")
Back to Journals » Clinical Ophthalmology » Volume 13
Safety and efficacy of twice daily administration of KPI-121 1% for ocular inflammation and pain following cataract surgery
Authors Kim T, Sall K , Holland EJ, Brazzell RK, Coultas S , Gupta PK
Received 30 August 2018
Accepted for publication 6 December 2018
Published 27 December 2018 Volume 2019:13 Pages 69—86
DOI https://doi.org/10.2147/OPTH.S185800
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Terry Kim,1 Kenneth Sall,2 Edward J Holland,3 R Kim Brazzell,4 Susan Coultas,4 Preeya K Gupta1
1Department of Ophthalmology, Duke University Eye Center, Durham, NC, USA; 2Sall Research Medical Center, Inc, Artesia, CA, USA; 3Department of Ophthalmology, Cincinnati Eye Institute, University of Cincinnati, Cincinnati, OH, USA; 4Kala Pharmaceuticals, Inc, Waltham, MA, USA
Purpose: KPI-121 is a nanoparticle suspension of loteprednol etabonate with improved ocular pharmacokinetics compared with marketed formulations. The efficacy and safety of KPI-121 1% ophthalmic suspension (INVELTYS™) dosed twice daily (BID) were evaluated in participants who had undergone cataract surgery.
Materials and methods: In two multicenter, randomized, double-masked, parallel-group, vehicle-controlled clinical trials, 386 participants with ≥ grade 2 anterior chamber cells (≥6 cells) on the day following routine cataract surgery were treated with KPI-121 1% and 325 participants were treated with placebo (vehicle); each group was dosed BID for 2 weeks. Primary efficacy endpoints were complete resolution of ocular inflammation by slit-lamp biomicroscopy and complete resolution of subject-rated ocular pain at Days 8 and 15 with no rescue medication before Day 15. Safety assessments included adverse events (AEs), visual acuity, intraocular pressure measurements, and evaluation of ocular AEs by slit-lamp biomicroscopy and dilated ophthalmoscopy.
Results: Both trials achieved statistical significance favoring KPI-121 1% BID for both primary efficacy endpoints. Combined data analysis showed that significantly more participants treated with KPI-121 vs vehicle achieved complete resolution of anterior chamber cells at Days 8 and 15 (P≤0.0001) and complete clearing of ocular pain at Days 4, 8, and 15 (P<0.0001). AEs were reported more frequently with vehicle than KPI-121.
Conclusion: KPI-121 1% ophthalmic suspension was effective in resolving postoperative ocular inflammation and pain when dosed BID for 2 weeks in patients following cataract surgery. KPI-121 was found to be safe and well tolerated in both trials.
Keywords: loteprednol etabonate, nanoparticle, mucus penetrating particles, postoperative ocular inflammation, pain
Introduction
Intraocular inflammation is an anticipated sequela of intraocular surgery, such as cataract removal and intraocular lens implantation, and is manifested principally as conjunctival injection, corneal edema, ciliary flush, and aqueous cells and flare. In general, trauma to the internal structures of the eye is accompanied by the production of prostaglandins and other vasoactive moieties, an increase in blood flow to the affected area, and extravasation of protein and cellular blood elements. Untreated inflammation may lead to complications such as cystoid macular edema and corneal scarring.1,2 Thus, managing and treating postoperative inflammation is an important goal following cataract surgery.
Current postoperative medication regimens commonly include topical corticosteroids.3 Treatment with corticosteroids is employed to reduce pain and discomfort, and to facilitate recovery of the blood–aqueous barrier. When administered at the time of surgery and during the immediate postoperative period, corticosteroids can reverse the clinical manifestations of inflammation.4 In the US, topical corticosteroids are routinely prescribed with four times daily (QID) dosing for at least 2 weeks following cataract surgery.
KPI-121 1% (INVELTYS™) was developed by Kala Pharmaceuticals, Inc. (Waltham, MA, USA) and recently approved by the US Food and Drug Administration (FDA) for the treatment of postoperative inflammation and pain following ocular surgery.5 KPI-121 1% contains loteprednol etabonate (LE) 1% formulated using a proprietary technology known as mucus penetrating particles (MPP). MPP utilizes nanoparticles of drug formulated to enhance penetration of loteprednol through the mucus layer of the tear film, resulting in increased penetration into ocular tissues.
LE was chosen because it is an ester corticosteroid and is rapidly absorbed into ocular issues. After exerting its effects, loteprednol is rapidly metabolized to inactive metabolites and has been reported to have fewer side effects than traditional glucocorticoids.6 Preclinical studies have shown that loteprednol formulated with MPP technology achieved higher ocular exposure than Lotemax® ophthalmic suspension 0.5% (Bausch + Lomb, Rochester, NY, USA), with peak concentrations approximately three-fold higher in ocular tissues and aqueous humor.7 The profile of KPI-121 has the potential to deliver potent anti-inflammatory activity with less frequent dosing, while still retaining the safety profile of loteprednol.
The purpose of the current research was to investigate the efficacy and safety of a twice daily (BID) dosing regimen of KPI-121 1% ophthalmic suspension compared to placebo (hereafter referred to as vehicle) in subjects who experienced inflammation following routine, uncomplicated cataract surgery. Herein, the results are presented for two randomized, double-masked, vehicle-controlled, parallel-group trials comparing the safety and efficacy of KPI-121 1% to vehicle in the treatment of subjects with postsurgical inflammation and pain following cataract surgery.
Materials and methods
Patients
These trials included two samples of patients who underwent routine, uncomplicated cataract surgery (eg, phacoemulsification with posterior chamber intraocular lens implantation, not combined with any other surgery), had ≥ grade 2 anterior chamber cells (ie, ≥6 cells), and had potential postoperative Snellen distance visual acuity (VA) by pinhole method of at least 20/200 in the study eye. The first trial (hereafter referred to as Trial 1) was conducted at 24 sites (Table S1) within the US between May 2014 and December 2014 and the second trial (hereafter referred to as Trial 2) was conducted at 35 sites (Table S2) within the US between June 2016 and March 2017. Each trial was designed and monitored in accordance with the ethical principles of Good Clinical Practice and the Declaration of Helsinki, and Institutional Review Board approval was obtained by each trial site. Written informed consent was obtained from all subjects at the time of their enrollment and prior to any screening evaluations for the trial.
Individuals of either sex or any race/ethnicity who were ≥18 years and were candidates for routine, uncomplicated cataract surgery were eligible for participation in the trials. Patients who required concurrent ocular or non-ocular therapy with any type of medication (eg, nonsteroidal anti-inflammatory drugs, mast cell stabilizers, antihistamines, or decongestants) within 2 days prior to surgery and for the duration of the study were excluded from the trial as were patients with a history of glaucoma, intraocular pressure (IOP) >21 mmHg at the screening or randomization visit(s), or were being treated for glaucoma in either eye. Also excluded were patients who: 1) had penetrating intraocular surgery in the study eye within 3 months or within 2 weeks in the fellow eye; 2) had selective laser trabeculoplasty within 3 months prior to surgery or had corneal refractive surgery, glaucoma surgery, or corneal transplantation (full thickness, anterior, or posterior) within a year prior to enrollment or were unstable and/or required medication; 3) had active uveitis in either eye or a diagnosis of any type of infection, disease, and/or severe/serious condition that would interfere with study drug effectiveness or study compliance.
Design and treatment
Both studies were randomized, double-masked, parallel-group trials. Randomization was stratified by trial site. In Trial 1, two concentrations and dosing regimens of KPI-121 vs vehicle were administered to one eye for 2 weeks. Subjects were randomized to one of four treatment arms in a 2:2:1:1 ratio: 1) KPI-121 0.25% QID, 2) KPI-121 1% BID, 3) vehicle A QID, 4) vehicle B BID. Following the completion of Trial 1, which demonstrated favorable safety and efficacy profiles for both KPI-121 treatment groups, Trial 2 was initiated to evaluate the safety and efficacy of KPI-121 1% BID compared with vehicle BID administered to one eye for 2 weeks.
As illustrated in Figure 1, subjects in both trials completed up to seven clinic visits (including the surgery day) over 18–33 days of total planned trial duration. Screening occurred between 14 and 1 day(s) prior to surgery, and subjects who met the preoperative screening inclusion/exclusion criteria were allowed to participate in the trial. At Day 0 (surgery), subjects underwent routine, uncomplicated cataract surgery according to normal procedures. Randomization occurred on the day following surgery (Day 1). Subjects who met the qualifying postoperative randomization criteria were eligible for randomization and initiated dosing with study treatment on that day. Subjects self-administered 1–2 drops in the study eye (defined as the surgery eye) for 14 days and recorded dosing information in a daily diary. Following initiation of study treatment dosing, subjects returned to the clinic to be evaluated at Days 4, 8, and 15. The last dose of study treatment was administered on the morning of the Day 15 visit, upon completion of the 14 days of dosing. Following the Day 15 visit, subjects were asked to return to the clinic once between Days 17 and 19 for follow-up and, following completion of all study procedures, were exited from the trial. Efficacy measures were performed on Days 4, 8, and 15. Adverse events (AEs) were assessed across the entire study period.
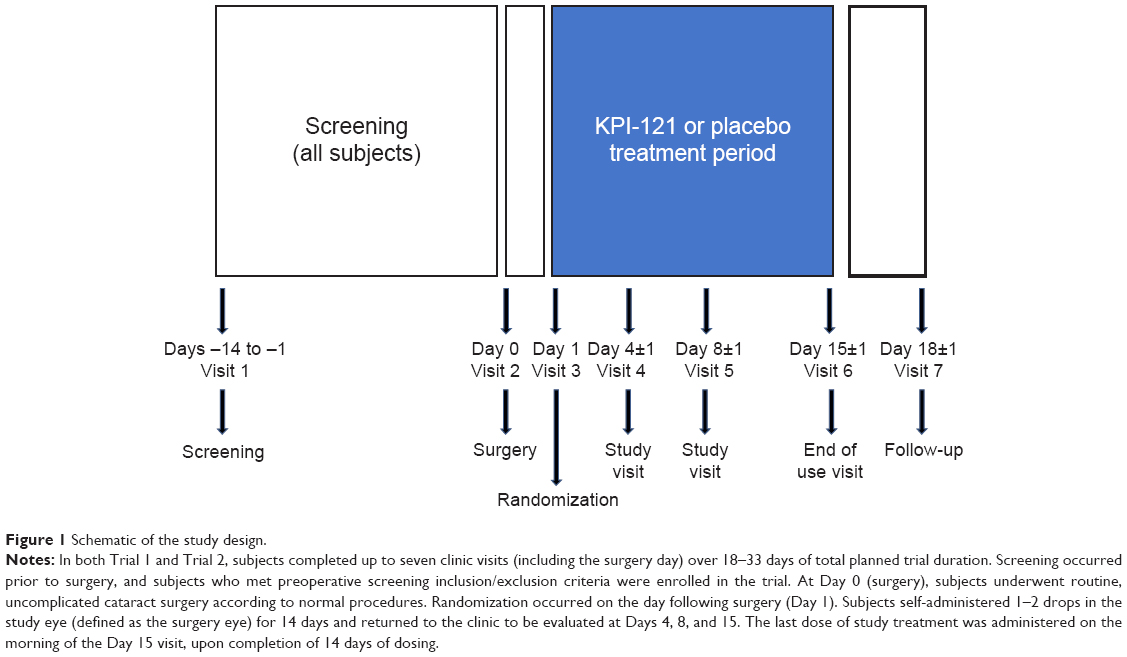 | Figure 1 Schematic of the study design. |
Primary efficacy endpoints
Anterior chamber cell grade (slit-lamp biomicroscopy)
Ocular inflammation was evaluated by an anterior chamber cell assessment of the study eye performed by the investigator via slit-lamp biomicroscopy. Investigators rated the subjects’ anterior chamber cell grade using a 5-point scale (0= no cells seen; 1=1–5 cells; 2=6–15 cells; 3=16–30 cells; 4=>30 cells). Complete resolution of inflammation was defined as an anterior chamber cell grade =0.
Subject-rated ocular pain assessment
Ocular pain was rated by the subject-rated ocular pain assessment using a 6-point scale (0= none; 1= minimal; 2= mild; 3= moderate; 4= moderately severe; 5= severe). Complete resolution of ocular pain was defined as a grade =0.
Safety evaluation
Safety assessments included evaluation of AEs, Snellen distance VA by pinhole method, IOP measurement, slit-lamp biomicroscopy (palpebral conjunctival erythema, corneal edema, hyphema, ciliary flush, and bulbar conjunctival injection), dilated ophthalmoscopy, and use of rescue medication.
AEs
An AE was defined as any untoward medical occurrence in a clinical investigation subject administered a pharmaceutical product that does not necessarily have a causal relationship with this treatment. Treatment-emergent AEs were defined as those events with onset after the first dose of study treatment or, if occurring prior to the first dose, worsened after administration of the first dose. The severity of AEs was classified as mild, moderate, or severe. The relatedness of AEs to the investigational product was also evaluated. Serious AEs were defined as ones resulting in death or that were life threatening, represented a persistent or significant incapacity or substantial disruption of the ability to conduct normal life functions, resulted in inpatient hospitalization or prolongation of existing hospitalization, a congenital anomaly/birth defect, or the occurrence of a significant medical event.
Snellen distance VA
Snellen distance VA was assessed at all study visits except the surgery visit (Day 0). VA measurement was performed with the Snellen eye chart using the pinhole at a distance of 20 feet (6 m).
IOP measurement
IOP measurements were performed at all study visits except the surgery visit (Day 0) using Goldmann applanation tonometry according to the investigator’s standard procedure.
Slit-lamp biomicroscopy
Slit-lamp biomicroscopy was also used as a safety measure to assess palpebral conjunctival erythema, corneal edema, hyphema, ciliary flush, and bulbar conjunctival injection at all study visits except the surgery visit (Day 0).
Dilated ophthalmoscopy
Dilated ophthalmoscopy assessment included evaluation of the optic nerve head for pallor and cupping (cup/disc ratio) and was performed at screening and on Day 15. For each subject, the investigator determined whether direct or indirect ophthalmoscopy should be used. Findings were recorded as normal or abnormal.
Use of rescue medication
At any time during the treatment period, any subject not responding adequately to the study treatment could be “rescued,” or placed on alternate therapy, at the investigator’s discretion. Any subject placed on rescue medication discontinued use of the study treatment and continued trial participation through the final visit (Day 17–19). Subjects who received rescue medication prior to Day 15 were considered treatment failures, but the need for rescue medication was not considered an AE. At some sites, anti-inflammatory medications were administered starting on Day 15 in the absence of anterior chamber cells as standard-of-care, and this was not considered as a rescue.
Statistical analyses
The primary population for all efficacy analyses was the intent-to-treat (ITT) population, which was defined as all randomized subjects with at least one post-baseline (baseline = Day 1) evaluation. The safety population was defined as all subjects who received at least one dose of the allocated study treatment and from whom at least one safety assessment was obtained after randomization. Imputation for missing data was not applied.
Data from the efficacy and safety evaluations conducted during Trials 1 and 2 were analyzed separately and pooled, given the similarity in study design and purpose. The two vehicle groups in Trial 1 were pooled for all analyses of Trial 1 findings, because preliminary analyses illustrated no statistically significant differences between the two vehicle groups for the primary efficacy endpoints (complete resolution of anterior chamber cells at Day 8 maintained through Day 15 without rescue medication prior to Day 15 [P=0.3130]; complete resolution of ocular pain at Day 8 maintained through Day 15 without rescue medication prior to Day 15 [P=0.6320]). Because the purpose of the present research was to evaluate KPI-121 1% BID compared with vehicle, the results from the KPI-121 0.25% QID group from Trial 1 are not summarized here. For pooled efficacy analyses, all vehicle groups (Trial 1 QID, Trial 1 BID, and Trial 2 BID) were pooled; for pooled safety analyses the Trial 1 and Trial 2 BID vehicle groups were pooled but the Trial 1 vehicle QID group was excluded from the analyses.
Primary efficacy endpoints in both trials were evaluated using hierarchical statistical testing using the chi-squared statistic, with the provision that testing would cease if any single hypothesis had a P-value >0.05. The primary endpoints were tested in the following sequence, comparing KPI-121 1% vs vehicle: 1) the difference in the proportion of study eyes with complete resolution of anterior chamber cells (grade =0) at postoperative Day 8 maintained through Day 15; 2) the difference in the proportion of study eyes with complete resolution of pain (grade =0) at postoperative Day 8 maintained through Day 15. Key non-primary endpoints were defined as the difference in the proportions of study eyes with: 1) complete resolution of anterior chamber cells (grade =0) at postoperative Day 15; 2) complete resolution of ocular pain (grade =0) at postoperative Day 4; and 3) complete resolution of ocular pain at postoperative Day 15. For each of these primary and key non-primary endpoints, subjects treated with rescue medication prior to Day 15 were considered as nonresponders.
Separate and pooled summaries were created for all safety measures: evaluation of AEs, VA, IOP, slit-lamp biomicroscopy, dilated ophthalmoscopy, and use of rescue medication.
Results
Patient disposition and demographics
Across the two trials, 900 subjects were enrolled and included in the ITT populations: 380 subjects in Trial 1 and 520 subjects in Trial 2. Of these 900 subjects, 386 subjects were treated with KPI-121 1% BID, 125 subjects were treated with KPI-121 0.25% QID, 325 subjects were treated with vehicle BID, and 60 subjects were treated with vehicle QID. As illustrated in Table 1, there were no significant differences in the demographics of the enrolled populations between the two trials, and there were no significant differences among the treatment groups within each trial. Mean (range) ages were 67.7 (38–89) years for the pooled KPI-121 1% BID group (N=386) and 69.4 (40–90) years for the pooled vehicle group (N=385). Both trials, considered individually as well as pooled, enrolled more females than males, with a majority of subjects being White and not Hispanic/Latino.
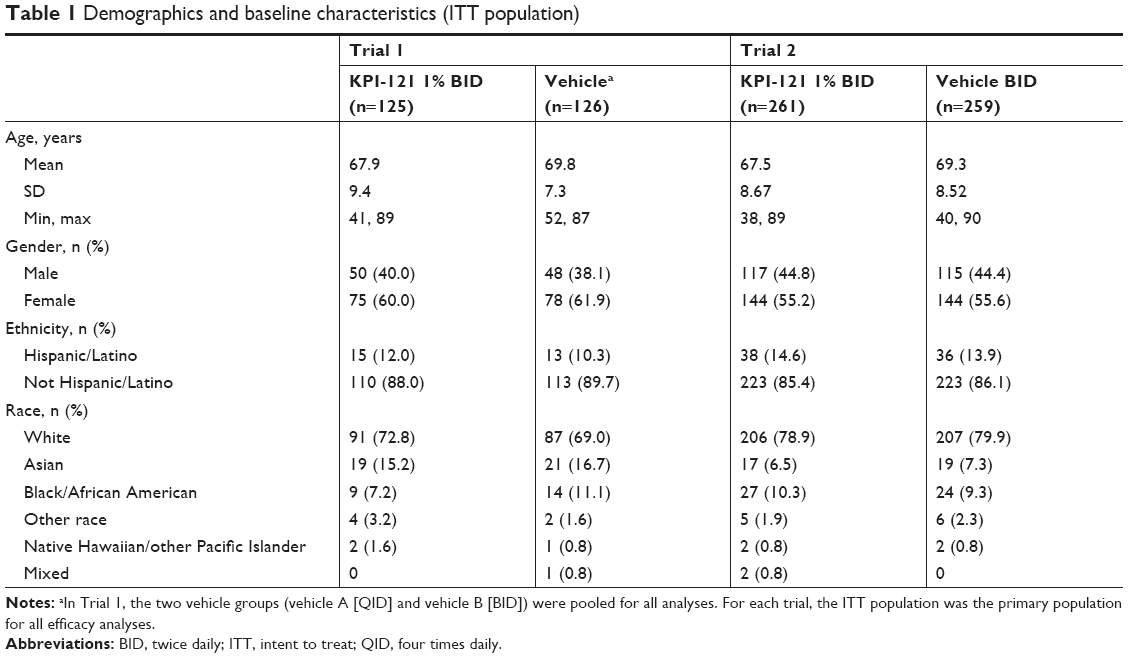 | Table 1 Demographics and baseline characteristics (ITT population) |
Efficacy
Statistically significant between-group differences were observed for both primary efficacy endpoints, in both Trials (Table 2).
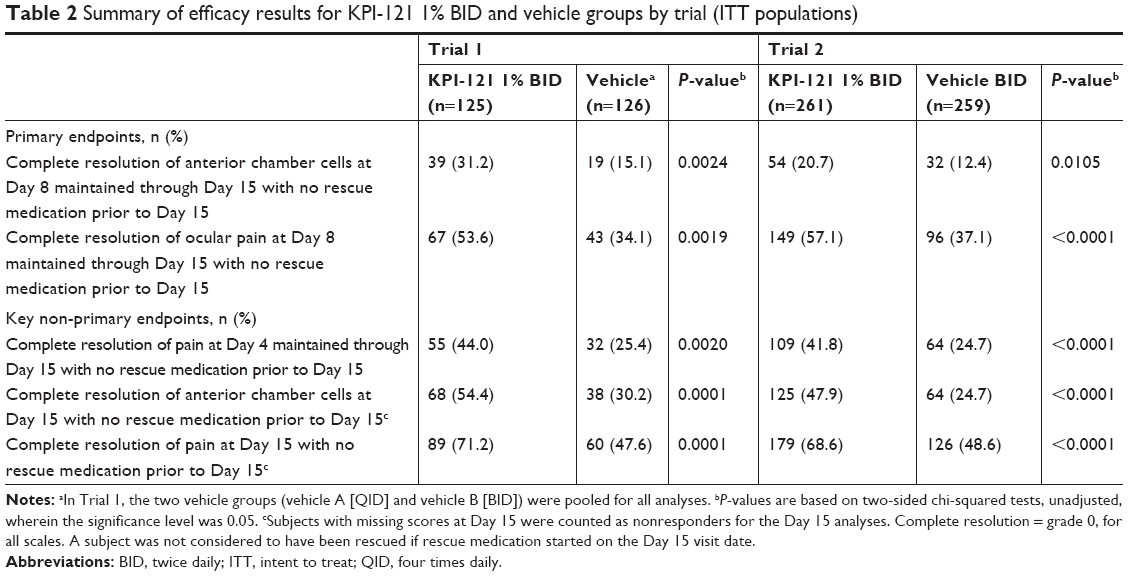 | Table 2 Summary of efficacy results for KPI-121 1% BID and vehicle groups by trial (ITT populations) |
Pooled analyses of the two trials also demonstrated statistically significant results favoring KPI-121 1% over vehicle for each primary and key non-primary endpoint, as summarized in Table 3. Results for these endpoints are presented graphically in Figure 2 (for responders on anterior chamber cells at Days 8 and 15) and Figure 3 (for responders on ocular pain at Days 4, 8, and 15).
 | Table 3 Summary of efficacy results (pooled ITT populations) |
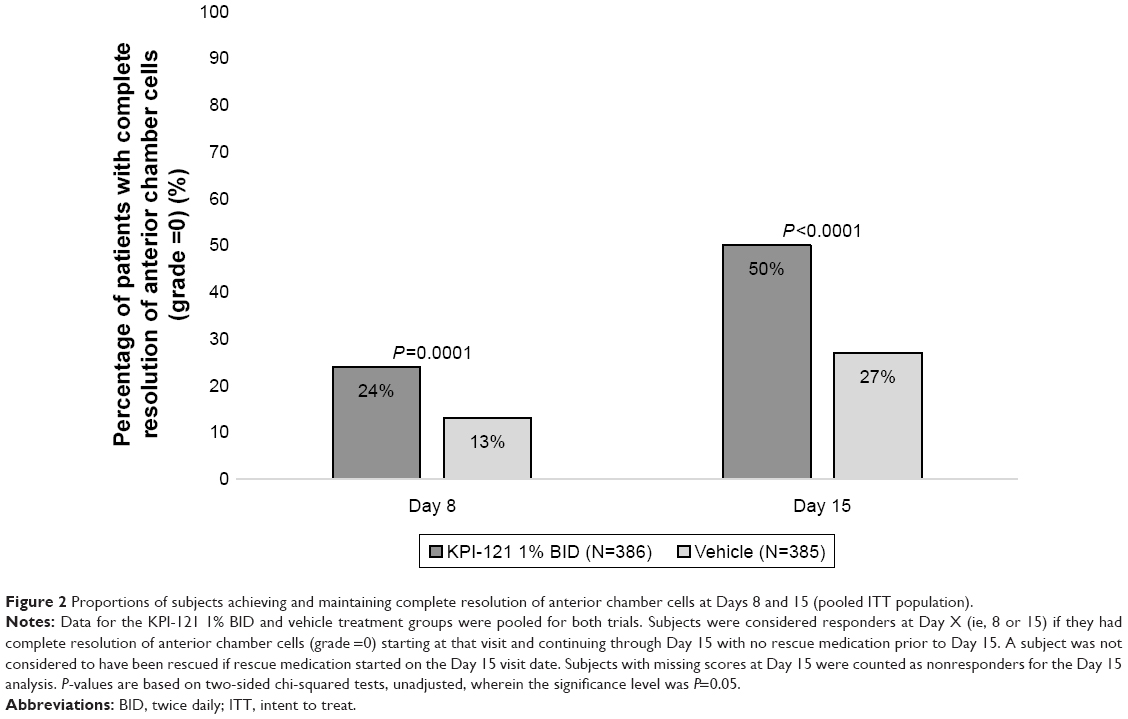 | Figure 2 Proportions of subjects achieving and maintaining complete resolution of anterior chamber cells at Days 8 and 15 (pooled ITT population). |
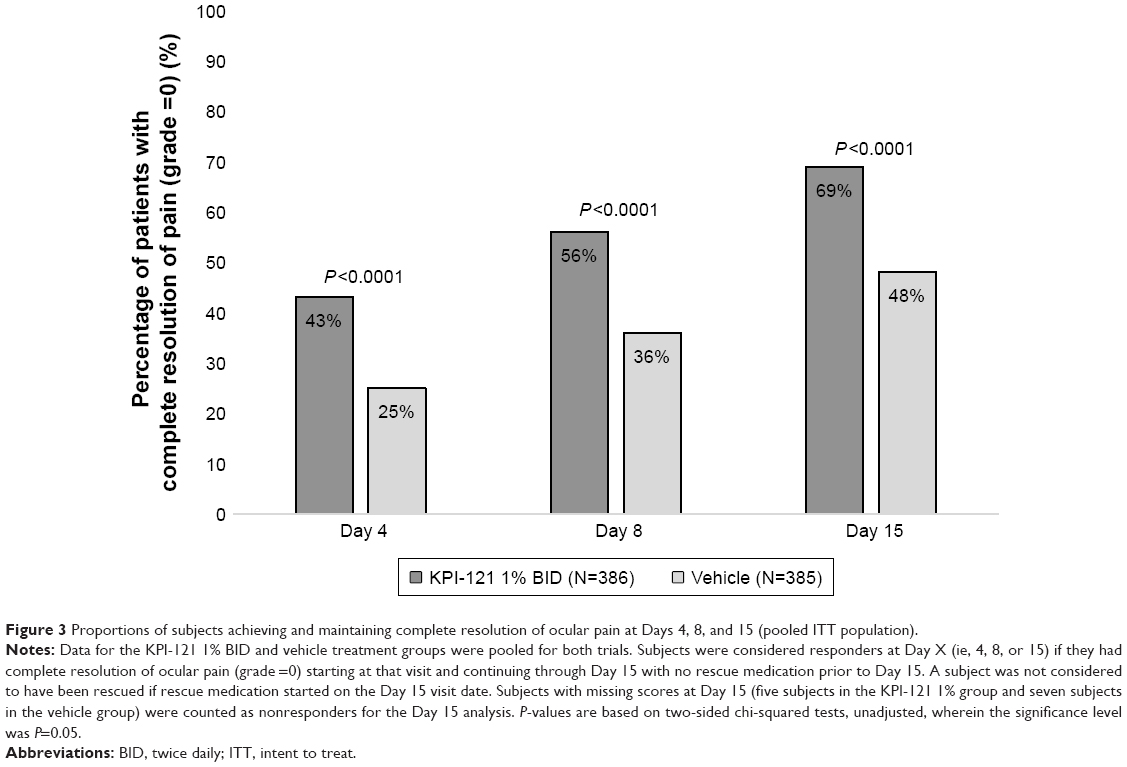 | Figure 3 Proportions of subjects achieving and maintaining complete resolution of ocular pain at Days 4, 8, and 15 (pooled ITT population). |
In Trial 1, complete resolution of anterior chamber cells at Day 8 and maintained through Day 15 with no rescue medication prior to Day 15 was demonstrated in 31.2% of subjects in the KPI-121 1% BID group, compared with 15.1% of subjects in the pooled vehicle group (between-group difference for percent responders: 16.1, 95% CI, 5.9–26.4, P=0.0024). Complete resolution of ocular pain at Day 8 and maintained through Day 15 with no rescue medication prior to Day 15 was demonstrated in 53.6% of subjects in the KPI-121 1% BID group, compared with 34.1% of subjects in the pooled vehicle group (between-group difference for percent responders: 19.5, 95% CI, 7.4–31.5, P=0.0019). Results for the key non-primary efficacy analysis were also significantly different favoring KPI-121 1% over vehicle for each analysis.
In Trial 2, complete resolution of anterior chamber cells at Day 8 and maintained through Day 15 with no rescue medication prior to Day 15 was demonstrated in 20.7% of subjects in the KPI-121 1% group, compared with 12.4% of subjects in the vehicle BID group (between-group difference for percent responders: 8.3, 95% CI, 2.0–14.7, P=0.0105). Complete resolution of ocular pain at Day 8 and maintained through Day 15 with no rescue medication prior to Day 15 was demonstrated in 57.1% of subjects in the KPI-121 1% group, compared with 37.1% of subjects in the vehicle BID group (between-group difference for percent responders: 20.0, 95% CI, 11.6–28.4, P<0.0001). Results for the key non-primary efficacy analysis were also significantly different favoring KPI-121 1% over vehicle BID for each analysis in both trials (Table 2).
Safety
AEs
In both Trial 1 and 2, considered individually and pooled, the incidence of AEs overall, treatment-related AEs, severe AEs, and AEs leading to discontinuation of treatment was lower in the KPI-121 1% BID group than the vehicle BID group (Tables 4 and 5). AEs were reported by 13.7% (53/386) and 17.5% (57/325) of subjects in the KPI-121 1% BID and vehicle BID groups, respectively. Most AEs were of mild or moderate severity. The most commonly reported (≥1% in any treatment group) preferred terms were eye pain, headache, photophobia, posterior capsule opacification, corneal edema, and nasopharyngitis (Tables 6 and 7).
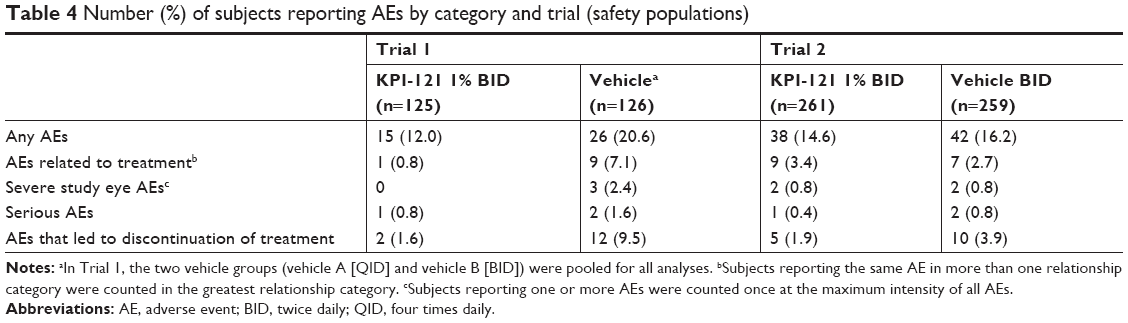 | Table 4 Number (%) of subjects reporting AEs by category and trial (safety populations) |
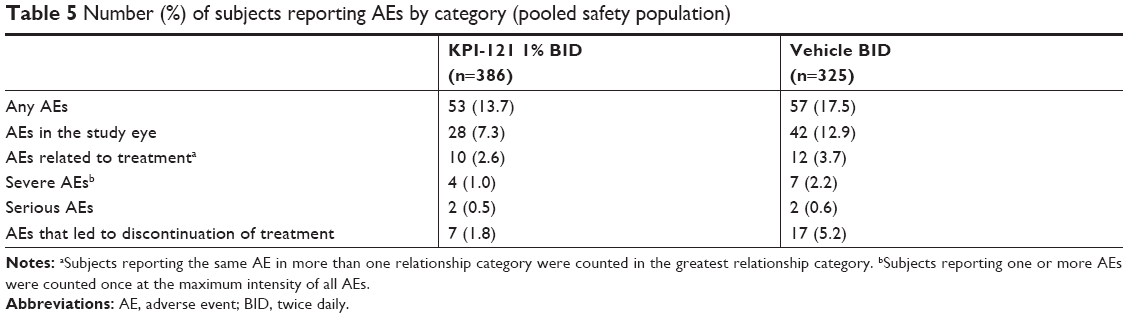 | Table 5 Number (%) of subjects reporting AEs by category (pooled safety population) |
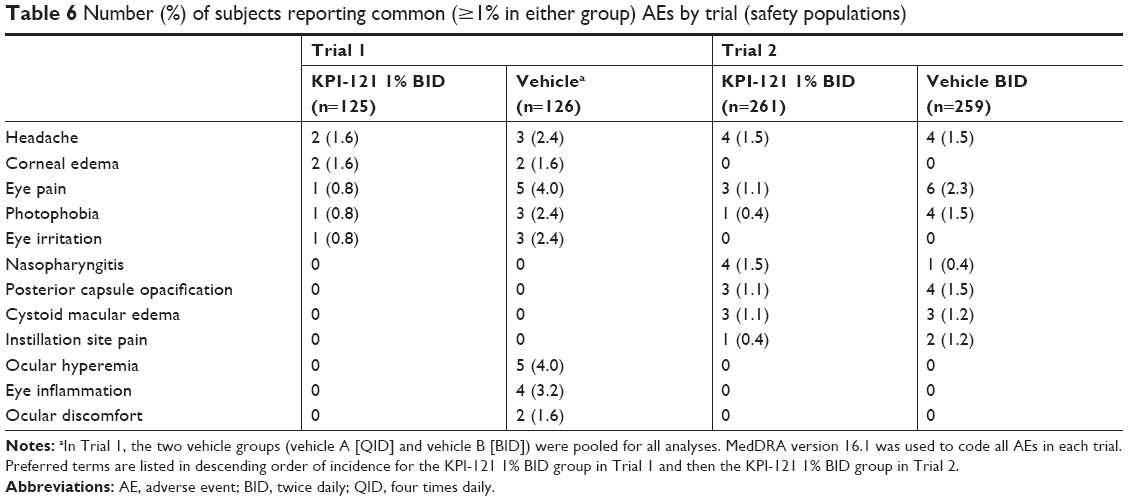 | Table 6 Number (%) of subjects reporting common (≥1% in either group) AEs by trial (safety populations) |
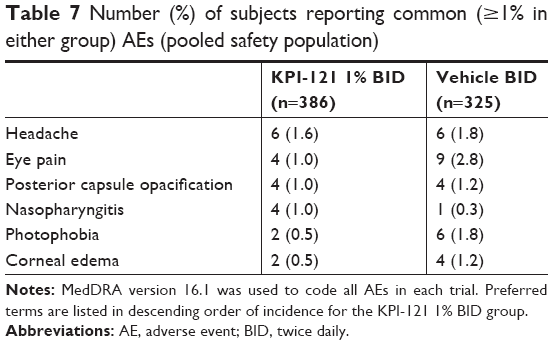 | Table 7 Number (%) of subjects reporting common (≥1% in either group) AEs (pooled safety population) |
VA
In both Trial 1 and Trial 2, a review of Snellen distance VA data revealed no clinically important differences between the KPI-121 1% BID group and the vehicle group in terms of mean changes from baseline in VA at any post-baseline assessment. Pooled summaries illustrated that changes from baseline in VA were negligible in both the KPI-121 1% BID and vehicle groups.
IOP
In Trial 1 and Trial 2, as well as in the pooled analysis of both trials, mean IOP measurements of study eyes at each scheduled visit as well as increased IOP were similar across treatment groups (Table 8). Additionally, IOP elevations of ≥10 mmHg compared with baseline and a raw IOP measurement of ≥21 mmHg in the study eye were experienced by 0.5% (2/386) and 0% (0/325) of subjects in the KPI-121 1% BID and vehicle BID groups, respectively. Using a very strict definition of IOP elevation, ≥5 mmHg increase and a raw IOP measurement of ≥21 mmHg in the study eye, again both groups experienced similar incidence, 1.3% (5/386) and 0.6% (3/325) in the KPI-121 1% BID and vehicle groups, respectively. These results indicate that no unexpected safety signal was observed relative to IOP following KPI-121 1% treatment.
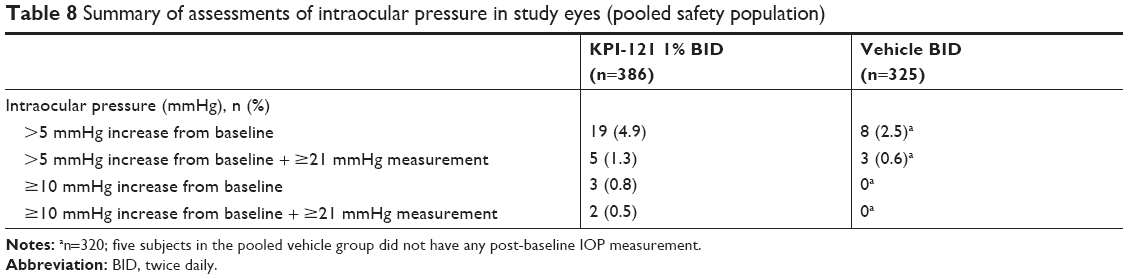 | Table 8 Summary of assessments of intraocular pressure in study eyes (pooled safety population) |
Slit-lamp biomicroscopy
In Trial 1 and Trial 2, as well as in the pooled analysis of both trials, no clinically important differences were observed based on safety evaluations of slit-lamp biomicroscopy (palpebral conjunctival erythema, corneal edema, hyphema, ciliary flush, and bulbar conjunctival injection) between the KPI-121 1% BID and vehicle groups, and no significant differences from baseline were observed in either group.
Dilated ophthalmoscopy
In Trial 1 and Trial 2, as well as in the pooled analysis of both trials, no clinically important differences between the KPI-121 1% BID and vehicle groups were observed based on evaluations from dilated ophthalmoscopy. There was a low prevalence of abnormal disc pallor and similar cup/disc ratio scores at both screening and Day 15 for KPI-121 1% BID and vehicle groups.
Use of rescue medication
Rescue medication was used at any time prior to Day 15 in Trial 1 by 14 patients (11.2%) in the KPI-121 1% group vs 48 patients (38.1%) in the vehicle group, and in Trial 2 by 45 patients (17.2%) in the KPI-121 1% group vs 96 patients (37.1%) in the vehicle group.
Discussion
Results from two double-masked, randomized, vehicle-controlled trials provide evidence of the efficacy of KPI-121 1% ophthalmic suspension administered twice daily for 2 weeks in the resolution of anterior chamber cells and ocular pain following cataract surgery. In both trials, a significantly greater proportion of subjects treated with KPI-121 1% BID compared to vehicle demonstrated a response to treatment, in terms of complete resolution of anterior chamber cells (ie, grade =0) at Days 8 and 15 (each maintained through Day 15 without use of rescue medication) and complete clearing (ie, grade =0) of ocular pain at Days 4, 8, and 15 (each maintained through Day 15 without use of rescue medication). Additionally, summaries of safety measures, including AEs, VA, IOP, slit-lamp biomicroscopy, dilated ophthalmoscopy, and use of rescue medication, demonstrated that KPI-121 1% BID was also found to be safe and well tolerated with no new or unexpected safety concerns observed during the trial.
The AE profile was composed primarily of events that were local to the site of treatment, mild in severity, reversible, and nonserious, and most reported preferred terms were consistent with those that would be expected in a population that had recently undergone routine, uncomplicated cataract surgery and received topical ophthalmic corticosteroid treatment. AEs recognized as common with corticosteroid use were reported with low incidence in the KPI-121 1% group. In the pooled analysis of AEs, the incidence of AEs was lower in the KPI-121 1% group than in the vehicle group for all AEs overall, treatment-related AEs, severe AEs, and AEs leading to discontinuation of treatment.
As demonstrated in these two studies, KPI-121 1% with MPP technology delivers a desirable combination of robust efficacy with twice daily dosing, a low overall incidence of AEs, and the favorable IOP-safety profile consistently demonstrated with LE.8–14 While the efficacy and safety of LE in treating postoperative inflammation and pain are well established,15–22 all currently available formulations require QID dosing. This is due at least in part to the eye’s innate ability to remove foreign material, including suspended drug particles, from the ocular surface. One of the key mechanisms of this removal involves mucin, the primary component of mucus, which provides a key protective role for the ocular surface. The cornea and conjunctiva are covered with a 3- to 40-μm layer of mucus.7,23–25 The outer layer of secreted and other mucins primarily traps and rapidly eliminates allergens, pathogens, and other particles (eg, corticosteroids in eye drops) while the cell-bound mucins of the inner glycocalyx protect the cornea from abrasive stress and turn over much less rapidly.7,26 The ability to penetrate the outer mucin matrix and reach the glycocalyx will likely increase the penetration and retention of drug particles on the ocular surface and enhance drug release to the underlying tissues.7 In order to penetrate the mucin meshwork effectively, particles must be sufficiently small in size (<500 nm) and have the ability to overcome the adhesive nature of the ocular mucus layer.27,28
MPP technology uses proprietary methods to create a novel nanoparticle formulation of LE designed to evade these mucus barriers. Coarse drug particles are milled in the presence of an MPP enabling surface-altering agent until the particle size is reduced to ~200–400 nm. In an ocular pharmacokinetic study in rabbits, a 0.4% suspension of LE-MPP produced peak concentrations approximately three-fold higher in ocular tissues (cornea, conjunctiva, iris/ciliary body, retina) and aqueous humor compared to Lotemax® suspension.7
KPI-121 1% is a novel suspension of MPP-coated LE nanoparticles formulated with excipients approved by FDA for ophthalmic use.7 LE was selected for the formulation based upon several criteria including high lipophilicity, high binding affinity for the glucocorticoid receptor, and extensive and predictable metabolism to inactive metabolites by tissue esterases.29
Conclusion
KPI-121 1% given BID was effective in resolving postoperative inflammation and pain following cataract surgery, based on analyses demonstrating statistical significance compared with vehicle on the primary and key non-primary efficacy endpoints of pain and inflammation. It was safe and well tolerated with no new or unexpected safety concerns observed during the trials. KPI-121 1% offers clinicians an efficacious BID treatment option for inflammation and pain after ocular surgery which provides significant benefit to patients while retaining the favorable safety profile of loteprednol.
Disclosure
This study was sponsored and funded by Kala Pharmaceuticals, Inc, which participated in the design of the study and oversaw its conduct, monitoring, and analysis. Dr Kim, Dr Sall, Dr Holland, and Dr Gupta are consultants for Kala Pharmaceuticals, Inc. Dr Brazzell and Dr Coultas are employees of Kala Pharmaceuticals, Inc. The abstract of this paper was presented at the 2018 American Society of Cataract and Refractive Surgery (ASCRS) Annual Meeting as a paper presentation with interim findings. The paper’s abstract was published in the “Paper Session Abstracts” on the ASCRS conference website: https://ascrs.confex.com/ascrs/18am/meetingapp.cgi/Paper/47263.
References
American Academy of Ophthalmology. Cataract in the adult eye: preferred practice patterns; 2016. Available from: https://www.aao.org/preferred-practice-pattern/cataract-in-adult-eye-ppp-2016. Accessed May 28, 2018. | ||
El-Harazi SM, Feldman RM. Control of intra-ocular inflammation associated with cataract surgery. Curr Opin Ophthalmol. 2001;12(1):4–8. | ||
Shoss BL, Tsai LM. Postoperative care in cataract surgery. Curr Opin Ophthalmol. 2013;24(1):66–73. | ||
Leopold IH. Update on antibiotics in ocular infections. Am J Ophthalmol. 1985;100(1):134–140. | ||
INVELTYS™ (loteprednol etabonate ophthalmic suspension 1%) [package insert]. Waltham, MA: Kala Pharmaceuticals Inc; 2018. | ||
Druzgala P, Hochhaus G, Bodor N. Soft drugs-10. Blanching activity and receptor binding affinity of a new type of glucocorticoid: loteprednol etabonate. J Steroid Biochem Mol Biol. 1991;38(2):149–154. | ||
Schopf L, Enlow E, Popov A, Bourassa J, Chen H. Ocular pharmacokinetics of a novel loteprednol etabonate 0.4% ophthalmic formulation. Ophthalmol Ther. 2014;3(1–2):63–72. | ||
Howes JF, Baru H, Vered M, Neumann RON. Loteprednol etabonate: comparison with other steroids in two models of intraocular inflammation. J Ocul Pharmacol Ther. 1994;10(1):289–293. | ||
Bartlett JD, Horwitz B, Laibovitz R, Howes JF. Intraocular pressure response to loteprednol etabonate in known steroid responders. J Ocul Pharmacol Ther. 1993;9(2):157–165. | ||
Holland EJ, Djalilian AR, Sanderson JP. Attenuation of ocular hypertension with the use of topical loteprednol etabonate 0.5% in steroid responders after corneal transplantation. Cornea. 2009;28(10):1139–1143. | ||
Ilyas H, Slonim CB, Braswell GR, Favetta JR, Schulman M. Long-term safety of loteprednol etabonate 0.2% in the treatment of seasonal and perennial allergic conjunctivitis. Eye Contact Lens. 2004;30(1):10–13. | ||
Rajpal RK, Digby D, D’Aversa G, Mah F, Hollander DA, Conway T. Intraocular pressure elevations with loteprednol etabonate: a retrospective chart review. J Ocul Pharmacol Ther. 2011;27(3):305–308. | ||
Sheppard JD, Scoper SV, Samudre S. Topical loteprednol pretreatment reduces cyclosporine stinging in chronic dry eye disease. J Ocul Pharmacol Ther. 2011;27(1):23–27. | ||
Sheppard JD, Comstock TL, Cavet ME. Impact of the topical ophthalmic corticosteroid loteprednol etabonate on intraocular pressure. Adv Ther. 2016;33(4):532–552. | ||
Comstock TL, Paterno MR, Singh A, Erb T, Davis E. Safety and efficacy of loteprednol etabonate ophthalmic ointment 0.5% for the treatment of inflammation and pain following cataract surgery. Clin Ophthalmol. 2011;5:177–186. | ||
Fong R, Leitritz M, Siou-Mermet R, Erb T. Loteprednol etabonate gel 0.5% for postoperative pain and inflammation after cataract surgery: results of a multicenter trial. Clin Ophthalmol. 2012;6:1113–1124. | ||
Holzer MP, Solomon KD, Sandoval HP, Vroman DT. Comparison of ketorolac tromethamine 0.5% and loteprednol etabonate 0.5% for inflammation after phacoemulsification: prospective randomized double-masked study. J Cataract Refract Surg. 2002;28(1):93–99. | ||
Lane SS, Holland EJ. Loteprednol etabonate 0.5% versus prednisolone acetate 1.0% for the treatment of inflammation after cataract surgery. J Cataract Refract Surg. 2013;39(2):168–173. | ||
Loteprednol Etabonate Postoperative Inflammation Study Group. A double-masked, placebo-controlled evaluation of 0.5% loteprednol etabonate in the treatment of postoperative inflammation. Ophthalmology. 1998;105(9):1780–1786. | ||
Rajpal RK, Fong R, Comstock TL. Loteprednol etabonate ophthalmic gel 0.5% following cataract surgery: integrated analysis of two clinical studies. Adv Ther. 2013;30(10):907–923. | ||
Stewart R, Horwitz B, Howes J, Novack GD, Hart K. Double-masked, placebo-controlled evaluation of loteprednol etabonate 0.5 for postoperative inflammation. J Cataract Refract Surg. 1998;24(11):1480–1489. | ||
Comstock TL, Sheppard JD. Loteprednol etabonate for inflammatory conditions of the anterior segment of the eye: twenty years of clinical experience with a retrometabolically designed corticosteroid. Expert Opin Pharmacother. 2018;19(4):337–353. | ||
King-Smith PE, Fink BA, Fogt N, Nichols KK, Hill RM, Wilson GS. The thickness of the human precorneal tear film: evidence from reflection spectra. Invest Ophthalmol Vis Sci. 2000;41(11):3348–3359. | ||
Prydal JI, Campbell FW. Study of precorneal tear film thickness and structure by interferometry and confocal microscopy. Invest Ophthalmol Vis Sci. 1992;33(6):1996–2005. | ||
Prydal JI, Artal P, Woon H, Campbell FW. Study of precorneal tear film thickness and structure using laser interferometry. Invest Ophthalmol Vis Sci. 1992;33(6):2006–2011. | ||
Mantelli F, Argüeso P. Functions of ocular surface mucins in health and disease. Curr Opin Allergy Clin Immunol. 2008;8(5):477–483. | ||
Olmsted SS, Padgett JL, Yudin AI, Whaley KJ, Moench TR, Cone RA. Diffusion of macromolecules and virus-like particles in human cervical mucus. Biophys J. 2001;81(4):1930–1937. | ||
Sigurdsson HH, Kirch J, Lehr C-M. Mucus as a barrier to lipophilic drugs. Int J Pharm. 2013;453(1):56–64. | ||
Samir A, Bodor N, Imai T. Identification of esterase involved in the metabolism of two corticosteroid soft drugs. Biochem Pharmacol. 2017;127:82–89. |
Supplementary materials
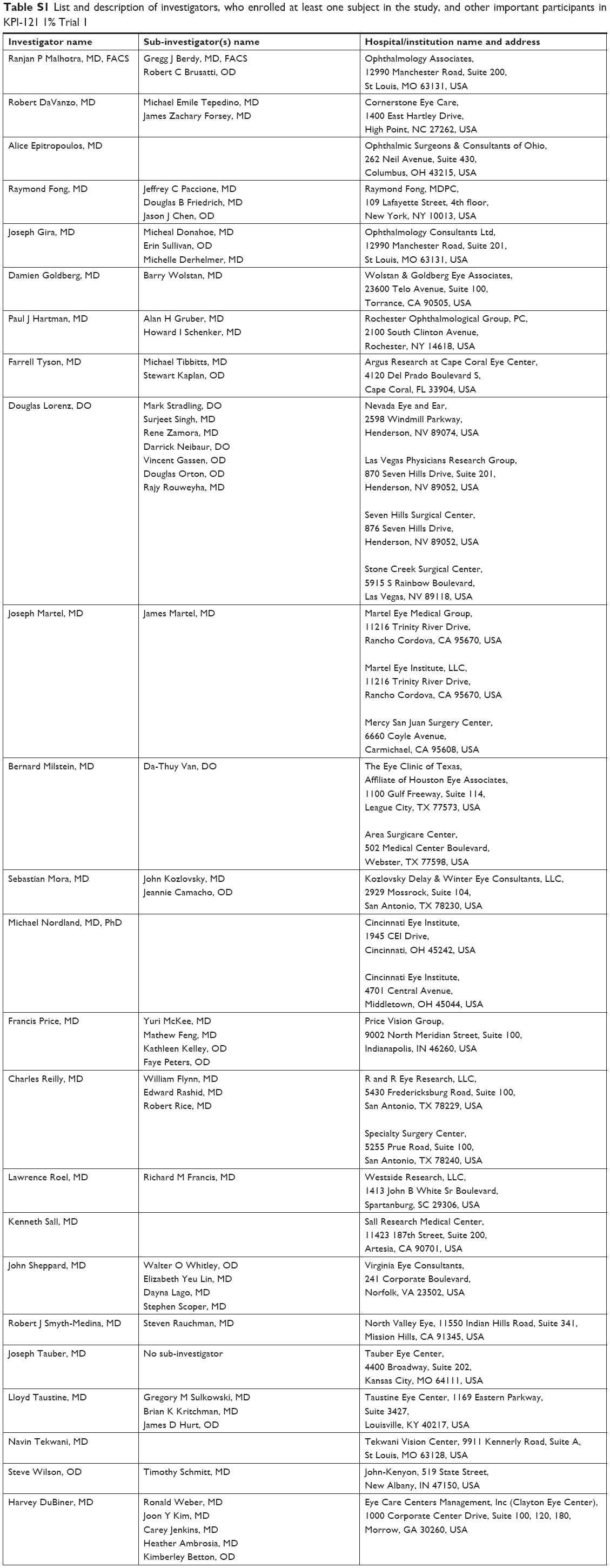 | Table S1 List and description of investigators, who enrolled at least one subject in the study, and other important participants in KPI-121 1% Trial 1 |
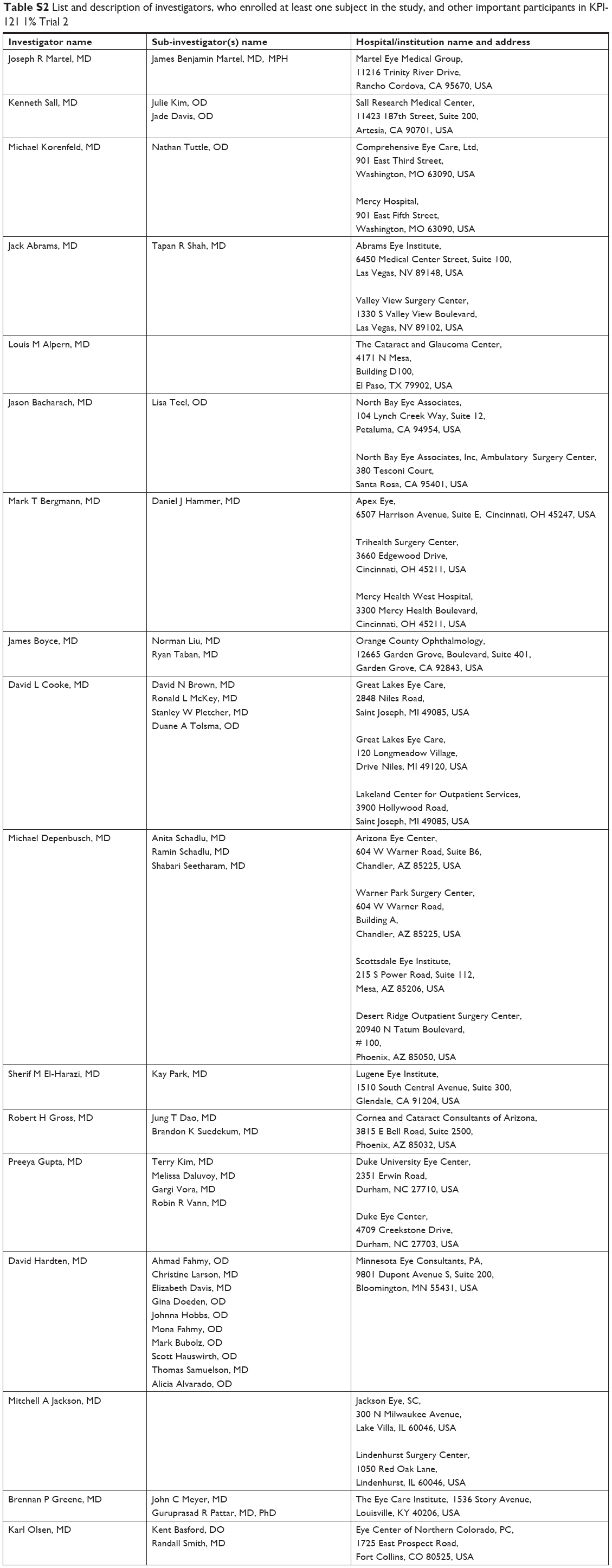 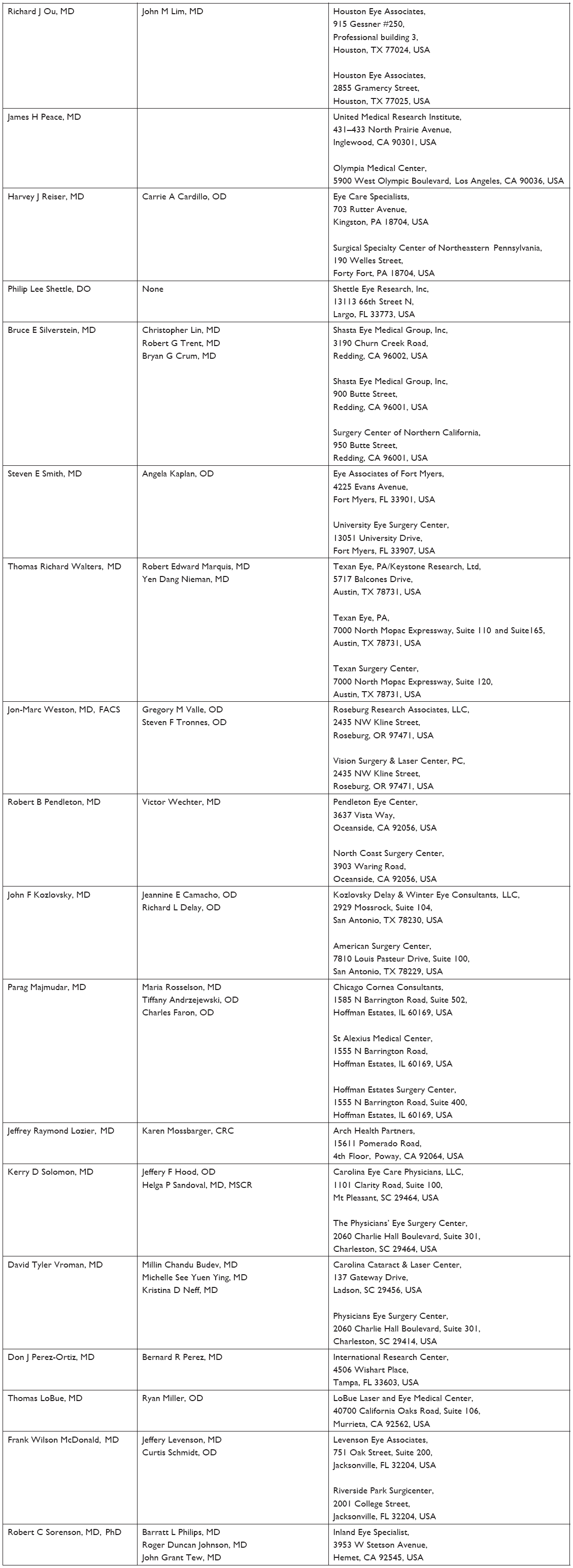 | Table S2 List and description of investigators, who enrolled at least one subject in the study, and other important participants in KPI-121 1% Trial 2 |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.