")
Back to Journals » Clinical Ophthalmology » Volume 13
Safety and efficacy of amniotic cytokine extract in the treatment of dry eye disease
Authors Yeu E , Goldberg DF, Mah FS, Beckman KA , Luchs JI , Solomon JD, White DE, Gupta PK
Received 30 January 2019
Accepted for publication 26 March 2019
Published 27 May 2019 Volume 2019:13 Pages 887—894
DOI https://doi.org/10.2147/OPTH.S203510
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Elizabeth Yeu,1 Damien F Goldberg,2 Francis S Mah,3 Kenneth A Beckman,4,5 Jodi I Luchs,6,7 Jonathan D Solomon,8 Darrell E White,9 Preeya K Gupta10
1Eastern Virginia Medical School, Virginia Eye Consultants, Norfolk, VA, USA; 2Wolstan & Goldberg Eye Associates, Torrance, CA, USA; 3Scripps Clinic Torrey Pines, La Jolla, CA, USA; 4Ohio State University, Columbus, OH, USA; 5Comprehensive Eye Care of Central Ohio, Westerville, OH, USA; 6Hofstra Northwell School of Medicine, Hempstead, NY, USA; 7South Shore Eye Care, Wantagh, NY, USA; 8Bowie Vision Institute, Bowie, MD, USA; 9Skyvision Centers, Westlake, OH, USA; 10Duke University Eye Center, Durham, NC, USA
Purpose: Evaluate the safety and efficacy of cryopreserved amniotic cytokine extract (ACE) in the treatment of subjects with dry eye disease (DED).
Patients and methods: This was a retrospective, multicenter, chart review of adult patients with DED that instilled cryopreserved ACE drops twice-daily for 4 or 12 weeks. Patients had corneal fluorescein staining (0–20 range) and/or a lissamine green conjunctival staining score (0–18 range) of ≥3 and ≤9 in at least 1 eye and a score ≥40 (0–100 range) of eye dryness/irritation on a visual analog scale (VAS). Following completion of a treatment course, medical records were reviewed from the initiation of therapy (baseline), and at post-treatment visits (4 weeks, 8 weeks, and 12 weeks). Patient records for visual acuity, adverse events, corneal fluorescein staining, conjunctival lissamine green staining, and symptom scores of ocular dryness/irritation were reviewed for each visit, as available. Safety and tolerability were assessed through the evaluation of patient-reported adverse events recorded in the medical records.
Results: A total of 54 eligible patients were identified at 7 clinical sites; 16 patients administered ACE drops for 4-weeks, and 38 patients instilled ACE drops for 12 weeks. Significant improvements in the mean changes from baseline were observed for corneal fluorescein staining, lissamine green staining, visual acuity (LogMar) and VAS ocular symptom scores at the 4-week post-treatment visit (p<0.01). Additional improvements continued out to the 12-week follow-up assessment visits. Two patients discontinued therapy due to reports of ocular burning or foreign body sensation.
Conclusion: The cryopreserved ACE formulation was well-tolerated and effective in reducing the clinical signs and symptoms of DED. Conduct of a vehicle-controlled prospective study is warranted.
Keywords: dry eye, amniotic membrane, amnion extract, cytokine, inflammation
Introduction
Dry eye disease (DED) is a common condition with multifactorial etiologies characterized by tear film instability and ocular surface inflammation.1 The clinical presentation of DED includes disruptions in the integrity of the corneal and conjunctival epithelium, a rapid tear break-up time, and reports of a range of symptoms associated with ocular discomfort and irritation, including dryness, grittiness, and foreign body sensation.1,2 Multiple subtypes of DED may occur within patients, including aqueous-deficient and evaporative forms of the disease, with overlap being common, as well as comorbidity with other ocular conditions, such as blepharitis.2,3 The prevalence of DED varies across studies due to differences in study population and diagnostic criteria, with estimates ranging from 5% to 50% of the study population based on the presence of symptoms with or without clinical signs.4 Risk factors that are commonly associated with DED include advancing age, female gender, meibomian gland dysfunction, a history of ocular surgery, contact lens wear, and exposure to low humidity or high airflow.4
A range of non-pharmacological and pharmacological therapies have been evaluated and recommended for the treatment of patients with DED through the use of staged-treatment algorithms based on the nature and severity of the patient’s condition. Inflammation of the ocular surface is a common factor across patients and is often a target for therapy, with treatment options including topical anti-inflammatory and/or immunomodulatory agents. One form of therapy that has been recommended for the treatment of DED includes amniotic membrane transplantation.3,5
In cases of severe conditions affecting the ocular surface that can result in alteration of the function and integrity of the tear film, including conjunctival scarring, trauma, and persistent epithelial defects, transplantation of amniotic membrane is recognized as a therapeutic treatment option. Amniotic membrane, the thin avascular inner layer of the fetal membranes, may be transferred to the ocular surface as a temporary graft to act as a bandage, shielding ocular tissue, as well as promoting healing of the damaged structures. The transparent amniotic membrane has been demonstrated to promote epithelialization, and possess anti-inflammatory, anti-angiogenic, and anti-fibrotic properties.3,6 Preserved amniotic membrane has been used to treat patients with DED, resulting in improvements in patient-reported ocular symptoms and clinical signs, as well as corneal nerve restoration.7–9
The improvements in ocular surface healing and comfort associated with amniotic membrane transplantation have been attributed in part to the anti-inflammatory properties of the tissue, inhibiting the expression of pro-inflammatory mediators and growth factors. Interest in the potential for improvements in wound healing and other applications of the properties of amniotic tissue has led to the development of extracts. Amnion-derived extract solutions have been demonstrated to contain combinations of cytokines, growth factors, and inhibitors of enzymes involved in tissue remodeling (matrix metalloproteinases).10,11
A cryopreserved eyedrop formulation of amniotic cytokine extract (ACE) was developed and launched in 2017 (Genesis, Ocular Science, Manhattan Beach, CA). The ACE formulation was prepared through a proprietary cryopreservation method using a compounding pharmacy and contains soluble mediators that have been shown to stimulate healing and reduce inflammation. This report presents the results of a retrospective analysis of the safety and efficacy of ACE drops for the treatment of patients with DED.
Patients and methods
This was a retrospective chart review designed to evaluate the safety and efficacy of the use of ACE in the treatment of patients with DED. The study was approved by the Duke University (Durham, NC, USA) Institutional Review Board (IRB) and Sterling IRB (Atlanta, GA, USA) as a central review board for the other clinical sites. A waiver was obtained for collection of informed consent and the authorization to use of protected health information. The study was performed in accordance with the Declaration of Helsinki and the Health Insurance Portability and Accountability Act. Patient medical records were evaluated at 7 clinical centers in the United States.
Study design and participants
The medical records of patients with symptomatic DED that were treated with ACE were reviewed. The study dates ranged from patients that initiated treatment on January 12, 2017, through March 21, 2018. Patient groups included 2 arms of treatment: topical bilateral administration of ACE drops twice daily for a 4-week treatment period, and patients that administered ACE drops twice daily for a 12-week treatment period. Retrospective data from medical charts were collected at the following times: prior to the initiation of ACE topical therapy (baseline), 4 weeks (±2 days), 8 weeks (±2 days), and 12 weeks (±2 days) after beginning ACE treatment.
The inclusion criteria for the study allowed for evaluation of patient medical records that were 18 years of age or older with a diagnosis of mild, moderate, or severe DED. In addition to a patient-reported history of DED for a period of at least 6 months, a clinical diagnosis of bilateral DED was required at the baseline assessment. Eligible participants had to have a total corneal fluorescein staining score of ≥3 and ≤9 out of a possible score of 0–20 in at least 1 eye or a conjunctival lissamine green staining score of ≥3 and ≤9 out of a possible score of 0–18 in at least 1 eye at baseline. Patients were also required to have a baseline symptom score of ≥40 for eye irritation/dryness on a visual analog scale (VAS) on a 0–100 scale. Only patients with a Snellen visual acuity of 20/200 or better were included in the study population. All patients that met inclusion criteria from the study sites were included.
Patients were excluded from the study if any of the following criteria were met: current diagnosis or history of seasonal or perennial allergic conjunctivitis, unstable macular disease, diagnosis of herpes keratitis, presence of chronic uveitis, prior history of corneal transplant refractive surgery, cataract surgery, glaucoma treatment surgical procedures (or laser treatment) within 3 months of the baseline evaluation, recent punctal plug placement (within 3 months), lagophthalmos or other clinically significant eyelid margin irregularity, clinically significant conjunctivochalasis, presence of pterygium, use of contact lenses, current or recent use (within 30 days of the baseline assessment) of lifitegrast ophthalmic solution 5% (Xiidra; Shire, Lexington, MA) or cyclosporine ophthalmic emulsion 0.05% (Restasis; Allergan, Irvine, CA), patients that were human immunodeficiency virus positive, women who were pregnant or breastfeeding, patients that had a current active ocular disease other than DED, or other serious or unstable medical condition. Patients that were on immunomodulator medications and omega-3 fatty acid supplements were included in the chart review if the dose was stable for 3 months prior to the baseline evaluation, and the dose did not change during the ACE treatment period. Patient data were not included if any other ophthalmic medications were used during the ACE treatment period.
Study assessments
Demographic information (gender) was collected at the baseline visit. Data were collected as part of the retrospective chart review for the following parameters (if recorded): visual acuity, symptom score assessed as the severity and frequency of eye dryness/irritation over the past week on a VAS scale (0–100 range with 0= no dryness/irritation and 100= severe dryness/irritation), total corneal fluorescein staining score (0–4 severity scale; 0= no punctate staining, 4= severe diffuse/coalescent macropunctate staining) for each region of the cornea (central, inferior, superior, medial, lateral) with a total possible score of 0–20, conjunctival lissamine green staining on a 0–3 scale (0= no punctate staining, 3= densely concentrated micropunctate staining; NEI scale) for each of 6 regions of the conjunctiva (temporal, nasal, 2 inferior regions, and 2 superior regions) with a total possible score of 0–18.12 Safety and tolerability of the ACE formulation was assessed through the evaluation of patient-reported adverse events recorded in the patients’ medical records.
Statistical methods
Descriptive statistics were calculated (mean values, standard deviation [SD] and 95% confidence limits) for chart review assessment parameters, including visual acuity (LogMAR), eye dryness/irritation VAS score, total corneal fluorescein staining score, and total conjunctival lissamine green staining score. Statistical analysis was performed to compare the visual acuity, VAS scores, and staining scores at the 4-week, 8-week, and 12-week post-treatment assessments to the baseline parameters. The analysis presented for the 4-week post baseline assessments combines data from both treatment arms (both the 4-week and 12-week treatment groups were on the same treatment regimen through week 4). The study eye for each patient was defined based on the eye that had the highest total corneal fluorescein staining score at the baseline assessment. The results of the analysis are presented for the study eye for all recorded parameters except for the eye dryness/irritation assessment where only one score was recorded for each patient.
Patient records without baseline or any post baseline assessments were excluded from the analysis. A paired t-test was used to analyze within-group changes from baseline. No adjustment was made to p-values due to multiple comparisons. Data analysis was conducted using Statistical Analysis System (SAS®) Software version 9.4 (SAS Institute, Cary, NC).
Results
A total of 54 patients met the eligibility criteria and were included in the study analysis; 16 patients were identified that had administered ACE drops for 4 weeks, and 38 patients were identified for the 12-week treatment period. The data that was identified from the medical records for eligible patients is presented for each endpoint; not all patients that were determined to be eligible for inclusion in the study analysis returned to the clinic for all the assessment visits. The majority of eligible patients were female for each group (10 out of 16 [62.5%] patients in the 4-week treatment group, and 29 out of 38 [76.3%] patients in the 12-week group) and overall (39 out of 54 total patients; 72.2%). The baseline characteristics of the patients by treatment group and for the overall study population, including mean (SD) scores for eye dryness/irritation, total corneal staining, total conjunctival staining, and visual acuity, are listed in Table 1. The baseline characteristics were generally similar between the overall study population and the 4-week and 12-week ACE treatment groups.
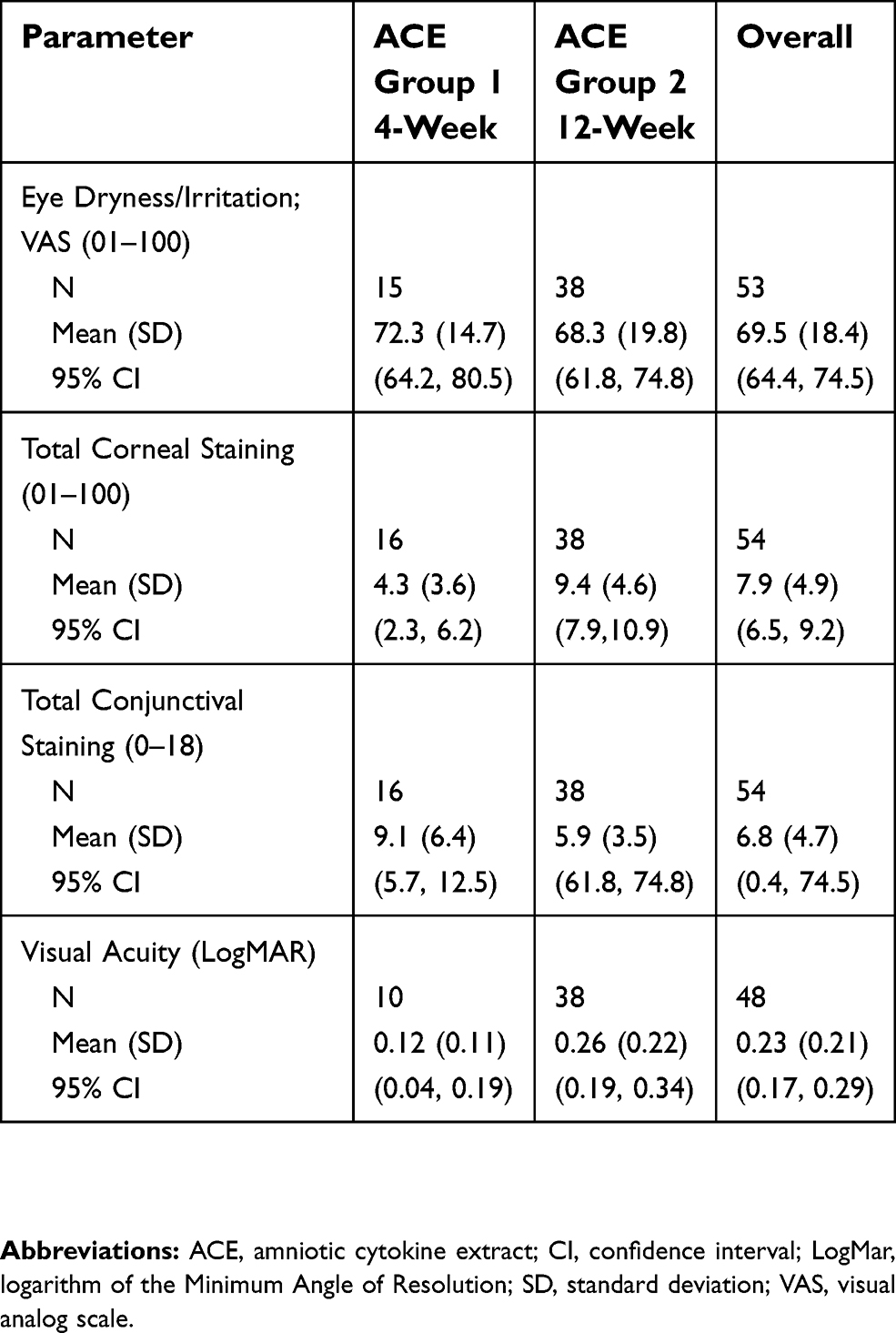 | Table 1 Baseline characteristics of the study population |
Efficacy assessments
Eye dryness/irritation
The mean (SD) change from baseline in VAS scores for eye dryness/irritation is presented by assessment time point in Figure 1. Significant improvements in patient-reported symptoms were observed at each post-baseline visit, including the 4-week evaluation, and continued to improve out to the 12 assessments (p<0.01). Analysis of the distribution of patient responses indicated that 81% of the subjects reported an improvement of at least 30% and 67% of the subjects reported an improvement of at least 40% at week 12.
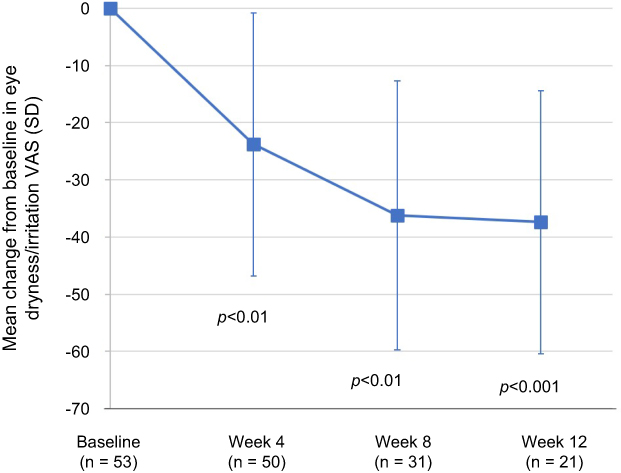 | Figure 1 The mean changes from baseline in the subject-reported symptoms of ocular dryness/irritation assessed by visual analog scale (VAS) on a 0–100 scale (0= no ocular dryness/irritation, 100= severe ocular dryness/irritation) are presented by visit. The mean changes from baseline were considered statistically significant at week 4 and week 8 (p<0.01), and week 12 (p<0.001). A paired t-test was used to analyze within-group changes from baseline. Abbreviations: SD, standard deviation; VAS, visual analog scale. |
Ocular surface staining
Mean (SD) change from baseline in total corneal fluorescein staining scores are presented at baseline and for each follow-up visit in Figure 2. Significant improvements in corneal staining were observed beginning at the 4-week assessment and improvements continued to the 12-week assessment (p<0.01). Examination of the distribution in total corneal staining scores indicated that an improvement of at least 50% was observed in 95% of the patients at week 12. Similar corresponding improvements in the mean (SD) total conjunctival lissamine green staining were observed; the mean change from baseline in conjunctival lissamine green staining is presented by follow-up visit in Figure 3. Significant improvements in the mean change from baseline in total conjunctival staining were observed at week 4, week 8, and week 12 assessments (p<0.01).
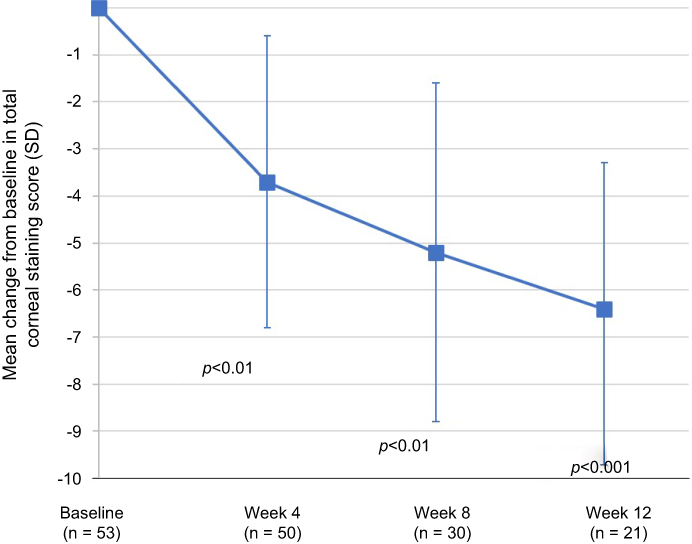 | Figure 2 The mean changes from baseline in the corneal fluorescein staining on a 0–20 scale (0–4 scale per region for each of the 5 regions of the cornea) are presented by visit. Analysis of the mean changes from baseline in corneal fluorescein staining indicated that the improvements were statistically significant at week 4 (p<0.01), week 8 (p<0.01), and week 12 (p<0.001). A paired t-test was used to analyze within-group changes from baseline. Abbreviation: SD, standard deviation. |
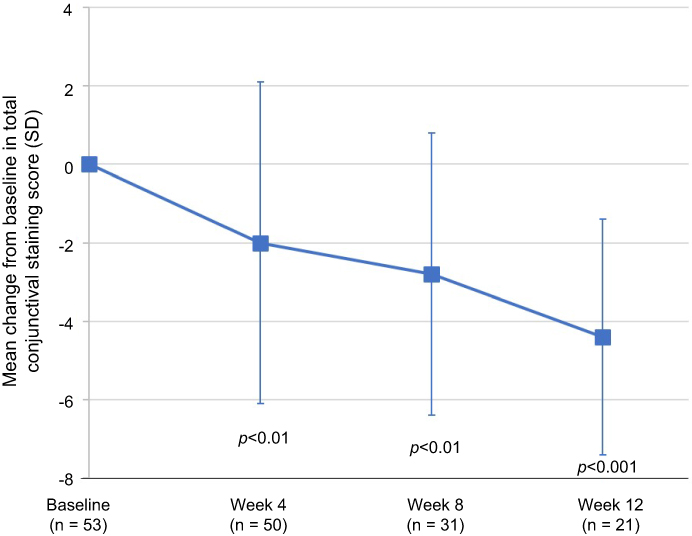 | Figure 3 The mean changes from baseline in the conjunctival lissamine green staining on a 0–18 scale (0–3 scale per region for each of the 6 regions of the conjunctiva) are presented by visit. Changes from baseline were determined to be statistically significant at week 4 (p<0.01), week 8 (p<0.01), and week 12 (p<0.001). A paired t-test was used to analyze within-group changes from baseline. Abbreviation: SD, standard deviation. |
Safety assessments
Visual acuity assessments generally remained stable or showed modest improvement throughout the treatment and follow-up period. The mean (SD) change from baseline in visual acuity scores (LogMAR) are presented by visit in Figure 4. Based on the review of patient medical records, 2 patients complained of ocular burning after instillation, with 1 patient discontinuing therapy, and 1 patient discontinued therapy due to a report of foreign body sensation. No other adverse events were reported during the ACE treatment or follow-up period.
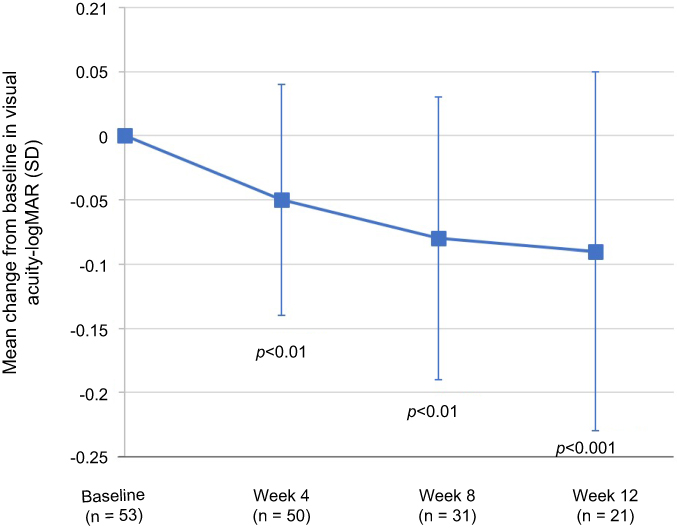 | Figure 4 The mean changes from baseline in visual acuity (LogMAR) are presented by visit. SD = standard deviation. The improvement from baseline at follow-up visits was statistically significant at week 4 (p<0.01), week 8 (p<0.01), and week 12 (p<0.001). A paired t-test was used to analyze within-group changes from baseline. Abbreviation: SD, standard deviation. |
Discussion
The ACE formulation used by patients in this study population is a cryopreserved solution containing over 120 cytokines, growth factors, and anti-inflammatory agents. The ACE solution is prepared using a proprietary cryopreservation technique to harvest a combination of biologically active molecules that contribute to wound healing and a reduction of inflammation. Key factors that have been identified in the extracts of amniotic tissue include prostaglandin E2 (PGE2),13 growth differentiation factor 11 (GDF11),14,15 thrombospondin-1,16 and WNT4.17 PGE2 has been identified as a major mediator found in mesenchymal stem cell extracts capable of modulating the immune response through the induction of differentiation of naïve T cells into regulatory T cells and the promotion of IL-10 production.13
The etiology of DED is complex with multiple pathways for initiation of the chronic inflammatory response that is characteristic of the condition.18 A chronic cycle of inflammation that occurs in patients with DED can be related to other ocular conditions or associated with damage that occurs to the ocular surface through tear film hyperosmolarity. The release of proinflammatory mediators, including cytokines and increase in expression of matrix metalloproteinase enzymes can enhance recruitment of immune cells to the ocular surface, further disrupting the tear film.19 Methods of treatment for DED designed to address inflammation include agents with a specific target as the method of action, such as calcineurin inhibitors (cyclosporine) and lymphocyte-function-associated antigen-1 (LFA-1) antagonists (lifitegrast), as well as topically applied corticosteroids which function as general anti-inflammatory agents.5,20
Although the precise mechanism of action of the ACE formulation in the treatment of patients with DED is unknown, a combined effect is likely due to the diverse complement of biologically active agents that have been identified as present in amniotic tissue extracts at physiological concentrations.11 Amniotic membrane extract preparations contain many of the similar properties to cryopreserved amniotic membrane. Amniotic tissue extracts have been evaluated for treatment related to corneal injury and wound healing.21 Pre-clinical evaluation has also been conducted in a murine model of DED.22 The results observed in this retrospective analysis on the use of ACE in the treatment of DED suggest that the benefits of amniotic membrane treatment for DED may be extended to the topical formulation of the extract.
Based on the minimal adverse events in the medical records evaluated in this retrospective study and the improvements observed in patient-reported symptoms, the ACE formulation appears to be well tolerated by patients with DED. Improvements in the clinical signs and symptoms of DED were observed by the majority of patients within 4 weeks of initiation of therapy, with improvements continuing throughout the 12-week follow-up period.
Limitations of the present study include the small size of the study population, the retrospective aspect of the study design, lack of the ability to monitor patient compliance with therapy, and the lack of a comparator control group. Additionally, the retrospective nature of the study did not control for the duration of treatment for all patients evaluated in the study; the duration of treatment for patients was based on the recommendations for each clinician. Conduct of a controlled, double-masked, prospective study would help to further elucidate the safety and efficacy profile of the ACE formulation. The ongoing improvement in signs and symptoms in both the 4-week and 12-week treatment groups of the current study suggests that evaluation beyond 3 months of treatment and follow-up may be necessary to determine the optimal treatment regimen.
Conclusion
The statistically significant improvements in the clinical signs combined with the reduction in symptoms observed in this retrospective review suggest that the ACE formulation is efficacious and well tolerated in patients with DED. Further evaluation in a larger, well-controlled prospective study is warranted to confirm the findings of this study and expand the understanding of the precise mechanism of action.
Acknowledgments
Writing and editorial assistance in preparation of the manuscript was provided by Kurt Brubaker of Bridge Over Brook, Inc., with funding provided by Ocular Science. This study was sponsored and funded by Ocular Science. Ocular Science participated in the design, data collection, data management, and data analysis of the study.
Disclosure
E Yeu, KA Beckman, JI Luchs, JD Solomon, DE White, and PK Gupta are consultants to Ocular Science. DF Goldberg, FS Mah and DE White have ownership interests in Ocular Science. E Yeu is consultant for: Alcon; Allergan; Aurea Medical; Avedro; Baush & Lomb; BioTissue; Bruder; EyePoint Pharm.; iOptics; Guidepoint; J&J Vision; LENSAR; Kala Pharm.; Merck; Mynosys; Novartis; Ocular Science; Ocular Therapeutix; Ocusoft; Omeros; Oyster Point Pharm.; Science Based Health; Shire; Sight Sciences; SightLife Surgical; Sun; TopCon; TearLab Corporation; TearScience & Zeiss. DF Goldberg reports equity from Ocular Science, during the conduct of the study and outside the submitted work. FS Mah reports equity from Ocular Science, during the conduct of the study; received grants from Allergan, reports personal fees from Shire/Takeda, outside the submitted work. KA Beckman was a consultant for Ocular Science, during the conduct of the study. JI Luchs reports personal fees including consultancy for allergan, received personal fees from Bausch and Lomb, Shire, Tear Lab, Aerie, Alcon, Ocular Sciences, Insightful Solutions, Kala, Eyevance Pharma, Calhoun Vision, Trefoil Therapeutics, RPS, Sun, and Omega Ophthalmics, outside the submitted work. PK Gupta reports personal fees from Ocular science, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. Craig JP, Nichols KK, Akpek EK, et al. TFOS DEWS II definition and classification report. TFOS DEWS II definition and classification report. Ocul Surf. 2017;15(3):276–283. doi:10.1016/j.jtos.2017.05.008
2. Wolffsohn JS, Arita R, Chalmers R, et al. TFOS DEWS II diagnostic methodology report. Ocul Surf. 2017;15(3):539–574. doi:10.1016/j.jtos.2017.05.001
3. Milner MS, Beckman KA, Luchs JI, et al. Dysfunctional tear syndrome: dry eye disease and associated tear film disorders – new strategies for diagnosis and treatment. Curr Opin Ophthalmol. 2017;27(Suppl 1):3–47. doi:10.1097/01.icu.0000512373.81749.b7
4. Stapleton F, Alves M, Bunya VY, et al. TFOS DEWS II epidemiology report. Ocul Surf. 2017;15(3):334–365. doi:10.1016/j.jtos.2017.05.003
5. Jones L, Downie LE, Korb D, et al. TFOS DEWS II management and therapy report. Ocul Surf. 2017;15(3):575–628. doi:10.1016/j.jtos.2017.05.006
6. Malhotra C, Jain AK. Human amniotic membrane transplantation: different modalities of its use in ophthalmology. World J Transplant. 2014;4(2):111–121. doi:10.5500/wjt.v4.i2.111
7. Cheng AM, Zhao D, Chen R, et al. Accelerated restoration of ocular surface health in dry eye disease by self-retained cryopreserved amniotic membrane. Ocul Surf. 2016;14(1):56–63. doi:10.1016/j.jtos.2015.07.003
8. McDonald MB, Sheha H, Tighe S, et al. Treatment outcomes in the dry eye amniotic membrane (DREAM) study. Clin Ophthalmol. 2018;12:677–681. doi:10.2147/OPTH.S162203
9. John T, Tighe S, Sheha H, et al. Corneal nerve regeneration after self-retained cryopreserved amniotic membrane in dry eye disease. J Ophthalmol. 2017;6404918:1–10. doi:10.1155/2017/6404918
10. Franz MG, Payne WG, Xing L, et al. The use of amnion-derived cellular cytokine solution to improve healing in acute and chronic wounds. Eplasty. 2008;8:188–199.
11. Steed DL, Trumpower C, Duffy D. Amnion-derived cellular cytokine solution. Eplasty. 2008;8:157–165.
12. Lemp MA. Report of the national eye institute/industry workshop on clinical trials in dry eyes. Clao. 1995;21(4):221–232.
13. Madrigal M, Rao KS, Riordan NH. A review of therapeutic effects of mesenchymal stem cell secretions and induction of secretory modification by different culture methods. J Trans Med. 2014;12:1–14. doi:10.1186/s12967-014-0260-8
14. Blau HM, Cosgrove BD, Ho ATV. The central role of muscle stem cells in regenerative failure with aging. Nat Med. 2015;21(8):854–862. doi:10.1038/nm.3918
15. Andersen RE, Lim DA. An ingredient for the elixir of youth. Cell Res. 2014;24(12):1381–1382. doi:10.1038/cr.2014.107
16. Schollhorn L, Bock F, Cursiefen C. Thrombospondin-1 as a regulator of corneal inflammation and lymphangiogenesis: effects on dry eye disease and graft immunology. J Ocul Pharmacol Ther. 2015;31(7):376–385. doi:10.1089/jop.2015.0020
17. Labus MB, Stirk CM, Thompson WD, Melvin WT. Expression of Wnt genes in early wound healing. Wound Repair Regen. 1998;6(1):58–64.
18. Bron AJ, De Paiva CS, Chauhan SK, et al. TFOS DEWS II pathophysiology report. Ocul Surf. 2017;15(3):438–510. doi:10.1016/j.jtos.2017.05.011
19. Nebbioso M, Del Regno P, Gharbiya M, et al. Analysis of the pathogenic factors and management of dry eye in ocular surface disorders. Int J Mol Sci. 2017;18(8):
20. Nebbioso M, Fameli V, Gharbiya M, et al. Investigational drugs in dry eye disease. Expert Opin Investig Drugs. 2016;25(12):1437–1446. doi:10.1080/13543784.2016.1249564
21. Murri MS, Moshirfar M, Birdsong OC, et al. Amniotic membrane extract and eye drops: a review of literature and clinical application. Clin Ophthalmol. 2018;12:1105–1112. doi:10.2147/OPTH.S165553
22. Xiao X, Luo P, Zhao H, et al. Amniotic membrane extract ameliorates benzalkonium chloride-induced dry eye in a murine model. Exp Eye Res. 2013;115:31–40. doi:10.1016/j.exer.2013.06.005
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.