Until relatively recently, the treatment options for those afflicted by pulmonary arterial hypertension were limited; today, a greater understanding of the pathophysiology behind this disease has led to several evidence-based therapies that can improve pulmonary function and quality of life for these patients. One of the primary mediators of pulmonary vascular tone is endothelin-1, which is a potent and long-lasting vasoconstrictor. Macitentan is a second-generation endothelin receptor antagonist that acts selectively as a pulmonary vasodilator without the significant side effects noted with previous endothelin receptor antagonists. This review focuses on the mechanism of action and pharmacokinetics of macitentan, as well as the adverse effects, efficacy, and clinical uses of macitentan in the clinical trials to date. In addition, the authors briefly review clinical trials currently underway to illustrate possible future directions for the use of macitentan.
Keywords: pulmonary arterial hypertension, macitentan, endothelin receptor antagonist
Introduction
Pulmonary arterial hypertension (PAH) is a progressive, debilitating disease characterized by elevated arterial blood pressure in the pulmonary circulation that left untreated, results in right ventricular failure and death. Its prevalence has been estimated to be between 15 and 50 cases per million and even with modern treatment, the prognosis remains guarded.1 In fact, data from the US Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL) showed 1- and 5-year survival of 87% and 57%, respectively, therefore, new and more effective therapeutic options are necessary to treat this severe and unrelenting disease.2 PAH is pathologically defined by a mean pulmonary arterial pressure ≥25 mmHg at rest and pulmonary vascular resistance of >3 Woods units, in the absence of either an elevated wedge pressure or cardiac output. It is distinguished as Group 1 according to the updated clinical classification of pulmonary hypertension and comprises a group of conditions and diseases with similar pathological findings, hemodynamic characteristics and in some cases treatment options (Table 1).3 For the purpose of this review we will focus our discussion on the clinical utility and evidence on macitentan in PAH.
 | Table 1 Updated classification of pulmonary hypertension |
The development of effective treatment for PAH has necessitated a clearer understanding of the pathophysiology of the disease. While the complete mechanism is still incompletely understood, it is clear that the development and maintenance of PAH is secondary to a dysregulation of vascular tone. Due to Poiseuille’s law, minute changes in vessel radius translate into much greater changes in vascular resistance. These changes are mediated by vascular smooth muscle, which is influenced by three vasoactive molecules released by endothelial cells: nitric oxide (NO) and prostacyclins, which induce vasodilation, and endothelins, which induce vasoconstriction. In healthy subjects, these mediators are in a dynamic balance to preserve an optimal pulmonary vascular tone.4 Several experiments in both animal models and humans have demonstrated that PAH is associated with reduced levels of prostacyclins and increased vasoreactivity to NO.5–7 Agents that target NO metabolism via phosphodiesterase type 5 (PDE-5) inhibition to increase cyclic guanosine monophosphate (cGMP) levels have shown great promise in long-term trials and are now an important part of PAH therapy.8 There has been extensive interest in developing treatments that target NO release or prostacyclin receptor activation but these efforts have run into difficulties with drug delivery and duration of action.4 There is also emerging technology of prostacyclin synthase gene therapy and cell-based therapy using native stem cells and engineered stem cells with enhanced prostacyclin production capacity and direct activation of the cGMP cascade.9 Endothelin represents another well-known target in the treatment of PAH.
Endothelins are 21 amino acid peptides, with three distinct isoforms and two known endothelin receptors (ETA and ETB); endothelin-1 (ET-1) has been found in increased levels in the plasma and pulmonary vascular endothelium of patients with PAH and has been implicated in the pathogenesis and progression of pulmonary vasoconstriction and eventual right ventricular failure in these patients.10–12 Endothelin receptor antagonist (ERAs) including Ambrisentan (Letairis®), Bosentan (Tracleer®), and Sitaxsentan (Thelin®) have been designed and tested in patients with PAH in randomized controlled clinical trials and have been shown to improve functional capacity, exercise capacity and delay disease progression in these patients.13–15 Of these, ambrisentan and sitaxsentan are ETA-selective ERAs, while bosentan has nonselective activity on endothelin receptors.
Macitentan (Opsumit®) is a novel orally active dual ERA, which was recently approved in both the European Union and US to delay disease progression and reduce hospitalizations in patients with PAH.
Mechanism of action
ET-1 is expressed constitutively by endothelial cells and secreted from the basal surface of the vascular endothelium, where it promotes both local vasoconstriction and cell proliferation of the underlying smooth muscle as well as fibroblast proliferation; changing tissue structure and inducing fibrosis.16,17 Secretion of ET-1 can be further promoted by hypoxia, shear stress, thrombin activity, or inhibited by the effect of NO.18,19 It is first manufactured in the lung endothelial cells as an inert precursor, which is then activated by ET-converting enzyme. It is then released in close proximity to the endothelial smooth muscle where it binds to endothelin receptors; plasma levels of ET-1 do not reflect the true paracrine activity of ET-1 on these cells.20,21 ET-1 promotes vasoconstriction and vascular remodeling by activating G-protein coupled endothelin receptors. The most important of the endothelin receptors are ETAR and ETBR.4 The former is found on smooth muscle cells, fibroblasts, and cardiac myocytes, while the latter is expressed on both smooth muscle and endothelial cells.22 Binding of ET-1 to ETAR causes G-protein activation and increased intracellular inositol triphosphate levels, which causes calcium release and vasoconstriction in muscle cells.23 In contrast, ET-1 binding to ETBR causes release of NO and prostacyclin, and inhibition of apoptosis for several cell lines, including vascular smooth muscle.24,25 Despite their apparent contrasts, there appears to be significant roles for both ETAR and ETBR in ET-1-mediated pulmonary arterial vasoconstriction. In rat models, the combination of ETAR and ETBR blockade resulted in maximal reduction in vasoconstriction by ET-1.26 Classically, endothelin receptors have been considered monomers that, when activated by ET-1, signal the release of intracellular calcium via G-coupled protein and downstream cellular signaling. Recent experimental observations have suggested synergy between ETAR and ETBR, however, leading to the interesting proposal of significant cellular interaction between these two receptors. It is possible that the downstream activity of ETAR and ETBR requires heterodimerization to activate the coupled G-protein, and that ERAs block this heterodimerization and prevent downstream activation by ET-1, or that under conditions of selective ETAR antagonism, ETBR is able to assume partial functions of its counterpart receptor.27
Macitentan is a second-generation potent dual ERA with tissue targeting properties, its chemical name is N-5-(4-bromophenyl)-6-(2-[5-bromopyrimidin-2-yl]oxyethoxy)-N-(propylsulfamoyl)pyrimidin-4-amine-N-’-propylsulfamide (Figure 1). Macitentan displays high affinity and sustained occupancy of the ETRs in human pulmonary arterial smooth muscle cells, with a 50-fold increased selectivity for the ETAR when compared to the ETBR subtype. Macitentan also demonstrates slower dissociation kinetics from endothelin receptors than other ERAs, which may underlie its observed differences in adverse effects from other ERAs.28
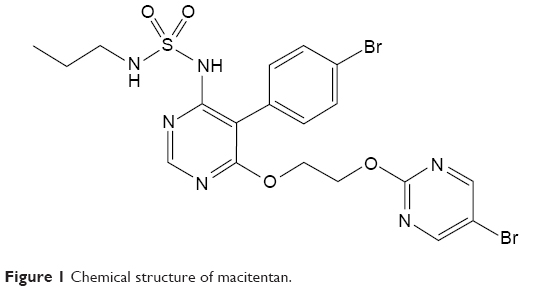 | Figure 1 Chemical structure of macitentan. |
Pharmacokinetics and safety profile of macitentan
Macitentan shares a similar side effect profile to other ERAs, but it possesses some important differences as well by virtue of its dissociation kinetics and chemical structure. It belongs to the sulfamide class, with a sulfur dioxide linked to two organic chains. It is metabolized by oxidative depropylation to ACT-132577 and by oxidative cleavage to ACT-373898; both of these reactions are catalyzed primarily by the cytochrome P450 (CYP3A4) system.29 Of these, only the former is metabolically active, and at steady state contributes about 40% to macitentan’s total potency.30 As its metabolism is driven by the CYP3A4 system, macitentan’s serum levels can rise significantly with strong inhibitors of the P450 system like ritonavir or ketoconazole, or fall precipitously with strong inducers like rifampin.31,32 Following the initial pharmacokinetics studies, it has been estimated that about 74% of macitentan’s ingested dose is bioavailable, this bioavailability is constant regardless of food intake, age, sex, or ethnicity.31,32 Greater than 99% of macitentan and its metabolites remains avidly bound to plasma proteins, and it is principally cleared by a combination of hepatic and renal pathways (50%), while fecal elimination accounts for another 25% of excretion. In terms of drug-drug interaction, a randomized, open-label crossover study by Sidharta et al33 found that coadministration of macitentan and warfarin did not change either the international normalized ratio levels or factor VII activity compared to controls. Also, the investigators found no significant changes in the pharmacokinetics of macitentan and sildenafil when administered together in healthy volunteers. Neither severe renal impairment nor hepatic impairment (Child–Pugh classes A, B, or C) were found to confer any additional risk with the drug.32 These were concluded after administering a single 10 mg dose of macitentan to sequential groups of eight subjects with mild, moderate, and severe liver disease and comparing the results to a control group without liver disease, although plasma levels were lower in patients with underlying liver disease, no significant difference in pharmacokinetics was found. Similar evaluation was done in patients with severe renal impairment, defined as creatinine clearance <30 mL/min. In this analysis, plasma concentration of macitentan and its metabolites was similar despite the presence of renal impairment. Even though clearance of the drug was slower, it was not considered relevant and therefore no dose adjustment was needed. Extrapolating these findings, which were done in patients without PAH, into the intended target population remains controversial. To date, no safety studies have been performed in patients with PAH and either severe renal or hepatic impairment; in the European Union, macitentan’s use is contraindicated in these patients as well as in patients with transaminases more than three times the upper limit of normal (ULN). In the US, the only absolute contraindication to the use of macitentan is pregnancy, though it does carry a warning regarding the potential for hepatotoxicity.
Macitentan, like other ERAs, is known to have teratogenic potential;34 all female patients of childbearing age on these agents should be on contraception when using these agents. ET-1 plays a vital role in fetal development; Kurihara et al35 demonstrated that fetal mice who were deficient in ET-1 or administered bosentan went on to develop hypertension and severe craniofacial abnormalities. Deficiency in ET-1 has also been implicated in premature closure of the ductus arteriosus in preclinical models.36 Finally, macitentan’s safety has not been studied in the pediatric population, and it is not approved for use in these patients.
There are three important adverse effects common to all ERAs: peripheral edema, hepatotoxicity, and anemia.37 Other noteworthy side effects of macitentan include mild nonspecific neurologic, cardiac, or respiratory disturbances. The drug design of macitentan was influenced by a desire to preserve the beneficial class effects of ERAs while avoiding the major limiting adverse effects of the older generation ERAs.
Peripheral edema is more common and pronounced in patients on ETAR-specific medications like ambrisentan than dual-receptor antagonists. Additionally, it appears to more commonly affect older patients.27 The pathophysiology behind this adverse effect is still incompletely understood, but it has been hypothesized to be a result of inhibition of ETAR or ETBR-mediated natriuresis, blockage of ETAR-induced myocardial contractility, or even by the direct effect of ERAs on capillary permeability.38–40 In one study of 145 patients on ambrisentan for PAH, peripheral edema was the reason for stopping drug therapy in 20 of the 50 patients who ended treatment due to side effects.41 In contrast, in the Study with an Endothelin Receptor Antagonist in Pulmonary Arterial Hypertension to Improve Clinical Outcome (SERAPHIN trial) no significant difference in the incidence of peripheral edema with macitentan compared to placebo was found (16% in the macitentan 3 mg group, 18.2% in the macitentan 10 mg group, and 18.1% in the placebo group). However, a trend toward developing peripheral edema was seen in older patients on subgroup analysis (30.3% in the macitentan 3 mg group, 25.9% in the macitentan 10 mg group, and 18.2% in the placebo group).42
Hepatotoxicity with ERAs can be idiosyncratic and severe enough to result in death; in fact, sitaxsentan was recently withdrawn from the market due to reports of fatal liver toxicity.43 The development of hepatotoxicity with the use of ERAs is likely due to antagonism of ETBR, which causes modulation of several hepatobiliary transporters (basolateral sodium-taurocholate cotransporting polypeptide, the bile salt export pump, and the organic anion-transporting polypeptides), which leads to accumulation of cytotoxic bile acids and cholestatic liver disease. More recent studies of bosentan-induced hepatotoxicity have also demonstrated that ETBR inhibition directly results in portal sinusoid constriction and further cholestasis.44 Similar to bosentan, macitentan has been shown to strongly inhibit hepatobiliary transport in vitro and has a high affinity for ETBR.45 However, the SERAPHIN trial did not report a higher incidence of liver transaminitis (>3× ULN) in the treatment arm compared to placebo (3.6% in the macitentan 3 mg group, 3.4% in the macitentan 10 mg group, and 4.5% in the placebo group). When transaminitis was present, though, it tended to be more severe in the patients treated with macitentan; transaminases >8× ULN were fivefold greater in the treatment arm (2.1%) compared to placebo (0.4%).42 Therefore, it is currently recommended to monitor liver enzymes at the initiation of treatment and then in the presence of clinical findings, and to discontinue macitentan therapy if patients develop sustained aminotransferase elevations, bilirubin elevations >2× ULN, or severe liver injury.
Anemia has been a commonly reported side effect in clinical trials of all of the ERAs. The anemia is generally mild, rarely required treatment discontinuation, and usually stabilizes after about 12 weeks of therapy.37 The etiology of this anemia is unclear, but it does not appear to be related to hemolysis or hemorrhage. Rather, it may be related to drug inhibition of an ETBR-mediated protection against red blood cell apoptosis,46 or more likely due to hemodilution as a result of increased fluid retention.47 A dose-dependent increased incidence of anemia was seen in the SERAPHIN trial (8.8% in the 3 mg group, 13.2% in the 10 mg group, and 3.2% in the placebo group). This anemia was severe enough to require drug discontinuation in two subjects, and resolved with cessation of therapy. Similar to liver enzymes, it is recommended to measure hemoglobin concentration before starting treatment with macitentan and as clinically indicated thereafter.
Finally, macitentan can cause a number of minor neurologic, cardiac, and respiratory disturbances. The most common neurologic complaint reported is headache (4.6% more when compared to placebo), while in terms of cardiorespiratory side effects there was an increased incidence of upper respiratory tract infections and nasopharyngitis in patients on macitentan (4%). In addition, there was a dose-dependent increase in the incidence of bronchitis with macitentan (8% at the 3 mg dose, 11.6% at the 10 mg dose, and 5.6% in the placebo group).42
Efficacy and clinical utility of macitentan
The initial studies with bosentan demonstrated an improvement in functional and hemodynamics parameter in subjects with PAH but also showed significant hepatotoxicity (14% of patients had levels >3× ULN).13–15 Similar studies examined ETAR-specific antagonists like sitaxsentan and ambrisentan and found therapeutic benefit but once more at the expense of severe, and (in the case of sitaxsentan) life-threatening adverse effects.46,48
Macitentan’s effectiveness was established in the pivotal SERAPHIN study in 2013.42 In this event-driven double-blind control study, investigators randomized in a 1:1:1 fashion 742 patients with proven diagnosis of PAH to either macitentan 3 mg/day, macitentan 10 mg/day, or placebo. Background PAH therapy was also allowed, including oral and inhaled prostanoid therapy, calcium channel blockers, or phosphodiesterase-5 inhibitors. Subjects included patients with idiopathic or heritable PAH (56.8%), pulmonary hypertension due to shunts (8.4%), HIV (1.4%), connective-tissue disease (30.5%), or drug or toxin exposure (3%). Hemodynamics and functional status were verified by right heart catheterization in all subjects (mean pulmonary artery pressure of 53.9±17.5 mmHg, mean pulmonary vascular resistance 12.8 Woods units) and by 6 minutes walk test (mean 350±100.2 meters; World Health Organization [WHO] class II–IV disease). The composite primary endpoint was time from initiation of therapy to the occurrence of the first PAH-related event (worsening of PAH, initiation of intravenous or subcutaneous prostanoids, atrial septostomy or lung transplantation) or all-cause mortality until the end of treatment. Prespecified secondary end points included the change from baseline to month 6 in the 6-minute walk distance, the percentage of patients with an improvement in WHO functional class at month 6, death due to PAH or hospitalization for PAH up to the end of treatment, and death from any cause up to the end of treatment and up to the end of the study. Following a median treatment duration of 115 weeks, 31.4% (n=76) patients taking macitentan 10 mg/day and 38% taking macitentan 3 mg/day (n=95) experienced a primary endpoint event, compared to 46.4% (n=116) of patients taking placebo (P<0.001). The beneficial effect of macitentan was mostly attributable to a lower incidence of clinical worsening of PAH, defined as >15% reduction in 6-minute walk distance, worsening of symptoms, and need for additional PAH treatment with a relative risk reduction of 45% for the primary composite endpoint in patients taking macitentan 10 mg/day and 30% in patients on 3 mg/day when compared to placebo (hazard ratio [HR] 0.55 [97.5% confidence interval {CI}, 0.39 to 0.76; P<0.001] and 0.70 [97.5% CI 0.52 to 0.96; P=0.01 by the log-rank test], respectively). The number needed to treat to avoid one primary endpoint at 2 years was six patients. Although benefit was shown in the secondary composite endpoint of death and hospitalizations due to PAH, this benefit was mainly driven by PAH-related hospitalization with a HR of 0.50 versus placebo in subjects taking macitentan 10 mg/day (97% CI, 0.34 to 0.75; P<0.001) and 0.67 versus placebo in subjects randomized to macitentan 3 mg/day (97% CI, 0.46 to 0.97; P<0.001). When isolated PAH-related mortality data was analyzed, there was a trend favoring the intervention arm versus placebo, however, it was not statistically significant (HR 0.44 [97% CI, 0.16 to 1.25] 10 mg/day and HR 0.87 [97% CI, 0.37 to 2.04] 3 mg/day P=0.07) (Table 2).
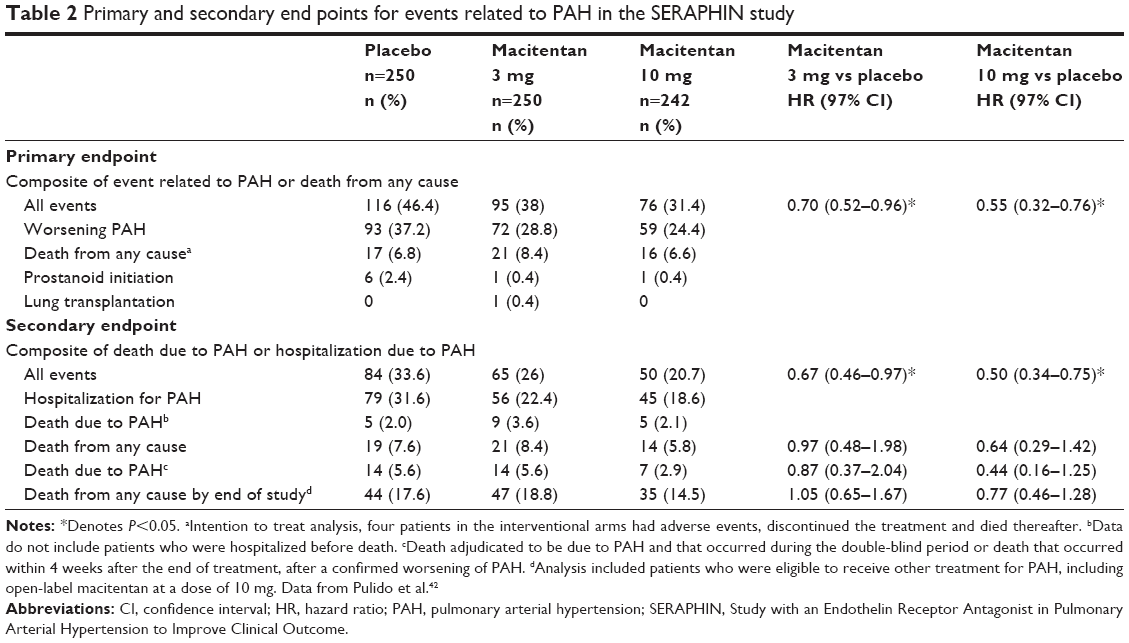 | Table 2 Primary and secondary end points for events related to PAH in the SERAPHIN study |
At 6 months of treatment, a subset of 145 subjects underwent hemodynamic assessment, which demonstrated reductions in pulmonary vascular resistance and improvements in cardiac index in the macitentan group but not in the placebo group. The WHO functional class of patients in each group was also examined; while all patients improved after 6 months of study from their baseline, there was a significant improvement in functional class in patients on macitentan compared to placebo (13% in placebo vs 20% at 3 mg [P=0.04] and 22% at 10 mg [P=0.006]). Finally, in assessments of the 6-minute walk distance, there was a mean decline in the placebo group of 9.4 m while the treatment groups both showed improvements in 6-minute walk distance (+7.4 m in the 3 mg arm and +12.5 m in the 10 mg arm). All of these benefits seemed to extend both to treatment-naïve and previously treated patients.42 Furthermore, a post hoc analysis by Channick et al49 showed that macitentan 10 mg/day was associated with a reduced risk of both; all-cause and PAH-related hospitalizations and no increase in the risk for hospitalizations for other causes. In 2013, the US Food and Drug Administration approved macitentan (Opsumit®) 10 mg once-daily for the treatment of PAH in order to delay disease progression and reduce hospitalizations. The EU Commission approved it for similar usage as monotherapy or combination therapy for PAH in adult patients with a WHO functional class of II to III. Given the drug’s known teratogenicity, it bears a boxed warning as category X, and the possibility of pregnancy should be excluded in any female patient who will be taking the drug.
While peripheral edema, hepatotoxicity, and anemia appear to be class effects of the ERAs, clinical trials of macitentan indicate that its unique pharmacokinetics decrease the incidence of these side effects significantly compared to previous ERAs. The 2013 SERAPHIN trial also illustrated macitentan’s efficacy in reducing the severity of PAH in both treatment-naïve patients and those on therapy, primarily in terms of disease progression and PAH-related hospitalizations. In the US, it can be used in patients with hepatic and renal impairment without adjustment, though it has known teratogenicity and should be absolutely avoided in patients who may be pregnant.
Future directions
An open-label extension of the SERAPHIN trial to examine the long-term effects of macitentan in patients with PAH is currently underway, labeled SERAPHIN-OL (NCT00667823). It is currently scheduled for completion in March 2017. There are also several trials underway to test macitentan’s efficacy in treating other related diseases and conditions, for which its use is currently off-label. It is currently being studied in the Phase II MERIT (macitentan in the treatment of inoperable chronic thromboembolic pulmonary hypertension) trial (NCT02060721) for its utility in chronic thromboembolic pulmonary hypertension. Also, the Phase II MELODY-1 and -2 (macitentan in combined pre- and postcapillary pulmonary hypertension due to left ventricular dysfunction) trials are prospective, multicenter, randomized controlled studies to evaluate the safety and tolerability of macitentan in pulmonary hypertension due to left ventricular dysfunction. Finally, macitentan is also being tested in congenital heart disease in the Phase III MAESTRO (macitentan in Eisenmenger syndrome to restore exercise capacity) trial, expected to conclude in March 2016. Macitentan is also under investigation for any utility in other processes involving the endothelin axis, including as combination therapy for glioblastoma multiforme, as treatment for digital ulcers in systemic sclerosis, or as treatment for portopulmonary hypertension.
Conclusion
PAH remains to be a significant disease with long-term burden and clinical implications. Important advances in PAH therapy have been made, including the development of ERAs. Macitentan is the newest ERA available for the treatment of PAH, and has been shown in clinical trials to be well tolerated and clinically effective in both treatment-naïve patients and those on background therapy. Like other ERAs, it can be orally dosed once daily, and has a similar cost to other members of its class. Unlike other ERAs, however, it does not appear to require dosage adjustments in patients with hepatic or renal impairment. Although head to head studies and long-term follow-up data are needed to further establish its beneficial effects and tolerability, current evidence suggests that macitentan has a more favorable side effect profile than older ERAs and may therefore be a more attractive treatment option for patients with PAH.
Disclosure
The authors report no conflicts of interest in this work.
References
Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J. 2007;30(1):104–109. | ||
Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest. 2012;142(2):448–456. | ||
Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D34–D41. | ||
Chester AH, Yacoub MH. The role of endothelin-1 in pulmonary arterial hypertension. Glob Cardiol Sci Pract. 2014;2014(2):62–78. | ||
Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327(2):70–75. | ||
Igari H, Tatsumi K, Sugito K, et al. Role of EDRF in pulmonary circulation during sustained hypoxia. J Cardiovasc Pharmacol. 1998;31(2):299–305. | ||
Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–1932. | ||
Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005;353(20):2148–2157. | ||
Gubrij IB, Martin SR, Pangle AK, Kurten R, Johnson LG. Attenuation of monocrotaline-induced pulmonary hypertension by luminal adeno-associated virus serotype 9 gene transfer of prostacyclin synthase. Hum Gene Ther. 2014;25(6):498–505. | ||
Galie N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. 2004;61(2):227–237. | ||
Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–1739. | ||
Stewart DJ, Levy RD, Cernacek P, Langleben D. Increased plasma endothelin-1 in pulmonary hypertension: marker or mediator of disease? Ann Intern Med. 1991;114(6):464–469. | ||
Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;358(9288):1119–1123. | ||
Galie N, Olschewski H, Oudiz RJ, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008;117(23):3010–3019. | ||
Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346(12):896–903. | ||
Peacock AJ, Dawes KE, Shock A, Gray AJ, Reeves JT, Laurent GJ. Endothelin-1 and endothelin-3 induce chemotaxis and replication of pulmonary artery fibroblasts. Am J Respir Cell Mol Biol. 1992;7(5):492–499. | ||
Wedgwood S, Dettman RW, Black SM. ET-1 stimulates pulmonary arterial smooth muscle cell proliferation via induction of reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2001;281(5):L1058–L1067. | ||
Boulanger C, Luscher TF. Release of endothelin from the porcine aorta. Inhibition by endothelium-derived nitric oxide. J Clin Invest. 1990;85(2):587–590. | ||
Russell FD, Skepper JN, Davenport AP. Evidence using immunoelectron microscopy for regulated and constitutive pathways in the transport and release of endothelin. J Cardiovasc Pharmacol. 1998;31(3):424–430. | ||
Channick RN, Sitbon O, Barst RJ, Manes A, Rubin LJ. Endothelin receptor antagonists in pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):62S–67S. | ||
Takahashi M, Matsushita Y, Iijima Y, Tanzawa K. Purification and characterization of endothelin-converting enzyme from rat lung. J Biol Chem. 1993;268(28):21394–21398. | ||
Sitbon O, Morrell N. Pathways in pulmonary arterial hypertension: the future is here. Eur Respir Rev. 2012;21(126):321–327. | ||
Pollock DM, Keith TL, Highsmith RF. Endothelin receptors and calcium signaling. FASEB J. 1995;9(12):1196–1204. | ||
Hirata Y, Emori T, Eguchi S, et al. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J Clin Invest. 1993;91(4):1367–1373. | ||
Shichiri M, Kato H, Marumo F, Hirata Y. Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension. 1997;30(5):1198–1203. | ||
Sauvageau S, Thorin E, Caron A, Dupuis J. Endothelin-1-induced pulmonary vasoreactivity is regulated by ET(A) and ET(B) receptor interactions. J Vasc Res. 2007;44(5):375–381. | ||
Trow TK, Taichman DB. Endothelin receptor blockade in the management of pulmonary arterial hypertension: selective and dual antagonism. Respir Med. 2009;103(7):951–962. | ||
Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS One. 2012;7(10):e47662. | ||
Bruderer S, Hopfgartner G, Seiberling M, et al. Absorption, distribution, metabolism, and excretion of macitentan, a dual endothelin receptor antagonist, in humans. Xenobiotica. 2012;42(9):901–910. | ||
Iglarz M, Binkert C, Morrison K, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J Pharmacol Exp Ther. 2008;327(3):736–745. | ||
Atsmon J, Dingemanse J, Shaikevich D, Volokhov I, Sidharta PN. Investigation of the effects of ketoconazole on the pharmacokinetics of macitentan, a novel dual endothelin receptor antagonist, in healthy subjects. Clin Pharmacokinet. 2013;52(8):685–692. | ||
Sidharta PN, van Giersbergen PL, Dingemanse J. Safety, tolerability, pharmacokinetics, and pharmacodynamics of macitentan, an endothelin receptor antagonist, in an ascending multiple-dose study in healthy subjects. J Clin Pharmacol. 2013;53(11):1131–1138. | ||
Sidharta PN, Dietrich H, Dingemanse J. Investigation of the effect of macitentan on the pharmacokinetics and pharmacodynamics of warfarin in healthy male subjects. Clin Drug Investig. 2014;34(8):545–552. | ||
Spence S, Anderson C, Cukierski M, Patrick D. Teratogenic effects of the endothelin receptor antagonist L-753,037 in the rat. Reprod Toxicol. 1999;13(1):15–29. | ||
Kurihara Y, Kurihara H, Suzuki H, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368(6473):703–710. | ||
Taniguchi T, Azuma H, Okada Y, Naiki H, Hollenberg MD, Muramatsu I. Endothelin-1-endothelin receptor type A mediates closure of rat ductus arteriosus at birth. J Physiol. 2001;537(Pt 2):579–585. | ||
Aversa M, Porter S, Granton J. Comparative safety and tolerability of endothelin receptor antagonists in pulmonary arterial hypertension. Drug Saf. 2015;38(5):419–435. | ||
Ge Y, Bagnall A, Stricklett PK, Webb D, Kotelevtsev Y, Kohan DE. Combined knockout of collecting duct endothelin A and B receptors causes hypertension and sodium retention. Am J Physiol Renal Physiol. 2008;295(6):F1635–F1640. | ||
Nagendran J, Sutendra G, Paterson I, et al. Endothelin axis is upregulated in human and rat right ventricular hypertrophy. Circ Res. 2013;112(2):347–354. | ||
Shapiro S, Pollock DM, Gillies H, et al. Frequency of edema in patients with pulmonary arterial hypertension receiving ambrisentan. Am J Cardiol. 2012;110(9):1373–1377. | ||
Condliffe R, Elliot CA, Hurdman J, et al. Ambrisentan therapy in pulmonary hypertension: clinical use and tolerability in a referral centre. Ther Adv Respir Dis. 2014;8(3):71–77. | ||
Pulido T, Adzerikho I, Channick RN, et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369(9):809–818. | ||
Galie N, Hoeper MM, Gibbs JS, Simonneau G. Liver toxicity of sitaxentan in pulmonary arterial hypertension. Eur Respir J. 2011;37(2):475–476. | ||
Ling L, Kuc RE, Maguire JJ, et al. Comparison of endothelin receptors in normal versus cirrhotic human liver and in the liver from endothelial cell-specific ETB knockout mice. Life Sci. 2012;91(13–14):716–722. | ||
Lepist EI, Gillies H, Smith W, et al. Evaluation of the endothelin receptor antagonists ambrisentan, bosentan, macitentan, and sitaxsentan as hepatobiliary transporter inhibitors and substrates in sandwich-cultured human hepatocytes. PLoS One. 2014;9(1):e87548. | ||
Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;169(4):441–447. | ||
Gabbay E, Fraser J, McNeil K. Review of bosentan in the management of pulmonary arterial hypertension. Vasc Health Risk Manag. 2007;3(6):887–900. | ||
Galie N, Badesch D, Oudiz R, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46(3):529–535. | ||
Channick RN, Delcroix M, Ghofrani HA, et al. Effect of macitentan on hospitalizations: results from the SERAPHIN trial. JACC Heart Fail. 2015;3(1):1–8. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.