")
Back to Journals » Journal of Blood Medicine » Volume 7
Ruxolitinib in the treatment of polycythemia vera: patient selection and special considerations
Authors Blum S, Martins F, Alberio L
Received 14 December 2015
Accepted for publication 29 June 2016
Published 23 September 2016 Volume 2016:7 Pages 205—215
DOI https://doi.org/10.2147/JBM.S102471
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin Bluth
Sabine Blum,* Filipe Martins,* Lorenzo Alberio
Service and Central Laboratory of Hematology, CHUV, University Hospital of Lausanne, Lausanne, Switzerland
*These authors contributed equally to this work.
Abstract: The discovery of JAK2 V617F mutation in the mid-2000s started to fill the gap between clinical presentation of polycythemia vera (PV), first described by Vaquez at the end of the 19th century, and spontaneous erythroid colony formation, reported by Prchal and Axelrad in the mid-1970s. The knowledge on this mutation brought an important insight to our understanding of PV pathogenesis and led to a revision of the World Health Organization diagnostic criteria in 2008. JAK–STAT is a major signaling pathway implicated in survival and proliferation of hematopoietic precursors. High prevalence of JAK2 V617F mutation among myeloproliferative neoplasms (>95% in PV and ~50% in primary myelofibrosis and essential thrombocythemia) together with its role in constitutively activating JAK–STAT made JAK2 a privileged therapeutic target. Ruxolitinib, a JAK 1 and 2 inhibitor, has already proven to be efficient in relieving symptoms in primary myelofibrosis and PV. In the latter, it also appears to improve microvascular involvement. However, evidence regarding its potential role in altering the natural course of PV and its use as an adjunct to current standard therapies is sparse. Therapeutic advances are needed in PV as phlebotomy, low-dose aspirin, cytoreductive agents, and interferon alpha are the only therapeutic tools available at the moment to influence outcome. Even though several questions are still unanswered, this review aims to serve as an overview article of the potential role of ruxolitinib in PV according to current literature and expert opinion. It should help hematologists to visualize the place of this tyrosine kinase inhibitor in the field of current practice and offer criteria for a careful patient selection.
Keywords: myeloproliferative neoplasms, polycythemia vera, ruxolitinib, JAK2 V617F, JAK2 JAK1 inhibitor
Introduction
Polycythemia vera (PV) is a myeloproliferative neoplasia (MPN) characterized by an absolute increase in red blood cell mass, reflecting a clonal stem cell disorder, variably associated with leukocytosis and/or thrombocytosis along with hepatosplenomegaly. Its first description by Louis Henri Vaquez,1 a French internist, dates back to more than a century ago (1892). His report narrating the case of a 40-year-old patient suffering from a special cyanosis accompanied by excessive and persistent erythrocytosis together with the series of cases published by William Osler a few years later served as the first clinically accurate descriptions of this pathology.2 Potentially severe vascular complications, both thrombotic and hemorrhagic, determine prognosis in the early years after diagnosis. Even more alarming are late events, such as leukemic and myelofibrotic transformations, which carry a bleak outcome. For instance, leukemic transformation has a median survival of ~2–5 months only, except for some rare cases cured after stem cell transplantation.3
Available treatments consist of phlebotomy, cytoreductive agents (hydroxyurea [HU] as first line, followed by busulfan or pipobroman), and interferon α (IFN-α). Age >60 years and a past medical history of thrombosis are the main risk stratifiers for treatment decision.4 They help guide the decision to initiate cytoreductive therapy in high-risk groups, depending on the estimated risk of vascular thrombotic events.5
The biological mechanism of the “myeloproliferative activity of the bone marrow” recognized by Dameshek6 in the early 1950s was identified almost 25 years later by Prchal and Axelrad7 as in vitro spontaneous erythroid colony formation in samples of patients suffering from PV. This observation along with a hypersensitivity to erythropoietin (EPO) of erythroid PV precursor cells8,9 focused research efforts on implicated signaling pathways, thus leading to the simultaneous discovery of a mutation at amino acid position 617 in the JAK2 by four groups.10–13 This mutation, which leads to a steadily increased JAK2 activity, explains cytokine hypersensitivity and is able to reproduce polyglobulia in mouse models.10–13 This phenomenon marked JAK2 V617F mutation as a main oncogenic event in PV pathogenesis and opened the gate to the idea of targeted tyrosine kinase inhibitors.
The first study on ruxolitinib (then called INCB018424) was released in 2010 by Quintás-Cardama et al,14 demonstrating a potential impact of JAK1 and JAK2 (JAK1/2) inhibition on the treatment of myeloproliferative neoplasms. Ruxolitinib was initially approved for the treatment of primary myelofibrosis (MF). It is now also the first oral JAK1/2 inhibitor approved in the setting of HU-intolerant or -resistant PV patients. Recent Phase II and III studies could demonstrate its efficacy on controlling symptoms and disease parameters, such as hematocrit or spleen size in PV.15,16 The potential ability of ruxolitinib to influence PV outcome and its transformation into secondary MF and/or acute myeloid leukemia remains to be proven. Since blasts in leukemic transformation are JAK2 V617F negative in a significant proportion of PV patients, a major impact of ruxolitinib on this phenomenon appears to be unlikely.17
PV clinical characteristics and paradigm of current treatment
PV is rare in patients under the age of 40 years (<5% of cases), with a median age at diagnosis of 60 years. It entails a small male predominance, with an incidence of about 1/100,000 individuals in Western countries.18,19 Only rare families with an inherited increased risk of developing PV have been described.20 An autosomal dominant transmission mechanism has been outlined by the recent discovery of germ line predisposition genes to familial myeloid neoplasms.21 A large retrospective study conducted on a cohort of 1,545 patients by an international working group showed that the natural course of PV is marked by a 15-year cumulative risk to develop secondary MF or acute leukemia of about 35% and 5%, respectively.22
Patients should be treated according to international guidelines or in clinical studies. Currently, the most frequently used international guidelines for PV treatment are those of the European Leukemia Net23 and the British Committee for Standards in Haematology.24 Clinical follow-up should be performed on a regular basis for disease symptoms assessment, using tools such as the MPN-10 score.25 Hematocrit control targeting less than 45%, as proven in the CYTO-PV trial, should be considered the aim of treatment, as it significantly decreased the rate of major thrombotic events compared to patients with a hematocrit between 45% and 50%.26,27 Some experts suggest a target <42% in females, but these data are currently not yet published. Phlebotomy remains the standard therapy in low-risk and some intermediate-risk patients, as long as it achieves hematocrit control without inducing severe thrombocytosis and/or excessive iron depletion. High-risk patients are generally treated with HU as first-line therapy. Pegylated IFN-α 2a is a good alternative with a reported 76% hematological response rate.28
Hematocrit control is a priority over the normalization of a possible concomitant thrombocytosis.29 Somewhat surprising and contrary to essential thrombocythemia (ET),30 decreased incidence of thrombotic events has not been linked to a definitive platelet count threshold neither at baseline nor during the course of the disease in PV.31 In PV, clear evidence for controlling thrombocytosis or leukocytosis in addition to the defined cutoff hematocrit level is lacking at the moment.
Patient treatment is currently based on the prevention of vascular complications, with late events such as secondary MF and leukemic transformation not being addressed. In this line of thinking, it is remarkable to see that no sufficiently powered trial is trying to evaluate the influence of ruxolitinib on thrombotic complications, as cardiovascular events account for almost half of the fatal outcomes reported in the ECLAP study.32,33 This seminal study demonstrated the value of aspirin for primary cardiovascular prevention in PV.33 Therefore, patients without any contraindications should receive low-dose aspirin therapy, given its favorable impact on cardiovascular outcome, by reducing nonfatal ischemic strokes and venous thrombotic events, without a significant rise in hemorrhagic events. IFN-α is mostly considered as a second-line therapy. A main concern regards its side effects. In some cases, it can be considered a front-line alternative, as, for example, in pregnant or young patients.24
Pathogenesis of PV and potential therapeutic targets
At present, our understanding of PV remains incomplete, with a lot of gaps still to be filled in order to obtain a whole picture. However, some important advances have been made. First, the spontaneous proliferation of erythroid precursors in the in vitro setting was observed in the mid-1970s.7 This phenomenon represented one of the minor diagnostic criteria in the 2008 World Health Organization definition of PV but does not figure any longer in the 2016 revised revision.5,34 During the 1980s, works of several groups pointed out the EPO hypersensitivity of PV erythropoietic progenitor cells from peripheral blood and in some cases even their EPO independency to form colonies.8,9 In the 1990s, this phenomenon was complemented by the discovery of increased reactivity to other cytokines, such as interleukin 1, interleukin 3, granulocyte macrophage colony-stimulating factor, and insulin-like growth factor 1.35,36
Biochemical background of V617F point mutation in JAK2 is a substitution of phenylalanine for valine at amino acid position 617 (V617F) of the JH2 domain. This gain of function mutation results in a permanent activation of JAK2.37 Because of the close interaction of the JAK–STAT pathway with the EPO receptor, this mutation offered the first explanation to the above-mentioned clinical observations. STAT, once phosphorylated by JAK2, works as an important transcription factor involved in survival, proliferation, and differentiation of hematopoietic precursors. In experimental models with knock-in mice expressing JAK2 V617F mutation, disease phenotype could be reproduced, verifying the hypothesis of its central part in pathogenesis.38
The high prevalence of JAK2 V617F mutation in PV (>95%) and other MPN, such as ET and primary MF (~50%), leads to a high level of suspicion that this event is a major early oncogenic event. Initiation of a clonal disease from a single cell harboring V617F as a sole mutation39 and development of a condition mimicking PV with secondary MF in mouse models as published in 2006 by Lacout et al38 suggest its driver role in initiation and evolution of the disease. However, Theocharides et al17 made the discovery that at the time of leukemic transformation, nine of their 17 patients presented JAK2 V617F-negative blasts. Microsatellite analysis along with clonality testing suggested a common JAK2 V617F-negative ancestor as a possible mechanism. Nussenzveig et al40 went further showing that somatic JAK2 V617F mutation was not the PV-initiating event by demonstrating the presence of homozygous wild-type pathological erythroid-forming colonies in patients harboring a mutated status. This work revealed allelic frequencies of <50% (using a real-time polymerase chain reaction assay) in three of ten patients. Reasonable suspicion about the fact that JAK2 V617F mutation is not the primary event is nowadays accepted, speaking against a single-hit theory.
JAK2 V617F homozygosity is found in 30% of PV patients and is a lot less frequent in ET. Scott et al41 suggested a double-hit theory leading to the transformation of heterozygous ET patients to PV phenotype. This phenomenon, although rare, highlights the possibility of a disease continuum between V617F-positive ET and PV.
Current murine models are still focused on the JAK2 V617F mutation to explain the phenotype and the natural history of the disease, without taking into account earlier events. Nonetheless, these models serve as a powerful tool to study the consequences of mutant JAK2 allelic changes and their role in MPN phenotypes.
Other interesting therapeutic targets have been identified by basic research. For instance, Yan et al42 and Walz et al43 demonstrated that the suppression of STAT5 expression was able to reverse the PV phenotype in murine models. When focusing on JAK2, we must recall that other mutations besides V617F, for example, in exon 12 have been discovered and account for ~4%–5% of cases.44 Exon 12 mutations of JAK2 correlate with a mainly erythrocytemic phenotype and are usually not associated with significant thrombocytosis or leukocytosis.44
Finally, the JAK2 V617F mutation can also be found in lymphoid lineages and even in endothelial precursor cells in patients with PV.45 Therefore, this mutation may play a direct role in thrombosis risk apart from provoking an elevated hematocrit level later on, as its presence has also been discovered in liver endothelial cells in some patients with Budd–Chiari syndrome and PV.46
Pharmacodynamics of ruxolitinib
Ruxolitinib acts by competing with adenosine triphosphate binding in the JAK2 catalytic site. Preclinical data demonstrate selective growth impairment of PV erythroid progenitor colonies through a dose-dependent mechanism of induced apoptosis and reduced cell proliferation. Inhibition of JAK provokes a downstream hypophosphorylation of STAT.47 In primary cultures, ruxolitinib preferentially suppresses erythroid progenitor colony formation from JAK2 V617F-positive patients (50% inhibitory concentration, IC50 =67 nM) as compared to healthy donors (IC50 =407 nM).14 However, this mutation is neither a specific nor an exclusive target of ruxolitinib as the drug also inhibits JAK1 and wild-type JAK2.14 This ubiquitary inhibition explains the hematological side effects, such as anemia and thrombocytopenia, by inhibiting both EPO and thrombopoietin signaling even in normal lineages.48 Further studies could help to individualize therapeutic concentrations of ruxolitinib.
Decreasing the levels of inflammatory cytokines, as demonstrated in murine models, may be another mechanism of action by ruxolitinib on symptom relief. Its significance in PV is certainly a lot lower than in MF, where the role of proinflammatory cytokines is more apparent. However, these cytokines are also detectable in PV patients and may serve as a stimulus to clonal erythroid growth. As JAK1 also plays a key role in the signaling of certain cytokines, its inhibition may also contribute to some therapeutic effect.
Long-term data in advanced cases of MPNs such as post-PV MF undergoing allogeneic transplantation are needed.49 Ruxolitinib may reduce treatment-related complications by improving symptoms in the pretransplant setting, but data on its influence on transplant outcome are sparse. Data suggesting some influence on bone marrow fibrosis in MF should encourage incorporating its evaluation in future trials.
Another important point is the mechanism of ruxolitinib resistance in PV clones. In contrast to BCR-ABL-positive MPNs, compensating point mutations in kinase domains have not been identified in humans suffering from JAK-2-positive neoplasms yet. This observation leads to alternative hypotheses. In particular, selective pressure of JAK1/2 inhibition may be easily overwhelmed by alternative pathway activation. An insufficient inhibition of STAT5 pathway is not likely, as the high potency of ruxolitinib to hypophosphorylate STAT5 has been demonstrated. Even with this potent blockage, complete hematological responses in the RESPONSE trial are observed in only 24% of patients, as opposed to a much higher rate in most tyrosine kinase inhibitor treatments for BCR-ABL-positive malignancies, highlighting the theory of alternative pathways.16
Pharmacokinetics of ruxolitinib
A pharmacokinetic study conducted on healthy subjects with radiolabeled ruxolitinib attested rapid absorption, and high solubility with peak plasma concentration reached in <2 hours. Food intake does not seem to impact significantly on drug bioavailability, which is >95%. In plasma, the drug is 97% protein bound, mostly with albumin. Plasma half-life is ~3 hours.50
Ruxolitinib is extensively metabolized by cytochrome 3A4 into less active metabolites and is subject to significant pharmacodynamic interactions that prescribers should be aware of. Modified dosing should be considered when ruxolitinib is prescribed in combination with CYP3A4 inhibitors, such as conazole-antimycotics (ketoconazole, fluconazole), or certain antibiotics, such as erythromycin. An estimated reduction of 50% should be considered with strong enzymatic inhibitors in light of data available in healthy volunteers.51 Pharmacodynamic properties seem to be less affected in the setting of concomitant inducers administration, such as rifampicin, and a dose adaptation is generally not indicated.
Active metabolites maintain a significant pharmacodynamic activity accounting for ~20% in laboratory assays. Daily intake does not lead to significant parent compound and drug metabolites accumulation. Urinary excretion of metabolites accounts for almost three-quarters of elimination route and the remaining quarter is found in feces, with <1% as an intact compound. Ruxolitinib exhibits both a monophasic and a biphasic decrease with a mean half-life of 2.3 hours and 5.8 hours, respectively.52 In the setting of mildly impaired renal function, concern regarding accumulation of active metabolites should lead to dose reduction, based on initial platelet count and close follow-up.51 Therapy should be significantly reduced or even completely avoided in patients with severe renal failure (creatinine clearance <15 mL/min) and in patients with baseline platelet count <100 G/L, according to available data and common expert opinion.51 Regarding hepatic dysfunction, Chen et al53 did not find a relationship between its severity and systemic exposure to ruxolitinib (area under the curve). In this case, starting dose based on platelet count should be reduced by ~50%.
RESPONSE to ruxolitinib
Ruxolitinib has proven its efficacy in the recently published Phase III RESPONSE trial.16 This study compared ruxolitinib to standard therapy in a cohort of 222 PV patients with unacceptable side effects of or inadequate response to HU. The latter was defined as a required dose ≥2 g/d or an inferior maximum tolerated dose resulting in hematotoxicity or failure to control leukocytosis, thrombocytosis, hematocrit level, or splenomegaly. Randomized in a 1:1 fashion, this patient cohort had a mean age of ~60 years, good performance status for the vast majority, and a long course of disease of 8–9 years since diagnosis. The studied treatment dose of ruxolitinib was 10 mg twice daily. The primary end point was hematocrit control and reduction of spleen size of at least 35% at 8 months after randomization. In the ruxolitinib arm, 60% and 38% of patients showed either hematocrit control or spleen volume reduction, respectively. Interestingly, only 21% of patients achieved the composite end point, indicating that one parameter is not closely related to the other. Spleen volume reduction was chosen as one primary end point because splenomegaly is associated with a decreased survival and with a higher symptom burden. Important symptom relief was reached in 49% of patients treated with ruxolitinib versus 5% in the control arm. Phlebotomy dependency was also decreased with supposed consecutive reduction in related side-effects (Tables 1–3). Both of these results represent a real value in clinical practice. While phlebotomy is very effective, its long-term feasibility is limited, due to the development of severe iron deficiency and its side effects both hematologically (eg, thrombocytosis) and systemically (eg, skin problems).
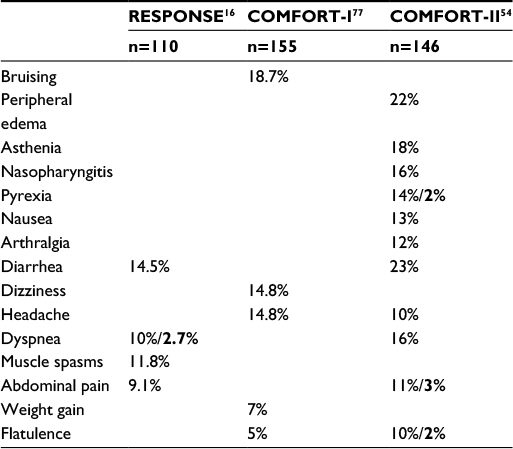 | Table 1 Adverse clinical events during ruxolitinib treatment Notes: RESPONSE: adverse events (all grades) with a frequency more than two times higher than in the best available therapy group. COMFORT-I/II: adverse events (all grades) with a frequency at least 5% higher than in the placebo group. Bold = Grades 3–4 (according to NCI CTCAE 3.0). |
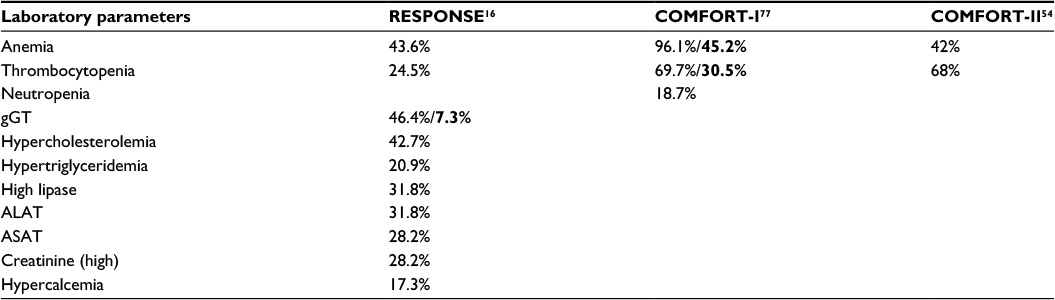 | Table 2 Adverse laboratory events during ruxolitinib treatment Notes: RESPONSE: laboratory parameters adverse events (all grades) with a frequency at least 5% (hematological) and 10% (nonhematological) higher than in the best available therapy. COMFORT-I/II: hematological adverse events (all grades) with a frequency at least 10% higher than in the placebo and best available therapy groups, respectively. Half of grades 3–4 anemias and thrombocytopenias occurred in the first 8 weeks of treatment initiation. Thrombocytopenia was the most important cause of dose interruption/modification in Phase III trials (40%–50%). Dose reduction is advocated for any hemoglobin <100 g/L and/or platelet count <75 G/L (it should be considered already if <100 G/L). Case series of “ruxolitinib withdrawal syndrome” defined as an acute worsening of disease-related symptoms, parameters, and splenomegaly of varying severity have been described in MF patients. Bold = Grades 3–4 (according to NCI CTCAE 3.0). Abbreviations: gGT, gamma-glutamyl transferase; ALAT, alanine aminotransferase; ASAT, aspartate aminotransferase; MF, myelofibrosis. |
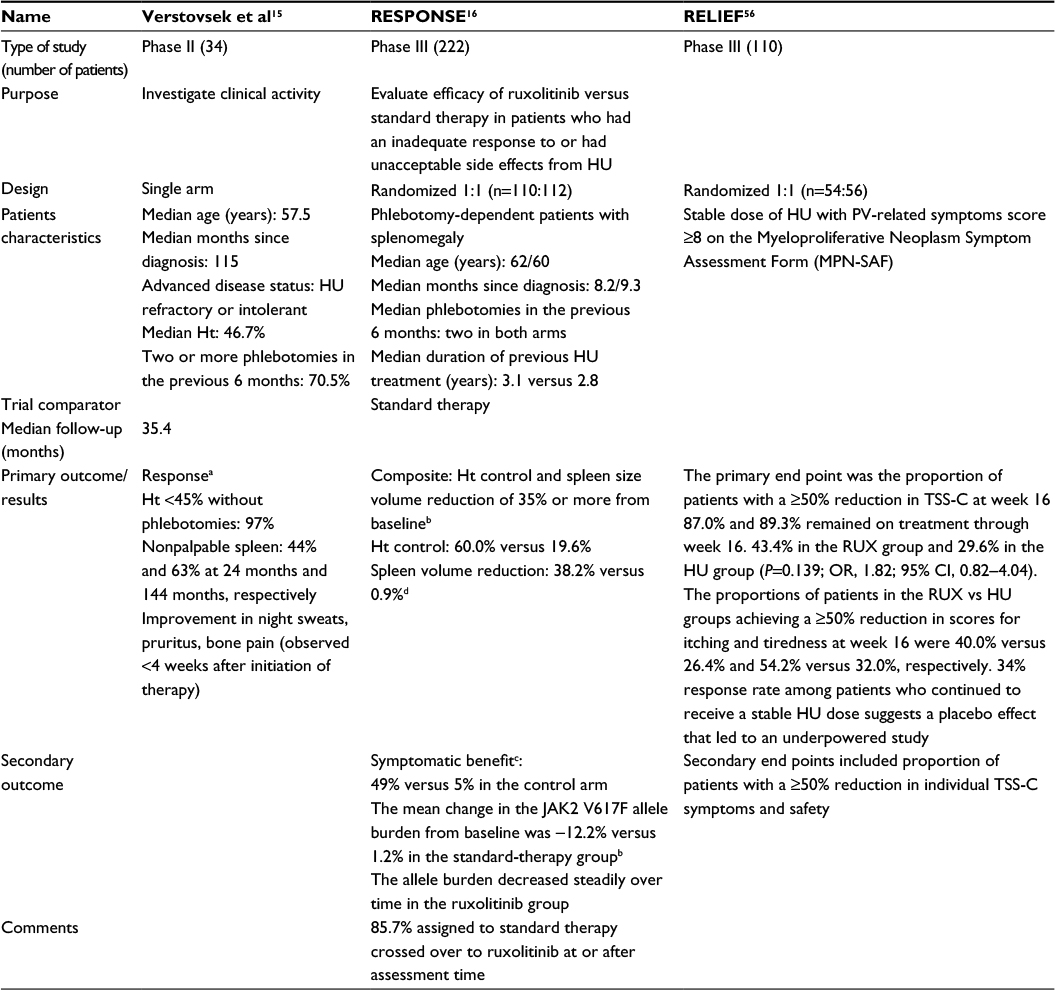 | Table 3 Summary of evidence in treating PV patients with ruxolitinib Notes: aAssessed by European Leukemia Net criteria. bAssessed at 8 months; spleen size measurement by means of centrally reviewed MRI or CT studies. cAssessed by Myeloproliferative Neoplasm Symptom Assessment Form (MPN-SAF total symptom score). dAt least one component of the primary end point occurred in 77.3% of patients in the ruxolitinib group. Abbreviations: HU, hydroxyurea; PV, polycythemia vera; TSS-C, cytokine total symptom score; RUX, ruxolitinib; OR, odds ratio; MRI, magnetic resonance imaging; CT, computed tomography; Ht, hematocrit; CI, contraindication. |
A point of criticism is the choice of the hematocrit level as the only surrogate for thromboembolic risk. A second criticism is the choice of a 35% spleen size reduction cutoff, as patients with important splenomegaly would still be symptomatic even after this relative downsizing. The 35% spleen size reduction was chosen, as it was already an end point in the COMFORT-II study.54 The rationale was based on the observation that a 50% reduction in spleen length was roughly equivalent to a 35% reduction in spleen volume.55 Another point is that 58% of patients in the standard arm were still on HU treatment at randomization. Despite the abovementioned limitations, the study demonstrated the efficacy of ruxolitinib (24% of complete hematological responses vs 1%) as attested by an impressive crossover of 86% of patients to ruxolitinib. Consequently, it is unlikely to obtain data on a potential effect on transformation rates into MF or acute myeloid leukemia during the ongoing cohort monitoring.
Side effects of ruxolitinib
Ruxolitinib is well tolerated in PV with 85% of patients still on treatment at 2 years of follow-up.16 Most frequent adverse events consist of myelosuppression, gastrointestinal toxicity, and an immunosuppression including an elevated risk of infection and reactivation of herpes viruses and other infectious agents controlled through cellular immunity. The most common side effect is bone marrow depression ranging from unilineage cytopenia to pancytopenia. Before starting therapy, a full blood count is needed and dose of ruxolitinib should be adapted depending on the platelet count. Under therapy, regular controls of full blood count are needed, as cytopenia is likely to develop. Dose adaptation or even therapy interruption may be required. Additional common side effects are vertigo, constipation, herpes zoster, and urinary tract infections. A high proportion of patients gained weight (Tables 1 and 2).
Practicians must be aware that cases of nonmelanoma skin carcinomas have been reported in patients treated with ruxolitinib. A causal relationship is not clearly established, due to previous use of other therapies, age of the concerned population, and the presence of premalignant lesions in some cases.
Sporadic life-threatening cases of progressive multifocal leukoencephalopathy due to JC virus reactivation have been associated to JAK signal inhibition, but as these are rare events, the prevalence of this serious condition cannot be quantitated.
Place of ruxolitinib in current clinical practice: for which patient and when?
The RESPONSE trial demonstrated similar efficacy in spleen size reduction after 1 year in PV patients as did the COMFORT-II study in MF patients.16,54 These two well-powered Phase III studies showed sufficient evidence for a significant benefit in patients suffering from disabling abdominal discomfort due to splenomegaly. Improvements of itching, constitutional symptoms (ie, night sweats), bone pain, and iron stores have also been demonstrated (Table 2). The above-mentioned clinical benefits should be the strongest reasons to initiate ruxolitinib treatment in the light of available data, provided that these symptoms persist under the standard treatment available.
Is there an indication to switch therapy in the case of a PV well controlled by HU but with persistence of disabling symptoms? The RELIEF study tried to answer the question by including PV patients on a stable dose of HU for at least 3 months with PV-related symptoms.56 This placebo-controlled randomized Phase IIIb study revealed a positive trend for a switch to ruxolitinib regarding symptom relief, but this trend did not prove statistically significant, and the abstract presentation at the 2014 American Society of Hematology has not yet been followed by a full publication.56 Symptoms were assessed according to the Myeloproliferative Neoplasm Symptom Assessment Form and the cytokine total symptom score and included evaluation of tiredness, itching, muscle aches, and night and daytime sweats. An unexpected placebo effect of 34% in symptoms improvement in the HU arm resulted in the underpowering of the study; a clear statistically significant difference could not be demonstrated. Nonetheless, after 4 months of treatment, individual symptoms such as itching and night sweats reached a median improvement from baseline of 40% and 68%, respectively.
The data on long-term outcome of PV patients treated with ruxolitinib are sparse. Most mature data come from MF cohorts in Phase III studies. A pooled analysis of overall survival (OS) from COMFORT-I and -II trials suggested an improved survival in ruxolitinib-treated arms (after 3 years of follow-up) with a crossover-corrected ratio of 0.29. Patients with intermediate- and high-risk features showed a survival benefit, although survival was only a secondary end point in both trials. These study results in MF patients should encourage future studies to address the question of survival improvement as a primary end point in PV patients treated with ruxolitinib, although pathogenesis, disease characteristics, and survival are different in this pathology.57,58
The ongoing Phase III trial RESPONSE-2 addresses hematocrit control in HU-intolerant or -resistant PV patients without palpable splenomegaly. The sponsoring pharmaceutical company has announced that this primary end point has been met. Some of its secondary end points include assessment of thrombosis and hemorrhagic and transformation events. First results addressing these questions are awaited for 2020. The British MAJIC randomized Phase II trial will also provide data on OS and thromboembolic event rate as secondary outcomes. Interestingly, histological (bone marrow biopsies) and molecular (JAK2 V617F burden) responses will also be examined.
Future study design in PV
A median survival of 15 years with a median age at diagnosis of 60 years makes treatment influence on survival of PV difficult to assess. Bone marrow changes, both event- and complication-free survival, and quality of life improvement may be end points of interest for future studies.59 Additionally, risk factors possibly associated with a shortened survival in PV (such as hematocrit levels, leukocytosis, and JAK2 V617F allele burden) might be used as best surrogate markers for improvement of OS.60 In particular, JAK2 V617F allele burden could be used to monitor treatment efficacy, as allele burden can reflect PV progression. However, allele burden reduction by treatment has not yet been demonstrated as a surrogate of better outcome. In a Phase II study by Verstovsek et al,15 JAK2 V617F allele burden reduction attained 20% in 42% of ruxolitinib-treated patients. In mice, gene expression level is correlated with disease phenotype, and allele burden control may have a different impact between individuals.61 High allele burden also seems to be a predictive risk factor for MF progression of PV, as it has been recently demonstrated, not only in mouse models but also in patients suffering from PV.60
Unfortunately, large studies are clearly designed to achieve fast drug approval rather than trying to offer best achievable evidence of both clinical and survival benefit. This approach impairs investigation of drug combinations and knowledge of disease behavior.
Future concepts and potential combination therapies
Intensifying response through combination therapies will certainly be one of the next steps in PV approach. Our knowledge of other active compounds in PV may be the best way to identify potentially synergistic drug associations. Beneficial IFN-α action, although unclear in many points, seems to ensue from a combination of direct effects on PV and normal stem cells in conjunction with immune-mediated response modulation. However, how exactly these mechanisms work together and how they can provoke a response rate of roughly 60% in patients suffering from PV are unknown.62 IFN-α signaling plays a role in effector CD4 lymphocyte survival and leads to decreased allele burden in some cases and also to polyclonal hematopoietic resurgence.63,64 Total dissociation in the two aforementioned events has been reported and is consistent with the existence of a proliferative/differentiating effect on normal quiescent stem cells on the one hand and a cytostatic action on PV progenitors on the other.65 Downstream signaling of IFN-α transduction through JAK1 and TYK2 allows putting forward the hypothesis that the combination of IFN-α with a selective JAK2 inhibitor could act in a complementary way. Feasibility, safety, and an impressive response in a patient treated with IFN-α concomitantly to ruxolitinib have been published in 2014.66
Possibly, elucidation of pathophysiological mechanisms will lead to the development of targeted therapy. PI3K/mTOR pathway demonstrated its implication in survival of JAK2-positive human cell lines as its inhibition showed an enhancement of ruxolitinib apoptotic activity, opening the way for future Phase II studies.67 Another interesting approach would be to target downstream important effectors of JAK such as STAT5. Furthermore, accumulation of reactive oxygen species as a consequence of JAK–STAT hyperactivity leads to DNA damage and genetic instability.10,68 Anti-inflammatory properties of ruxolitinib could also be exploited in the future.69 Other pathological circumstances are being discovered, as, for example, change of microenvironment in JAK2 V617F-positive MPNs with reduction of sympathetic nerve fibers and Nestin+ mesenchymal cells in the bone marrow of these patients. These discoveries are leading to upcoming drugs other than JAK inhibitors with influence on allele burden, as, for example, beta-mimetics, studied currently in Phase II in Switzerland (NCT02311569). MPN cells are held responsible for destroying cells of the microenvironment, an effect that could be proven reversible in a mouse model under beta-mimetic treatment.70
Other potential JAK inhibitors
Highly selective JAK2 V617F inhibition is a logic alternative to JAK1/2 inhibition and is now currently under investigation. Fedratinib, a selective JAK2 inhibitor, showed encouraging symptomatic benefit in patients suffering from MF, including post-PV and post-ET patients, according to the interim analysis of the JAKARTA-2 study released as an abstract in 2013.71 A previously published Phase I study conducted by Pardanani et al72 showed high normalization rates of platelet and leukocyte counts in MF. However, the development of fedratinib was stopped in 2013 while in Phase III, when the US Food and Drug Administration ordered immediate interruption of all ongoing studies with this molecule for safety reasons as a consequence of reports that study subjects were developing Wernicke’s encephalopathy.73
Other JAK2 inhibitors are on their way, such as momelotinib, which appears to exert a beneficial effect on splenomegaly and on anemia in patients with MF,72 pacritinib, which is currently in Phase II and III studies,74 and CHZ868.75
Summary
While ruxolitinib seems to be a potent drug to treat symptoms of splenomegaly and cytokine effects, these problems are less prominent in PV than in MF patients. Phlebotomy, low-dose aspirin, and cytoreduction with HU or IFN-α will remain the backbone of treatment for the time being, as indicated by current guidelines from the European Leukemia Net23 and the British Committee for Standards in Haematology.24
As JAK2 mutations do not seem to be the first mutational event in the development of PV and since blast cells in leukemic transformation often do not show the JAK2 V617F mutation, the most feared PV complication, transformation to acute myeloid leukemia, will probably not be influenced by ruxolitinib treatment. Finally, in patients undergoing stem cell transplantation, it cannot be recommended to pretreat them with ruxolitinib outside clinical studies.
There are certainly PV patients benefiting from ruxolitinib; however, these patients must be carefully selected. Ruxolitinib’s place in current practice should be directed toward disease-related symptoms relief after failure of standard treatment. Of particular note, the beneficial effect of ruxolitinib is not limited to patients with a positive mutational status, indicating that the drug is not specific for the PV clone. In fact, ruxolitinib is a good drug to address symptomatic burden, but less so the allelic one. Disease-related problems, such as itching, can effectively be treated with ruxolitinib, if they do not disappear with standard treatments, such as HU.
In PV, in which life expectancy can approach a near-normal range, the primary treatment goal is still prevention of thromboembolic events that account for most of the deaths in this pathology. As yet, no study on ruxolitinib has addressed the question of its influence on thrombotic events, and ruxolitinib cannot be considered as a first-line treatment from this point of view either. However, a meta-analysis from COMFORT-I, COMFORT-II, and RESPONSE trials (including 750 patients) provided data showing a significant reduction in thrombosis rate among patients treated with ruxolitinib with a risk ratio of 0.45 in comparison to placebo and best available therapies.76 Despite similar risk ratio, subgroup analysis of arterial and venous thrombosis did not reach statistical significance in this heterogeneous patient group containing both MF and PV patients. It is difficult to conclude if this potential benefit impacts more on the arterial or venous system or both, without future trials addressing this specific question in PV patients as well as MF patients only.
Another question to address is the longevity of symptom relief. If the expected benefit will be limited to only 1–3 years, ruxolitinib may not be a good option, at least in the beginning of PV treatment. Further studies on long-term outcome are needed to address this question. Additionally, treatment interruption among study patients was quite frequent, not only due to loss of response but also due to toxicity, allowing the rebound phenomenon of cytokine effects to become a real problem, even in PV.
Conclusion
A concerted discussion between a patient and his/her hematologist, balancing costs, side effects, and expected benefit from therapy, has to precede prescription. The patient has to be thoroughly informed about ruxolitinib side effects, particularly as the drug does not seem to have disease-modulating action and OS improvement is marginal, if existent at all. Careful management of myelosuppressive, infective, and gastrointestinal side effects with a thorough blood count survey and regular follow-up are the cornerstones of good practice. A final but not trivial point to consider is that treatment costs are quite high.
Disclosure
The authors report no conflicts of interest in this work.
References
Vaquez H. Sur une forme speciale de cyanose s’accompanant d’hyperglobulie excessive et persistante. CR Soc Biol (Paris). 1892;4:384. | ||
Osler W. Chronic cyanosis with polycythemia and enlarged spleen: a new clinical entity. 1930. Am J Med Sci. 2008;335(6):411–417. | ||
Tam CS, Nussenzveig RM, Popat U, et al. The natural history and treatment outcome of blast phase BCR-ABL- myeloproliferative neoplasms. Blood. 2008;112(5):1628–1637. | ||
Tefferi A. Annual clinical updates in hematological malignancies: a continuing medical education series: polycythemia vera en essential thrombosis: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86(3):292–301. | ||
Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14–22. | ||
Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6(4):372–375. | ||
Prchal JF, Axelrad AA. Letter: bone-marrow responses in polycythemia vera. N Engl J Med. 1974;290(24):1382. | ||
Casadevall N, Vainchenker W, Lacombe C, et al. Erythroid progenitors in polycythemia vera: demonstration of their hypersensitivity to erythropoietin using serum free cultures. Blood. 1982;59(2):447–451. | ||
Cashman J, Henkelman D, Humphries K, Eaves C, Eaves A. Individual BFU-E in polycythemia vera produce both erythropoietin dependent and independent progeny. Blood. 1983;61(5):876–884. | ||
James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. | ||
Baxter EJ, Scott LM, Campbell PJ, et al; Cancer Genome Project. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. | ||
Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. | ||
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | ||
Quintás-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. | ||
Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 Inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer. 2014;120(4):513–520. | ||
Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. | ||
Theocharides A, Boissinot M, Girodon F, et al. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;110(1):375–379. | ||
Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2014;55(3):595–600. | ||
Phekoo KJ, Richards MA, Moller H, Schey SA, South Thames Haematology Specialist Committee. The incidence and outcome of myeloid malignancies in 2,112 adult patients in southeast England. Haematologica. 2006;91(10):1400–1404. | ||
Skoda R, Prchal JT. Lessons from familial myeloproliferative disorders. Semin Hematol. 2005;42(4):266–273. | ||
Saliba J, Saint-Martin C, Di Stefano A, et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat Genet. 2015;47(10):1131–1140. | ||
Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874–1881. | ||
Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European Leukemia Net. J Clin Oncol. 2011;29(6):761–770. | ||
McMullin MF, Wilkins BS, Harrison CN. Management of polycythaemia vera: a critical review of current data. Br J Haematol. 2016;172(3):337–349. | ||
Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol. 2012;30(33):4098–4103. | ||
Tefferi A, Elliott M. Thrombosis in myeloproliferative disorders: prevalence, prognostic factors, and the role of leukocytes and JAK2V617F. Semin Thromb Hemost. 2007;33(4):313–320. | ||
Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33. | ||
Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27(32):5418–5424. | ||
Kremyanskaya M, Mascarenhas J, Hoffman R. Anagrelide hydrochloride and ruxolitinib for treatment of polycythemia vera. Expert Opin Pharmacother. 2015;16(8):1185–1194. | ||
Cortelazzo S, Finazzi G, Ruggeri M, et al. Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med. 1995;332(17):1132–1136. | ||
Berk PD, Goldberg JD, Donovan PB, Fruchtman SM, Berlin NI, Wasserman LR. Therapeutic recommendations in polycythemia vera based on polycythemia vera study group protocols. Semin Hematol. 1986;23(2):132–143. | ||
Landolfi R, Marchioli R. European collaboration on low-dose aspirin in polycythemia vera (ECLAP): a randomized trial. Semin Thromb Hemost. 1997;23(5):473–478. | ||
Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114–124. | ||
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. | ||
Dai CH, Krantz SB, Dessypris EN, Means RT Jr, Horn ST, Gilbert HS. Polycythemia vera. II. Hypersensitivity of bone marrow erythroid, granulocyte-macrophage, and megakaryocyte progenitor cells to interleukin-3 and granulocyte-macrophage colony-stimulating factor. Blood. 1992;80(4):891–899. | ||
de Wolf JT, Hendriks DW, Esselink MT, Halie MR, Vellenga E. The effects of IL-1 and IL-4 on the EPO-independent erythroid progenitor in polycythaemia vera. Br J Haematol. 1994;88(2):242–246. | ||
Marchioli R, Vannucchi AM, Barbui T. Treatment target in polycythemia vera. N Engl J Med. 2013;368(16):1556. | ||
Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood. 2006;108(5):1652–1660. | ||
Lundberg P, Takizawa H, Kubovcakova L, et al. Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. J Exp Med. 2014;211(11):2213–2230. | ||
Nussenzveig RH, Swierczek SI, Jelinek J, et al. Polycythemia vera is not initiated by JAK2V617F mutation. Exp Hematol. 2007;35(1):32–38. | ||
Scott LM, Scott MA, Campbell PJ, Green AR. Progenitors homozygous for the V617F mutation occur in most patients with polycythemia vera, but not essential thrombocythemia. Blood. 2006;108(7):2435–2437. | ||
Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood. 2012;119(15):3539–3549. | ||
Walz C, Ahmed W, Lazarides K, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCRABL1 and JAK2V617F in mice. Blood. 2012;119(15):3550–3560. | ||
Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468. | ||
Ishii T, Bruno E, Hoffman R, Xu M. Involvement of various hematopoietic-cell lineages by the JAK2V617F mutation in polycythemia vera. Blood. 2006;108(9):3128–3134. | ||
Sozer S, Fiel MI, Schiano T, Xu M, Mascarenhas J, Hoffman R. The presence of JAK2V617F mutation in the liver endothelial cells of patients with Budd-Chiari syndrome. Blood. 2009;113(21):5246–5249. | ||
Wernig G, Kharas MG, Okabe R, et al. Efficacy of TG101348, a selective JAK2 inhibitor, in treatment of a murine model of JAK2V617F-induced polycythemia vera. Cancer Cell. 2008;13(4):311–320. | ||
Mascarenhas JO, Cross NC, Mesa RA. The future of JAK inhibition in myelofibrosis and beyond. Blood Rev. 2014;28(5):189–196. | ||
Alchalby H, Kroger N. Allogeneic stem cell transplant vs. Janus kinase inhibition in the treatment of primary myelofibrosis or myelofibrosis after essential thrombocythemia or polycythemia vera. Clin Lymphoma Myeloma Leuk. 2014;14(suppl):S36–S41. | ||
Swaim SJ. Ruxolitinib for the treatment of primary myelofibrosis. Am J Health Syst Pharm. 2014;71(6):453–462. | ||
Chen X, Williams WV, Sandor V, Yeleswaram S. Population pharmacokinetic analysis of orally-administered ruxolitinib (INCB018424 Phosphate) in patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF) or post-essential thrombocythemia myelofibrosis (PET MF). J Clin Pharmacol. 2013;53(7):721–730. | ||
Shilling AD, Nedza FM, Emm T, et al. Metabolism, excretion, and pharmacokinetics of [14C]INCB018424, a selective Janus tyrosine kinase 1/2 inhibitor, in humans. Drug Metab Dispos. 2010;38(11):2023–2031. | ||
Chen X, Shi JG, Emm T, et al. Pharmacokinetics and pharmacodynamics of orally-administered ruxolitinib (INCB 0184 24 phosphate) in renal and hepatic impairment patients. Clin Pharmacol Drug Dev. 2014;3(1):34–42. | ||
Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047–4053. | ||
Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. | ||
Mesa R, Vannucchi AM, Yacoub A, et al. The efficacy and safety of continued hydroxyurea therapy versus switching to ruxolitinib in patients with polycythemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Blood. 2014;124(21):3168. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3-year follow-up of COMFORT-I. Haematologica. 2015;100(4):479–488. | ||
Vannucchi AM, Kantarjian HM, Kiladjian JJ, et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. 2015;100(9):1139–1145. | ||
Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755–761. | ||
Shirane S, Araki M, Morishita S, et al. Consequences of the JAK2V617F allele burden for the prediction of transformation into myelofibrosis from polycythemia vera and essential thrombocythemia. Int J Hematol. 2015;101(2):148–153. | ||
Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111(8):3931–3940. | ||
Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112(8):3065–3072. | ||
Passegue E, Ernst P. IFN-alpha wakes up sleeping hematopoietic stem cells. Nat Med. 2009;15(6):612–613. | ||
Prchal JF. Interferon and PV stem cells. Inside Blood. 2011;118(6):1429–1430. | ||
King KY, Baldridge MT, Weksberg DC, et al. Irgm1 protects hematopoietic stem cells by negative regulation of interferon signaling. Blood. 2011;118(6):1525–1533. | ||
Bjørn ME, de Stricker K, Kjær L, Ellemann K, Hasselbalch HC. Combination therapy with interferon and JAK1-2 inhibitor is feasible: proof of concept with rapid reduction in JAK2V617F-allele burden in polycythemia vera. Leuk Res Rep. 2014;3(2):73–75. | ||
Szymanska J, Smolewski P, Majchrzak A, Cebula-Obrzut B, Chojnowski K, Trelinski J. Pro-apoptotic activity of ruxolitinib alone and in combination with hydroxyurea, busulphan, and PI3K/mTOR inhibitors in JAK2-positive human cell lines. Adv Clin Exp Med. 2015;24(2):195–202. | ||
Marty C, Lacout C, Droin N, et al. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia. 2013;27(11):2187–2195. | ||
Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood. 2012;119(14):3219–3225. | ||
Mendez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–834. | ||
Pardanani A, Harrison CN, Cortes JE, et al. Results of a randomized, double-blind, placebo-controlled phase III study (JAKARTA) of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis (MF). Blood. 2013;122(21):393. | ||
Pardanani A, Laborde RR, Lasho TL, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia. 2013;27(6):1322–1327. | ||
Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1(5):643–651. | ||
Verstovsek S, Dean JP, Cernohous P, et al. Pacritinib, a dual JAK2/FLT3 inhibitor: an integrated efficacy and safety analysis of phase ii trial data in patients with primary and secondary myelofibrosis (MF) and platelet counts ≤100,000/µl. Blood. 2013;122(21):395. | ||
Meyer SC, Keller MD, Chiu S, et al. CHZ868, a type II JAK2 inhibitor, reverses type I JAK inhibitor persistence and demonstrates efficacy in myeloproliferative neoplasms. Cancer Cell. 2015;28(1):15–28. | ||
Samuelson BT, Vesely SK, Chai-Adisaksopha C, Scott BL, Crowther M, Garcia D. The impact of ruxolitinib on thrombosis in patients with polycythemia vera and myelofibrosis: a meta-analysis. Blood Coagul Fibrinolysis. 2016;27:648–652. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.