")
Back to Journals » Open Access Journal of Clinical Trials » Volume 6
Safety and tolerability of an intravenously administered alpha1-proteinase inhibitor at an increased infusion rate: a novel, randomized, placebo-masked, infusion rate-controlled, crossover study in healthy adults
Authors Ngo LY, Haeberle A, Dyck-Jones J, Gelmont D, Yel L
Received 20 February 2014
Accepted for publication 3 April 2014
Published 23 June 2014 Volume 2014:6 Pages 55—61
DOI https://doi.org/10.2147/OAJCT.S62754
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Leman Yel, M.D
Views: 900
Leock Y Ngo,1 Adam Haeberle,1 Jacqueline Dyck-Jones,1 David Gelmont,1 Leman Yel1
1Baxter Healthcare Corporation, Westlake Village, CA, USA
Purpose: Alpha1-proteinase inhibitor (A1PI) is indicated for chronic augmentation therapy in adults with emphysema due to congenital deficiency of A1PI. An intravenous infusion rate of 0.04 mL/kg/minute is currently recommended for the A1PI product, Glassia®. This randomized, placebo-masked, rate-controlled, crossover study was designed to evaluate the safety and tolerability of A1PI administration at an increased infusion rate.
Patients and methods: A total of 30 healthy male and female subjects aged 19–61 years were enrolled. Each subject received simultaneous intravenous infusions of A1PI (Glassia®) and placebo (human albumin 2.5%) administered through a single infusion site on two separate treatment periods. Subjects were randomized in a 1:1 ratio to receive either test treatment (A1PI 0.2 mL/kg/minute + placebo 0.04 mL/kg/minute), or reference treatment (A1PI 0.04 mL/kg/minute + placebo 0.2 mL/kg/minute) on Day 1. On Day 15, subjects received the other treatment regimen in a crossover sequence.
Results: A total of 36 adverse events (AEs), regardless of causality, were reported; all were non-serious and of mild intensity, with headaches and dizziness occurring most frequently (12 [33.3%] and three [8.3%] of 36 AEs, respectively). Only seven AEs in six subjects were assessed as related to study treatment: with two AEs reported in two subjects treated with the 0.2 mL/kg/minute rate compared with five AEs in four subjects treated with the 0.04 mL/kg/minute rate.
Conclusions: This study demonstrated the safety and tolerability of an A1PI product at an increased infusion rate (0.2 mL/kg/minute) resulting in a shorter infusion duration in healthy subjects.
Keywords: A1PI, Glassia, administration rate, Alpha-1 antitrypsin, ATT
Introduction
Alpha1-antitrypsin/proteinase inhibitor (A1PI) is a glycoprotein, which is synthesized in the liver and found in protective concentrations in the lungs of healthy individuals. In the absence of A1PI, neutrophil elastase, released by lung macrophages and neutrophils, damages protein components of the alveolar wall, leading to elastin breakdown and the loss of lung elasticity and lung tissue.1 Excessive elastolytic activity underlies progressive emphysema in patients with low plasma A1PI levels.2 A1PI augmentation therapy specifically aims to restore the protease-antiprotease balance in lung tissue of such patients and to attenuate the progression of pulmonary emphysema.
Currently, there are four US Food and Drug Administration (US FDA)-approved preparations of A1PI obtained from human plasma, which differ in purification method, concentration, and dosage form. The infusion duration varies from 15 to 75 minutes according to the products’ A1PI concentration and approved infusion rate. Among them, Glassia® (Kamada Ltd, Israel), a relatively new purified A1PI in a liquid formulation, ready-to-use preparation, which contains 2% active A1PI in a phosphate buffered saline solution, was approved for patients with emphysema due to congenital deficiency of A1PI, at an infusion rate of 0.04 mL/kg/minute; however, this infusion rate leads to a long infusion duration. An increased infusion rate will lead to a shorter infusion duration and thereby potentially improves the quality of life of patients who require Glassia® augmentation therapy on a weekly basis. The aim of this study was to demonstrate the safety and tolerability of Glassia® administration at an increased infusion rate of 0.2 mL/kg/minute, compared to the conventional infusion rate of 0.04 mL/kg/minute. To ensure that neither subjects nor clinical personnel were aware whether the rapid or standard infusion rate of A1PI product was being administered, a novel placebo-masked, infusion rate-controlled study design was developed. The primary focus of this study design was the masking of the infusion rate of the A1PI solution by simultaneously infusing a placebo solution at the opposite infusion rate.
Materials and methods
Study design
This was a randomized, placebo-masked, rate-controlled, crossover study designed to evaluate the safety and tolerability of A1PI administration at an increased infusion rate. The study was approved by an investigational review board (MidLands IRB, Overland Park, Kansas, USA) and conducted at a single investigative site (Quintiles Phase I Services, Kansas City, Kansas, USA) in accordance with local regulations and International Conference on Harmonisation Good Clinical Practice guidelines, which are based upon principles that have their origin in the Declaration of Helsinki.3 This study was registered with the US National Institutes of Health website (ClinicalTrials.gov) as NCT01651351.
A1PI (Alpha1-Proteinase Inhibitor, Human) (Glassia®) is a sterile, ready-to-use, stabilizer-free, preservative-free, liquid preparation of purified 2% active A1PI in a phosphate buffered saline solution. The product is derived from a large human plasma pool. The US FDA-approved dose of 60 mg A1PI/kg (3 mL/kg) was administered in this study. Human albumin 5% (Baxter Healthcare Corporation, Westlake Village, CA, USA) diluted to 2.5% solution with normal saline was used as placebo, because of its similar appearance, protein content, and viscosity compared to the study product (thus ensuring maintenance of the blind). Placebo was administered at a volume of 3 mL/kg (equivalent to the volume of A1PI infusion).
As A1PI deficiency is a rare disorder, the conduct of a clinical study to determine the safety of Glassia® at an infusion rate of 0.2 mL/kg/minute in the patient population would not be practicable. The product is provided as a ready-to-infuse solution, so it was not possible to concentrate the product to decrease infusion duration. For this reason, a more rapid infusion rate was assessed to ensure provision of an adequate A1PI dose over a shorter period of time than is currently possible with the licensed infusion rate of 0.04 mL/kg/minute.
The participation of healthy volunteers allows for a population that is both naïve to A1PI augmentation therapy and ideal to detect potential adverse events (AEs) resulting from an increased infusion rate. Healthy volunteers have never been exposed to A1PI products and thus are naïve to possible AEs that may occur with infusion therapy. They also tend to be more prompt to report AEs compared to treatment-experienced patients with alpha-1 antitrypsin deficiency. Additionally, healthy volunteers have normal physiological levels of A1PI in the range of 20 to 53 μM, compared to patients with A1PI deficiency whose circulating A1PI levels are 8 μM or less. Following exogenous A1PI product administration, circulating A1PI levels are elevated to a level much higher than physiological levels. Thus, AEs would be more likely to be experienced in healthy subjects in the presence of excessive A1PI levels.
Subjects were randomized in a 1:1 ratio to one of two treatment groups as described in Table 1. Each subject received simultaneous intravenous infusions of A1PI (Glassia®) and placebo (human albumin 2.5%) administered through a Y-connector at a single infusion site on two separate treatment days (Figure 1). Subjects were randomized in a 1:1 ratio to receive either test treatment (A1PI 0.2 mL/kg/minute + placebo 0.04 mL/kg/minute), or reference treatment (A1PI 0.04 mL/kg/minute + placebo 0.2 mL/kg/minute) on Day 1. As this was a double-blinded study, an independent pharmacist prepared and labeled the infusion solutions for each subject. On Day 15, subjects received the other treatment regimen in a crossover sequence. The simultaneous infusion design ensured that the blind was maintained as the placebo masked the standard versus (vs) rapid infusion rates of the A1PI product. All infusions were administered intravenously at the predefined rates via infusion pumps. Each subject acted as his/her own control for the comparison between standard vs fast infusion, and the bias in AE reporting upon repeated dosing vs first dosing was reduced with the randomized sequence.
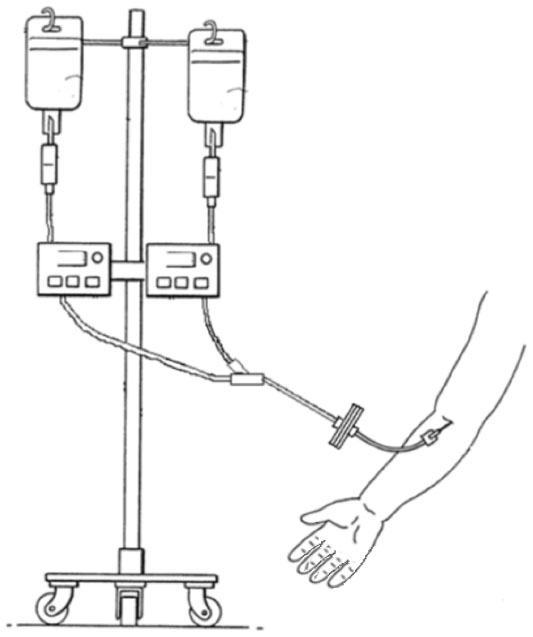 | Figure 1 Placebo-masked, infusion rate controlled investigational product administration. |
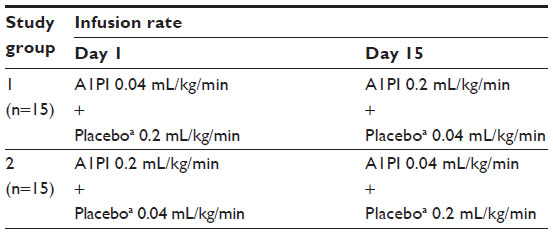 | Table 1 Treatment assignment and study design |
Subject population
Subjects volunteered to participate in this study. The investigator enrolled subjects according to predefined eligibility criteria. The sponsor was not involved in subject selection. Healthy subjects of either sex aged between 18 to 65 years were eligible for inclusion in the study. Subjects were required to have no clinical evidence of acute and/or chronic disease and no clinically significant abnormalities on hematology panel, clinical chemistry panel, urinalysis, or electrocardiogram at the time of screening. All subjects must have demonstrated a negative drug test at screening and agreed to refrain from heavy alcohol consumption and the use of narcotic drugs or illegal substances during study participation. Subjects with positive virus serology (hepatitis virus A, B, or C; parvovirus B19 [PVB19]; or human immunodeficiency virus type 1/2 [HIV]) at the time of screening, as well as those with a history of hypersensitivity or adverse reactions following blood transfusions, documented immunoglobulin A deficiency, or uncontrolled hypertension, were excluded from study participation. Pregnant or nursing women were excluded from study participation.
Outcome measures and safety assessments
The outcomes measured in this study were related to the safety and tolerability of increased infusion rate. The main outcome measures were the number of infusions with AEs that occurred during or within a) 1 hour, b) 24 hours, and c) 72 hours of completion of an infusion and the numbers of infusions which were interrupted, discontinued, or administered at a reduced infusion rate due to AEs.
The simultaneous infusion of the A1PI preparation and placebo did not allow AEs to be unquestionably ascribed to either A1PI or placebo. For this reason, any observed AE which had been assessed by the investigator as related to treatment was conservatively attributed to A1PI.
After completing all screening procedures, subjects who were eligible to participate in the study returned to the investigative site for study product administration on Day 1 and Day 15, and for longer term safety follow-up on Day 29 and Day 105. On each treatment day (Day 1 and Day 15), physical examination and clinical laboratory evaluations (hematology, chemistry, and urinalysis) were performed on each subject prior to dosing to ensure the subject was in good health. Subjects were monitored for safety and tolerability on-site throughout the duration of the study product administration up to 1 hour after completion of both infusions, and via telephone follow-up approximately 72 hours postinfusion to ensure potential AEs and use of concomitant medications were recorded. Vital signs (respiratory rate, pulse, body temperature, and systolic/diastolic blood pressure) were measured on each treatment day within 30 minutes prior to the start of study product administration, and at 15 minutes following completion of each of the 0.2 mL/kg/minute and the 0.04 mL/kg/minute infusions. Subjects returned to the investigative site 2 weeks (Day 29) after the last treatment for safety assessments, including physical examination, vital signs, clinical laboratory evaluations, AEs, and concomitant medications. Post-treatment viral serology and nucleic acid tests (NAT) were also performed during the Day 29 visit, to ensure no seroconversion had occurred compared to baseline assessments. Following this study visit, subjects returned to the investigative site approximately 3 months after the last treatment (Day 105) for the final viral serologic follow-up testing. AEs and concomitant medications were not actively solicited during the period between the Day 29 and Day 105 visits, but were recorded if reported by the subject.
Statistical analysis
Safety data were summarized using descriptive statistics, including mean, standard deviation, minimum, median, and maximum values. As is typical for safety studies, no hypothesis testing or power calculations were planned or performed. All available data from subjects receiving at least one infusion of either A1PI or placebo were included in the safety analyses. No adjustment or imputation was utilized for missing values.
Results
Study population
Of the 35 enrolled volunteers, 30 healthy subjects who met the eligibility criteria started the study. The median age of subjects who participated in the study was 24 years (range, 19 to 61 years), with 23 males (76.7%) and seven females (23.3%). Fifteen subjects were Caucasian (50.0%), 14 were African-American (46.7%), and one was Asian (3.3%). Two subjects were Latino (6.7%). The subjects’ weight ranged from 53.2 to 114.7 kg, with a median of 74.4 kg. The median body mass index was 25.7 kg/m2 (range, 19.5 to 31.5 kg/m2). The demographic characteristics of subjects in each study group were similar.
Exposure to investigational product
All 30 subjects completed both infusions of A1PI and placebo in two separate treatment periods. No infusions were interrupted or discontinued, and no infusions were administered at a reduced rate due to AEs. The duration of infusions at the 0.2 mL/kg/minute rate ranged from 12 to 15 minutes, whereas the duration of infusion at the rate of 0.04 mL/kg/minute ranged from 64 to 76 minutes.
During treatment period 1 (Day 1), the initial six subjects received A1PI and placebo infusions containing approximately 20 mL less than the planned volume to be administered due to an oversight of the tubing dead space. As a result, approximately 86% to 92% of the planned infusion volume was administered. For these six subjects, the infusion solutions for treatment period 2 (Day 15) were prepared in the same manner to match the corresponding infusion volumes in treatment period 1, thereby ensuring accurate comparison of data between periods. All subsequent subjects received their planned infusion volume.
Safety and tolerability
No serious adverse events and no severe AEs occurred following infusion with the investigational product, and no subjects discontinued due to AEs. All AEs reported during the study were of mild intensity. As A1PI and placebo were administered simultaneously into the same vein, no distinction could be made between the two preparations. It was therefore decided to conservatively designate all AEs assessed by the investigator as related to treatment as being related to A1PI. A relationship to placebo was, however, theoretically feasible.
Adverse events
A total of 36 AEs were reported in 16 subjects. No clinically relevant trends were noted between female and male subjects with respect to the frequency of AEs reported during the study. The frequency of AEs was determined to be lower for subjects receiving the rapid 0.2 mL/kg/minute A1PI infusion than those receiving the standard 0.04 mL/kg/minute A1PI infusion (Table 2).
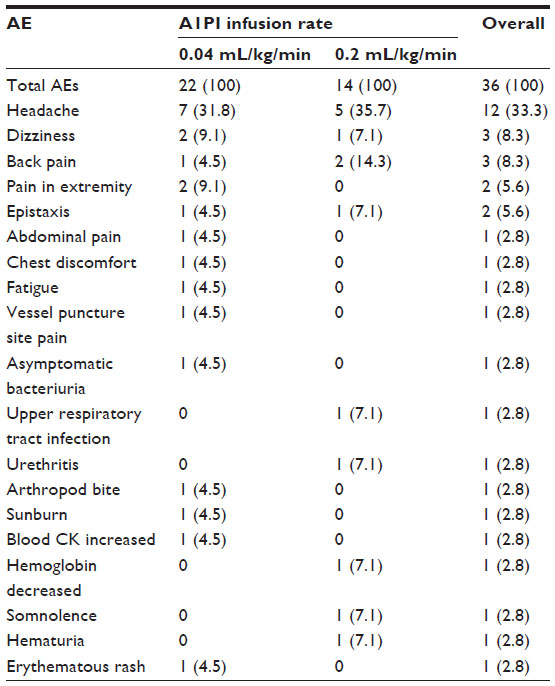 | Table 2 Number (%) of all adverse events reported, regardless of causality |
Of the 36 AEs, seven events reported in six subjects were assessed by the investigator as related to treatment. Treatment-related AEs also occurred slightly less frequently with the 0.2 mL/kg/minute A1PI infusion (two AEs in two [6.7%] subjects) than with the 0.04 mL/kg/minute A1PI infusion (five AEs in four [13.3%] subjects). Related AEs of dizziness and headache occurred in one (3.3%) subject each for the 0.2 mL/kg/minute infusion. Following administration of the 0.04 mL/kg/minute A1PI infusion, related AEs included chest discomfort, fatigue, pain in extremity, dizziness, and headache, each occurring in one (3.3%) subject. All related AEs began during an infusion or within 72 hours after the completion of an infusion.
Infusions associated with AEs
Overall, the number of infusions with AEs that began either during the infusion or within 1, 24, and 72 hours after its completion was lower for the 0.2 mL/kg/minute infusion rate than the 0.04 mL/kg/minute infusion rate. No clinically relevant trends were noted between female and male subjects with respect to the number of infusions associated with AEs. Of the 60 infusions administered during the study, 12 infusions were associated with 13 AEs; five AEs occurred during five infusions at the 0.2 mL/kg/minute rate and eight AEs occurred during seven infusions at the 0.04 mL/kg/minute rate. The number (proportion) of infusions with AEs that began during or within 1, 24, and 72 hours of completion of an infusion are summarized by study group and infusion rate in Table 3.
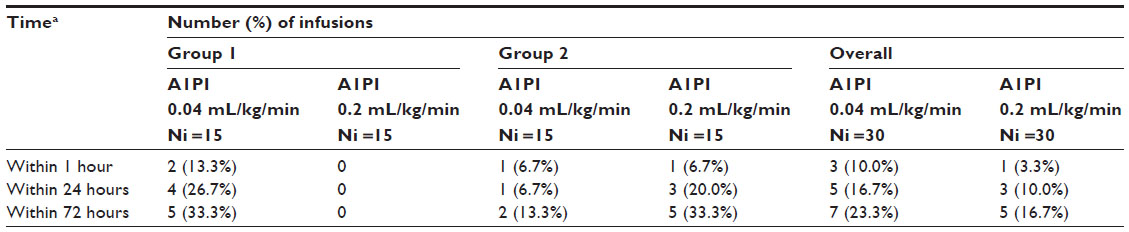 | Table 3 Summary of infusions associated with adverse events |
Clinical and laboratory evaluations
Viral serology tests for hepatitis virus A, B, and C and HIV were negative for all subjects. Levels of PVB19 immunoglobulin G and immunoglobulin M were within normal limits for all subjects (except one, who tested positive for PVB19 at baseline, prior to receiving an infusion with the study product). Virus NAT results were negative in all subjects.
No abnormalities were noted with respect to neutrophil counts, nor any trends of treatment-emergent abnormal neutrophil elevations. Three abnormal laboratory results were assessed as clinically significant. A 26-year-old male subject was reported to have increased plasma creatine phosphokinase (1,339 U/L; normal range 32–294 U/L) attributed to strenuous exercise, which returned to within the normal range 1 week later. One 58-year-old male subject developed hematuria, associated with a decrease in hemoglobin, 2 weeks after the first infusion. This AE, attributed to chronic prostatitis, persisted to the end of the study. A 21-year-old female subject was found to have asymptomatic bacteriuria and was referred for antibiotic treatment (after the end of the study). All three of these AEs were mild and assessed as unrelated to study treatment. All other changes in urinalysis, hematology, and blood chemistry values were not clinically significant.
Discussion
Glassia® has previously been shown to be comparable to other licensed products4–7 with respect to efficacy and safety. This study was carried out to investigate the safety and tolerability of Glassia® when administered at a faster than approved infusion rate, at 0.2 mL/kg/minute, providing a protein load as high as 4 mg/kg/minute. The more rapid infusion rate would enable patients to decrease their weekly A1PI infusion time by approximately five fold (eg, from 75 minutes to 15 minutes in a patient weighing 80 kg), thus potentially improving their quality of life.
The study presented here used a novel and unique study design to mask the infusion rate of the investigational product by simultaneously administering placebo intravenously. This ensured maintenance of the blind, thus reducing bias of clinical study investigators, infusion nurses, and study subjects. By providing study subjects with two infusion fluids administered simultaneously, one at a slower and one at a more rapid rate, all subjects required a comparable length of time for the procedure, and the infusion rate for the active treatment was effectively masked. This study design offers a better method of preserving the blind and the integrity of data compared with the more commonly used approach that involves study product administration by study nurses at different infusion rates, and thus has a higher chance of accidental/erroneous unblinding.8 Previous studies have employed a double-dummy design to evaluate the effect of different infusion rates of drug products on the safety and tolerability of patients.9,10 In these double-dummy studies, each patient received two infusions (one containing active drug and the other placebo solution such as 5% dextrose solution) sequentially over two separate treatment periods. While this type of study design offers a means to mask different infusion rates to both clinical staff and patients, the overall infusion duration for a given infusion visit is prolonged as a result of sequential administrations of active and placebo infusions. The simultaneous infusion approach used in this study enabled the shortest possible treatment time for study subjects while maintaining the double-blind.
The decision to use 2.5% albumin as a placebo was made as it has similar color and flow characteristics to the A1PI product, with a comparable protein concentration. Albumin also foams when agitated, similar to A1PI. No other placebo solution was acceptable to maintain the blind in the study presented here, as physiological saline, Ringer’s lactate solution, and other crystalloid solutions have lower viscosity, are colorless and transparent, and do not contain protein, all physical differences which would have been obvious to both subjects and clinical staff. Albumin has also been used as placebo in other A1PI studies, with a 2% albumin solution administered in a comparative study demonstrating that A1PI augmentation therapy delayed the progression of emphysema compared to placebo.11,12
The simultaneous administration of the A1PI product and placebo did not allow AEs to be unquestionably ascribed to either the investigational product or placebo; for this reason, it was decided to take a conservative approach and consider any AEs which were assessed by the investigator as being related to treatment to be attributed to A1PI. Numbers of AEs were lower among subjects who received the product at the faster infusion rate (0.2 mL/kg/minute) than the slower rate (0.04 mL/kg/minute), thus demonstrating the safety and tolerability of the more rapid infusion rate of the investigational product compared with that of the currently approved rate (0.04 mL/kg/minute) in healthy adult subjects. Only 26.7% of subjects reported AEs after receiving the more rapid 0.2 mL/kg/minute infusion rate, a percentage which is similar to that reported elsewhere in patients diagnosed with A1PI deficiency.5,13
There were no unexpected AEs. The most commonly reported AEs following administration of Glassia® are known to be headache and dizziness, and this finding was confirmed by the results of the present study. All AEs were mild in severity. Furthermore, no treatment-emergent viral infections were reported, as confirmed by negative viral serology and NAT. In addition, no subjects experienced hypersensitivity to the investigational product. We believe that potential adverse effects of high rate (ie, volume and protein load) would be similar in patients with A1PI deficiency. Therefore, we do not expect a different AE profile, and consider these results relevant to the A1PI-deficient population.
The results reported here compare favorably with other studies examining the safety profile of other A1PI augmentation products, where AEs have been reported at rates between 7% and 21% in patients with A1PI deficiency,5 with headache and dizziness/fainting occurring at much higher rates (47% and 17%, respectively) among patients receiving occasional or weekly A1PI infusions.13 In addition, in this study we did not observe AEs such as pyrexia, urticaria, and nausea/vomiting, unlike some previous studies.13,14
The investigational product does not require reconstitution, as it is a ready-to-use liquid. In addition to this advantage, we have shown in this study that it can be safely administered at a higher rate, thereby reducing the infusion time by five fold. For patients with A1PI deficiency who need to be maintained on A1PI augmentation therapy on a weekly basis, a significant reduction in the infusion time would potentially improve the patients’ quality of life.
Patients with A1PI deficiency can develop the first signs and symptoms of lung disease as early as their mid-20s, but symptoms typically first develop between 30 and 40 years of age in smokers and later (50–60 years) in nonsmokers, which is older than the median age of subjects in the current study population.15,16 A recent study with a similar A1PI product (Aralast NP®, Baxter Healthcare Corporation) in patients with type 1 diabetes demonstrated that A1PI administration was well tolerated and safe in both pediatric (8–15 years) and adult (16–35 years) subjects.17 Additionally, the AE profile observed in this study with healthy volunteers is consistent with that reported in A1PI-deficient subjects. Thus, these observations suggest that the AE profile associated with A1PI administration is not age dependent and that the findings reported in this study are relevant for the A1PI-deficient patient population.
Conclusion
This study used a novel, placebo-masked, infusion rate-controlled study design to demonstrate the safety and tolerability of an A1PI product at an increased infusion rate (0.2 mL/kg/minute), resulting in a shorter infusion duration.
Acknowledgments
We would like to thank Dr David R Mathews and his clinical study team for their assistance in the conduct of the study. We would also like to express our appreciation to Darby Stephens and the study team for their contributions to the execution of the study, Pnina Strauss for her review of the manuscript, and Clair Firth for preparation of the manuscript.
Disclosure
Leock Y Ngo, Jacqueline Dyck-Jones, David Gelmont, and Leman Yel are employees of Baxter Healthcare Corporation. Adam Haeberle was employed by Baxter Healthcare Corporation at the time of the study. All authors except Adam Haeberle own stocks or shares in Baxter Healthcare Corporation. David Gelmont holds a patent in Baxter Healthcare Corporation. Both the study and the manuscript were funded in their entirety by Baxter Healthcare Corporation. The authors report no further conflicts of interest in this work.
References
Eriksson S. Alpha1-antitrypsin deficiency: lessons learned from the bedside to the gene and back again. Historic perspectives. Chest. 1989;95(1):181–189. | |
Cole RB, Nevin NC, Blundell G, Merrett JD, McDonald JR, Johnston WP. Relation of alpha-1-antitrypsin phenotype to the performance of pulmonary function tests and to the prevalence of respiratory illness in a working population. Thorax. 1976;31(2):149–157. | |
International Conference on Harmonisation. ICH harmonised tripartite guideline: guideline for good clinical practice [webpage on the Internet]. E6(R1). June 10, 1996. Available from http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf. Accessed April 4, 2014. | |
Sandhaus RA, Stocks J, Rouhani FN, Brantly M, Strauss P. Biochemical efficacy and safety of a new, ready-to-use, liquid alpha-1-proteinase inhibitor, GLASSIA (alpha1-proteinase inhibitor (human), intravenous). COPD. 2014;11(1):17–25. | |
Stocks JM, Branity M, Pollack D, et al. Multi-center study: the biochemical efficacy, safety and tolerability of a new α1−proteinase inhibitor, Zemaira. COPD. 2006;3(1):17–23. | |
Stocks JM, Brantly ML, Wang-Smith L, et al. Pharmacokinetic comparability of Prolastin®-C to Prolastin® in alpha1-antitrypsin deficiency: a randomized study. BMC Clin Pharmacol. 2010;10:13. | |
Louie SG, Sclar DA, Gill MA. Aralast: a new alpha1-protease inhibitor for treatment of alpha-antitrypsin deficiency. Ann Pharmacother. 2005;39(11):1861–1869. | |
Lund PE, Wassbäck G, Thomas O, Carlsson T, Schött U. Comparison of two infusion rates of antithrombin concentrate in cardiopulmonary bypass surgery. Perfusion. 2010;25(5):305–312. | |
Gelfand EW, Hanna K; IGIV-C Increased Maximum Infusion Rate Study Group. Safety and tolerability of increased rate of infusion of intravenous immunoglobulin G, 10% in antibody-deficient patients. J Clin Immunol. 2006;26(3):284–290. | |
Ellis ME, al-Hokail AA, Clink HM, et al. Double-blind randomized study of the effect of infusion rates on toxicity of amphotericin B. Antimicrob Agents Chemother. 1992;36(1):172–179. | |
Dirksen A, Dijkman JH, Madsen F, et al. A randomized clinical trial of alpha(1)-antitrypsin augmentation therapy. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1468–1472. | |
Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009;33(6):1345–1353. | |
Stoller JK, Fallat R, Schluchter MD, et al. Augmentation therapy with alpha1-antitrypsin: patterns of use and adverse events. Chest. 2003;123(5):1425–1434. | |
Wencker M, Banik N, Buhl R, Seidel R, Konietzko N. Long-term treatment of alpha1-antitrypsin deficiency-related pulmonary emphysema with human alpha1-antitrypsin. Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL)-alpha1-AT-study group. Eur Respir J. 1998;11(2):428–433. | |
American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818–900. | |
Fregonese L, Stolk J. Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. Orphanet J Rare Dis. 2008;3:16. | |
Ehlers MR, Gottlieb PA, Herold K, et al. Alpha-1 antitrypsin therapy in new-onset type 1 diabetes: interim results from Part I of the RETAIN study [webpage on the Internet]. Presented at: European Association for the Study of Diabetes, Berlin, Germany, October 1–5, 2012. Available from: http://www.immunetolerance.org/professionals/publications/alpha-1-antitrypsin-therapy-new-onset-type-1-diabetes-interim-results-par. Accessed April 4, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.