")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
Ruxolitinib in myelofibrosis: to be or not to be an immune disruptor
Authors Manduzio P
Received 6 September 2016
Accepted for publication 4 January 2017
Published 13 February 2017 Volume 2017:13 Pages 169—177
DOI https://doi.org/10.2147/TCRM.S121683
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Palma Manduzio
Department of Haematology and Oncology, Haematology With BMT, IRCCS, Casa Sollievo della Sofferenza, Foggia, Italy
Abstract: Primary myelofibrosis (PMF) is a myeloproliferative neoplasm classified according to the 2016 revision of World Health Organization Classification of Tumors and Haematopoietic and Lymphoid Tissue. Ruxolitinib is an oral inhibitor of Janus kinase approved in the USA for the treatment of intermediate or high-risk PMF and approved in Europe for the treatment of splenomegaly and constitutional symptoms of the disease. More recently, case reports described serious opportunistic infections in this neoplasm treated with ruxolitinib. Research studies demonstrated the immunological derangement of this compound mainly based on T, dendritic, and natural killer cell defects. The purpose of this review of the literature was to analyze the relationship among ruxolitinib, immune system and bacterial, viral, fungal, and protozoan infections. A literature search was conducted using PubMed articles published between January 2010 and November 2016. The efficacy of drug in patients with PMF was demonstrated in two phase III studies, Controlled MyeloFibrosis Study with ORal Jak inhibitor Treatment (COMFORT-I and COMFORT-II). Grade 3 and 4 neutropenia were recognized in 7.1% and 2% of patients in the ruxolitinib and placebo arm of COMFORT-I. Grade 3 or 4 neutropenia or leukopenia were observed in 8.9% and 6.3% of ruxolitinib treated patients of 5-year follow-up of COMFORT-II. In addition, leukocyte subpopulations, lymphocyte functions, or antibody deficiency were not documented in either of the studies. The complex interactions between ruxolitinib, bone marrow, immune system, and infections in PMF need further investigation, robust data from a randomized clinical trial, registry, or large case-series.
Keywords: myelofibrosis, JAK/STAT pathway, immune system, infections, inflammation
Introduction
Myeloproliferative neoplasms (MPNs) according to the fourth edition of the World Health Organization (WHO) Classification of Tumors of the Haematopoietic and Lymphoid Tissues and the 2016 revision represent a group of heterogeneous diseases, which include polycythemia vera (PV), primary myelofibrosis (PMF), and essential thrombocythemia (ET).1–3
At the “early stage” of PMF, mandatory major criteria include megakaryocytic proliferation and atypia, clonal mutations of genes, such as Janus kinase 2 (JAK2), calreticulin (CALR), thrombopoietin receptor, or myeloproliferative leukemia virus oncogene (MPL). The minor criteria, confirmed in two determinations, include at least one of following: anemia, palpable splenomegaly, increased lactate dehydrogenase (LDH), and leukocytosis. In “overt PMF” mandatory major criteria include clonal mutations and megakaryocytic atypia associated with diffuse or dense increase of reticulin fibrosis or collagen fibrosis or osteosclerosis. The minor criteria, confirmed in two determinations, include at least one of the following: anemia, splenomegaly, increased LDH, and leukoerythroblastosis.1–3
Pathogenetically, PMF represents a stem cell-derived MPN associated with ineffective erythropoiesis and extramedullary hematopoiesis, cytokine-mediated stromal changes, including fibrosis and constitutional symptoms, poor prognosis due to thrombohemorrhagic complications and acute myeloid leukemic transformation. Lower risk of acute myeloid leukemia and better prognosis were recognized in PV and ET.4,5
The Mayo Clinic Dynamic International Prognostic Scoring System-plus (DIPSS-plus), clinical and cytogenetic risk score discriminates 4 risk groups of PMF: low, intermediate 1 and 2, high-risk disease in the presence of 0, 1, 2 or 3, 4 or more factors. Age, constitutional symptoms, hemoglobin, peripheral blasts, leukocytes, platelets, karyotype, and transfusion dependency influence median overall survival (OS) of the disease. In fact, the OS varies from 15.4 to 1.3 years.6 Currently, molecular genetic DIPSS-independent factors are being studied to better stratify the outcome of MF.7,8
Hematopoietic stem cell transplantation (HSCT) represents a curative approach reserved to a small group of intermediate-2 or high-risk PMF patients. However, novel agents targeting MPN molecular mechanisms, such as JAK, are available in clinical practice and trials to improve constitutional symptoms, splenomegaly, and quality of responses.9
Mutations of JAK2, CALR, and MPL are mutually exclusive in the pathogenesis of myeloproliferative Philadelphia-negative neoplasms. In this regard, 10%–15% of PMF are reported “triple negative” for JAK2, CALR, and MPL mutations; 50%–60% of PMF are JAK2 mutated, 20%–25% and 6%–7% are CALR and MPL mutated, respectively.2
Janus kinase signal transducers and activators of transcription pathway (JAK/STAT) transmit information from outside on the DNA of the cell. JAKs are associated with growth factor receptors (eg, erythropoietin receptor and thrombopoietin receptor). After binding of interferon or interleukin, cytokine receptors recruit JAKs, which phosphorylate the receptor protein and bind STAT proteins. STATs initiate transcription of target genes involved in cell growth, differentiation, and apoptosis.9 Specifically, JAK2V617F somatic mutation leads to constitutive activation of JAK/STAT pathway. The mechanism of CARL mutations or more rare mutations of epigenetic modifiers are still uncertain.10,11
Four JAK family members and seven STATs are recognized. The JAK family has a key role in myeloid and lymphoid cell proliferation and differentiation. JAK1 mediates the effect of proinflammatory cytokines: interleukin-2 (IL-2), interleukin-6 (IL-6), and tumor necrosis factor alpha (TNF-alpha). JAK2 mediates differentiation, proliferation, and avoidance of apoptosis. Ruxolitinib, an oral inhibitor of JAK1 and JAK2, is associated with reduction in the levels of inflammatory markers: IL-6, TNF-alpha, and C-reactive protein (CRP).12,13 Inflammatory cytokines are considered responsible for the constitutional symptoms (night sweats, pruritus and fever, fatigue, and weight loss) of myelofibrosis (MF).14
Ruxolitinib (JAKAFI, Incyte Corporation; JAKAVI, Novartis) is approved in the USA for the treatment of intermediate or high-risk disease and in Europe for the treatment of splenomegaly or constitutional symptoms. In addition, this drug is not specific for the mutated form of JAK2 and inhibits both the wild-type and JAK2V617F. The efficacy of this compound in patients with PMF was demonstrated in two phase III studies, Controlled MyeloFibrosis Study with ORral Jak inhibitor Treatment (COMFORT-I and COMFORT-II).15,16
More recently, case reports described serious opportunistic infections in ruxolitinib-treated PMF patients and research studies demonstrated the immunological derangement of this treatment.
Methods
The purpose of this review of the literature was to analyze the relationship among ruxolitinib, immune system, and infections. A literature search was conducted using PubMed for articles published from January 2010 to November 2016 using the terms “myelofibrosis and infections or myelofibrosis and immune system”, “ruxolitinib and infections or ruxolitinib and immune system”. Only articles published in English were considered. The contribution of ruxolitinib to the immune balance is focused on bacterial, viral, fungal, and protozoan infections.
Results
In COMFORT-I study, ruxolitinib was compared with placebo in intermediate-2 and high-risk MF. In COMFORT-II, ruxolitinib was compared with the best available therapy in intermediate-2 and high-risk MF. More specifically, COMFORT-I is a double-blind trial, included 309 patients with International Prognostic Scoring System (IPSS) high-risk or intermediate-2 MF, palpable spleen of at least 5 cm, and platelet count of at least 100,000/mm3. COMFORT-II is an open-label phase III trial, which enrolled 219 patients considering the same criteria of inclusion of the previous trial. In both the trials, the starting dose of drug was 15 or 20 mg twice daily based on platelet count >200,000/mm3 or between 100,000 and 200,000/mm3. Drug was reduced appropriately in patients with renal or hepatic impairment or platelet count <100,000/mm3. In both studies, reduction in spleen size was confirmed by imaging at 24 and 48 weeks of treatment.15,16
In the first month of the treatment of both trials, grade 3 or 4 anemia or thrombocytopenia were reported. These cytopenias result from the inhibitory effects of drug on JAK2 and erythropoietin/thrombopoietin signals.17 These adverse events (AE) can be managed adequately by prompt red blood cell transfusions and gradual tapering of dose due to “withdrawal syndrome” and rebound of inflammatory cytokines.18–20
Grade 3 and 4 neutropenia were recognized in 7.1% and 2% of patients in the ruxolitinib and placebo arm of COMFORT-I trial. Urinary infections and herpes zoster were documented in patients treated with ruxolitinib in COMFORT-I.15 More recently, 5-year follow-up of COMFORT-II noted grade 3 or 4 neutropenia or leukopenia in 8.9% and 6.3% of ruxolitinib-treated patients. Urinary tract infections, pneumonia, herpes zoster, sepsis, and tuberculosis (TBC) infections were observed in 24.6%, 13.1%, 11.5%, 7.9%, and 1% of ruxolitinib subgroup of patients, respectively.16
Leukocytes subpopulations and functions or antibody deficiency were not documented in both studies. Interestingly, Theocharides et al recognized that omozygous calreticulin mutations in PMF lead to acquired myeloperoxidase deficiency.21
Ruxolitinib and immune system: dendritic cell, natural killer, and T regulatory
The immune “orchestra” includes innate and adaptive arms. The former includes anatomic barriers, antimicrobial molecules, such as complement and cellular components: neutrophils, eosinophils, basophils, mast cell, natural killer (NK), monocytes/macrophages, and dendritic cells (DC). The latter includes B lymphocytes, CD4+ T-helper lymphocytes (Th1, Th2, Th17, and T regulatory [T reg]) and CD8+ cytotoxic T lymphocytes.
Figure 1 summarizes the complex relationship between the drug and the immune “orchestra”. More recently, Heine et al demonstrated that ruxolitinib affects DC differentiation, phenotype, and function leading to impaired T-cell activation.22
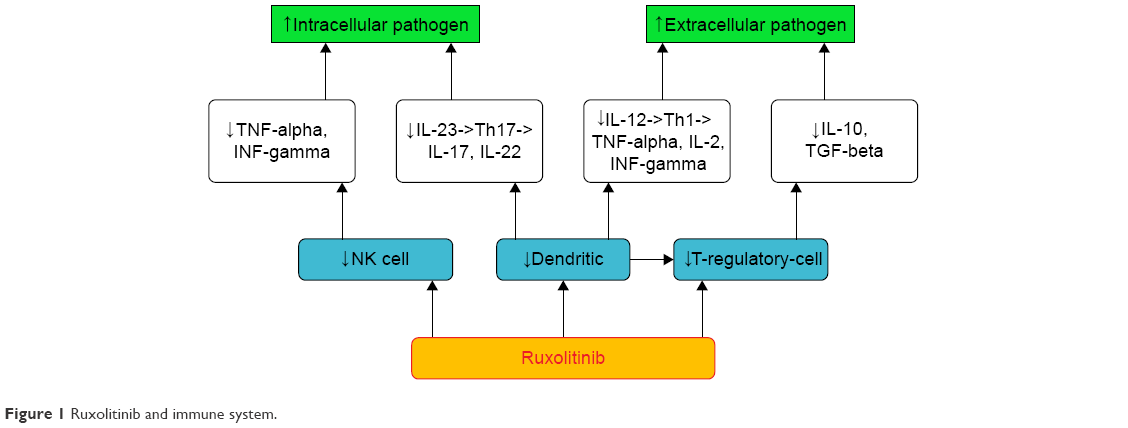 | Figure 1 Ruxolitinib and immune system. |
DC are important antigen presenting and phagocytic cells. They induce CD8+ T cells to destroy infected and neoplastic cells. In addition, DC control adaptive response producing IL-12 and IL-23, cytokines that drive Th1 and Th17 lymphocytes phenotypes. More specifically, Th1 cells produce cytokine interferon gamma (INF-gamma), IL-2, and TNF-alpha. TNF-alpha improves Th1 against intracellular pathogens. Th17 secrete IL-17 and IL-22, key cytokines against extracellular bacteria. More mature DC mediate the induction of T-reg cells.
In addition, Rudolph et al published very interesting data on the mechanism of ruxolitinib impairment of DC migration and inhibition of Rho-associated coil kinase.23
Schonberg et al reported a reduction of NK in ruxolitinib-treated patients.24 NK are lymphocytes, immune effector cells known to eliminate both virus and cancers and produce important inflammatory cytokines, such as INF-gamma and TNF-alpha.
Massa et al observed a rapid and long-lasting decrease of T-reg cell in patients treated with the drug.25 Keohane et al noted that T-reg cells are reduced in MPN patients compared with healthy, especially in those treated with ruxolitinib.26 Finally, Parampalli Yajnanarayana et al recognized a decreased T reg, Th1, and Th 17 in ruxolitinib exposure.27
T-reg cells control viral, fungal, and protozoan infections and are involved in moderating inflammation and maintaining self-tolerance. In fact, they produce inhibitory cytokines such as IL-10 and transforming growth factor-beta (TGF-beta) that promote fibrosis, affect the function, and induce apoptosis of T-effector lymphocytes.
Beyond immune derangement
Al-Ali et al28 recently reported an open-label, multicenter, single-arm phase IIIb expanded-access study in patients with MF. This study included patients treated with this compound outside a clinical trial. The analysis included a large cohort of 1,144 intermediate and high-risk MF patients, including 163 intermediate-1 diseases. Among nonhematologic AE, grade 3 or 4 neutropenia was reported. Herpes zoster and influenza were observed in 3.6% and 3% in the whole cohort of intermediate and high-risk MF, respectively. One case of hepatitis B reactivation was documented in intermediate-1 subgroup.28
MF is a rare myeloproliferative of elderly patients, it may be a primary disease or secondary transformation of PV/TE. PMF patients who undergo ruxolitinib treatment may have or develop defects involving innate and adaptive immune system, more specifically T, dendritic, and NK cells. In addition, these patients may have functional hyposplenism and agranulocytosis. Severe and prolonged neutropenia is a well-recognized risk factor for infections, such as neoplastic dysfunctional granulocytes, including myeloperoxidase deficiency. In summary, biological studies are consistent with the assumption of a combination of immune defects.22–27 Obviously, previous treatments, patient age and comorbidities, and environmental exposure may influence the risk of infections.29–31
Recently, a Cochrane study pointed out the small number of patients included in COMFORT-I and COMFORT-II.32 Therefore, more robust data are mandatory to answer the question of possible immune derangement of ruxolitinib treatment in MF. Randomized clinical trial (RCT) data or longer follow-up studies are not always available for rare diseases.33 Hultcrantz et al observed a higher mortality rate due to infections in patients diagnosed with MPNs than that of matched controls in Sweden between 1973 and 2005. In addition, big data from registry or large case series should be analyzed considering the 2016 revision of WHO.34
Bacterial, viral, fungal, and protozoan infections
Table 1 summarizes bacterial infections recently reported during JAKAFI/JAKAVI treatment.35–43 Bacterial infections are recognized early and late after treatment. The majority of case reports describe TBC disseminated disease, including Pott’s disease. The possible explanation of TBC reactivation during ruxolitinib may be the impairment of DC and IL-12 production, a key cytokine involved in the transcription of INF-gamma. In addition, ruxolitinib induces depression of Th1 lymphocyte responses and production of INF-gamma and TNF-alpha. Specifically, the former activates macrophage to produce reactive oxygen and the latter plays a critical role in protection against TBC.
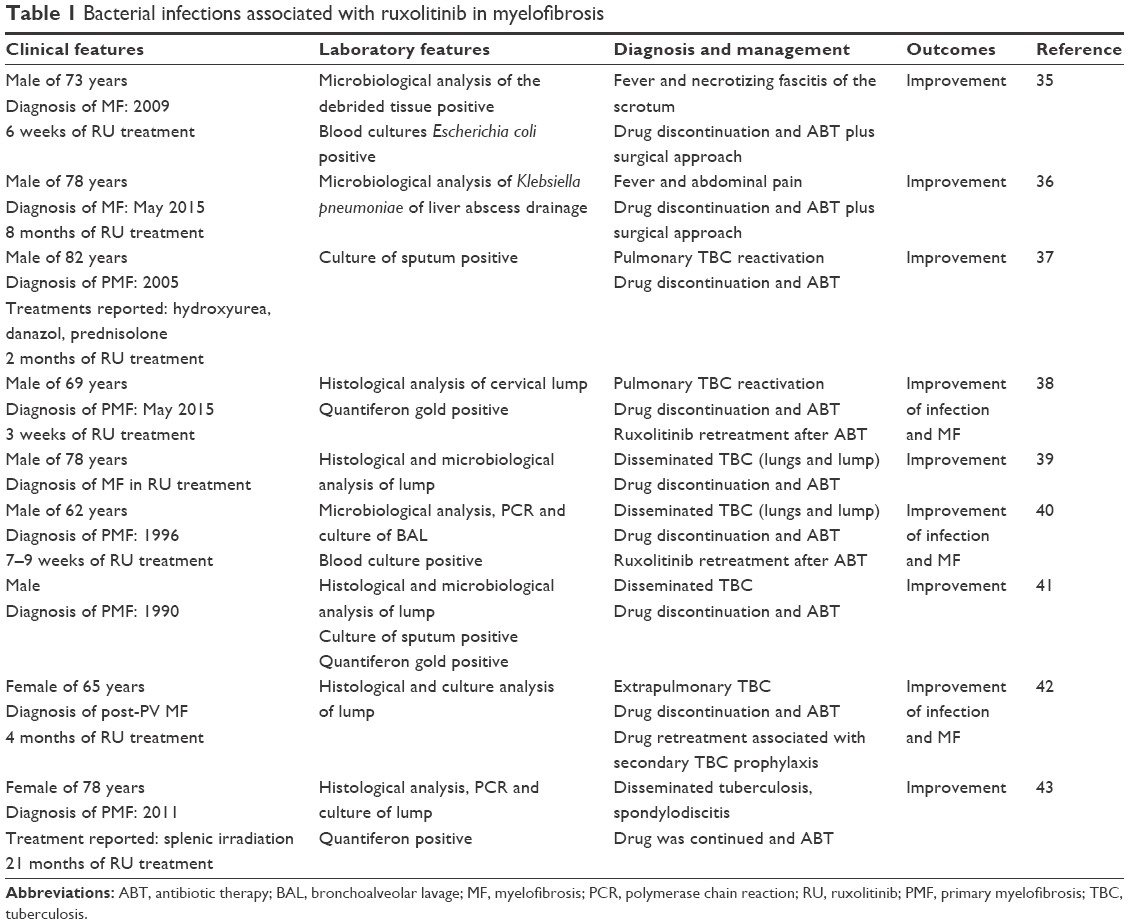 | Table 1 Bacterial infections associated with ruxolitinib in myelofibrosis |
Certainly, the screening for latent TBC must be considered if epidemiological risk factors are significant. Quantiferon TBC test may be negative due to immune dysregulation of disease or treatments. Furthermore, mild symptoms and low CRP are probably due to the reduction of inflammatory cytokines. Therefore, prompt and accurate physical examination and decisions are mandatory in this setting of patients. Indeed, patients with firmly suspected or documented active TBC should be isolated in single rooms using airborne precautions. Interestingly, some authors demonstrated that the drug may be safely administered in serious infections providing optimal monitoring of disease and TBC treatment.42,43
Viral infections were recognized early and late after this treatment (Table 2).44–50 The majority of case report describes herpes simplex virus (HSV) possible reactivation and hepatitis B virus (HBV) reactivation. The possible explanations include impairment of NK cells, which have a key role in controlling herpes infections, especially when T cells are low and a reduction of T-reg protective against virus occurs. Acyclovir/valacyclovir HSV prophylaxis may be considered in selected cases, for example, low CD4+ lymphocyte count. Frequently immunocompromised patients with defects in cell-mediated immunity experienced more severe and disseminate HSV than those with agammaglobulinemia.
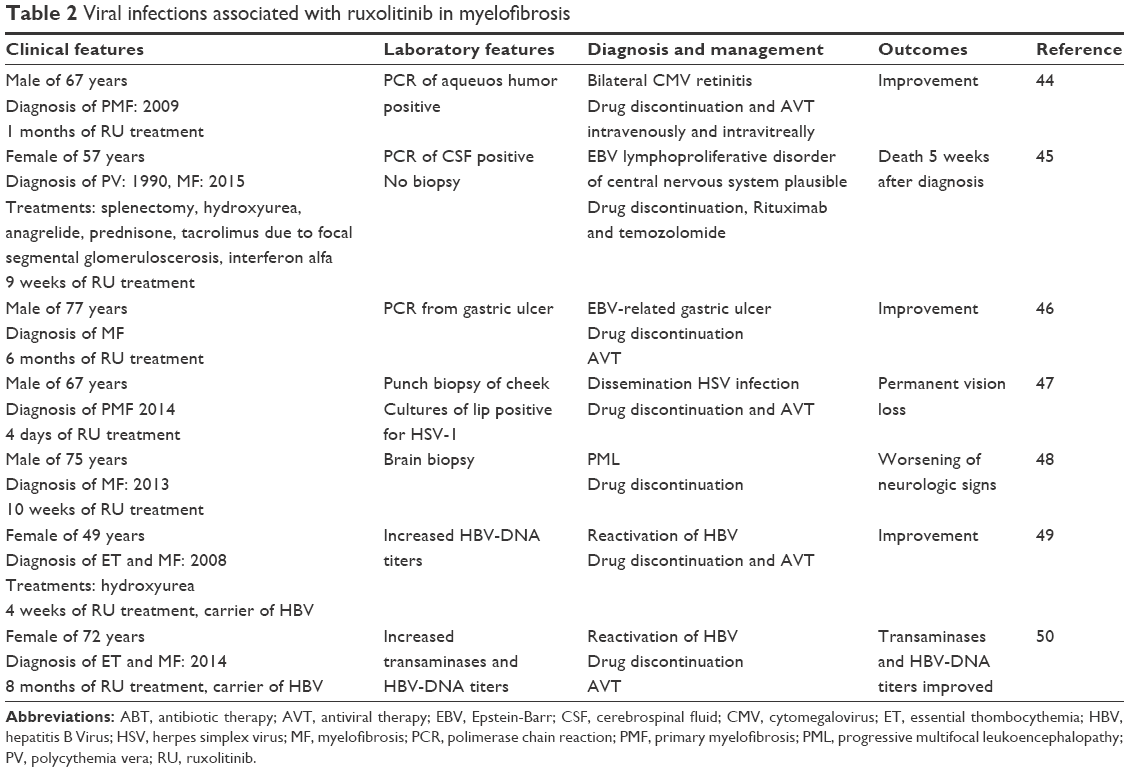 | Table 2 Viral infections associated with ruxolitinib in myelofibrosis |
In addition, surveillance of HBV markers and viral load are important due to the high incidence of latent HBV and reactivation during steroids or immunosuppression. More than one third of the world population has been infected with HBV, 350 million people present chronic infection, and the majority live in Southwest Asia and the Western Pacific regions. Reactivation of hepatitis B after steroids, chemotherapy and immunosuppressive therapy is a recognized complication. The timeframe of reactivation and rate varies from 20% to 50%, while in the HSCT setting this rate increases further. Interestingly, HBV-DNA contains corticosteroid elements stimulating virus replication.51
Most HBV reactivation occurs in hepatitis B surface Australia antigen (HbsAg)-positive patients and more rarely in patients without Australia but with antibodies against hepatitis B core antigen (anti-Hbc) and/or Australia.51 In addition, the host immune system may affect on hepatocellular damage and viral clearance, such as viral factors. Therefore, a prompt antiviral prophylaxis should be considered in high risk of HBV reactivation hematological patients according to the American Association for Study of Liver Diseases and European guidelines.52,53 However, another important point is a careful clinical monitoring due to the incidence of prophylactic drug resistance, such as an adequate monitoring of HBV-DNA and transaminases considering the occurrence of viral replication before evidence of hepatitis. In addition, the exact duration of prophylaxis may be guided by monitoring the immune reconstitution by flow cytometry, for example.
Epstein–Barr virus (EBV) lymphoproliferative disorder represents a life-threatening complication after HSCT with a major risk factor for profound T-cell depletion.54 Probably, previous treatments and comorbidities may justify the fatal possible case of EBV-lymphoproliferative of central nervous system as reported in Table 2.
Progressive multifocal leukoencephalopathy is due to reactivation of John Cunningham polyomavirus. At present, rare data are reported on the epidemiology of disease in non-HIV setting and its treatment.55
Fungal and protozoan infections after ruxolitinib are summarized in Table 3.56–59 Toxoplasma is commonly related to cat exposure. In addition, Toxoplasma gondii release kinase that interact with JAK–STAT pathway and drug downregulate cytokines, including TNF-alpha, which play an essential role in controlling intracellular fungal pathogen. Toxoplasmosis is likely to be an underestimated complication after allogeneic stem cell transplantation with a high mortality rate.
 | Table 3 Fungal and protozoan infections associated with ruxolitinib in myelofibrosis |
Furthermore, Pneumocystis jiroveci (PJP) is responsible for severe infections in HSCT patients. Rare data are reported in non-HSCT setting. For clinical point of view, serum diagnosis in immunocompromised hematological patients sometimes varies independently from reactivation.
PJP (formerly Pneumocystis carinii) pneumonia is a rare fungal infections observed in patients with a disrupted immune system, including HIV and HSCT recipients.60 A recent meta-analysis of RCTs indicates that prophylaxis of PJP pneumonia in immunocompromised non-HIV-infected patients is useful when the risk of disease is >3.5% in adults.61 Rare data were recognized in non-HSCT setting. Therefore, a physician should consider trimethoprim/sulfamethoxazole evaluating its tolerability and activity against PJP and other opportunist pathogens, such as toxoplasma, encapsulated bacteria organisms, such as Streptococcus pneumoniae, Haemophilus influenzae, and gram negative.
Cryptococcus is a yeast-like environmental fungus responsible for pneumonia or meningoencephalitis, especially in immunocompromised patients, such as HIV with CD4+ lymphocyte count inferior to 200/mm3.62–64 Interestingly, a case report presented in Table 3 describes the possibility of reintroduction of ruxolitinib after the prompt treatment of fungal infections.57
Conclusions
The complex interactions between the drug, bone marrow, and immune system (innate and adaptive arms) and infections (bacterial, viral, fungal, and protozoan) need further clinical and translational studies to confirm the significant influence of this relationship.
Biological studies are consistent with the assumption of a combination of immune defects mainly based on T, dendritic, and NK cell defects and dysfunctional granulocytes. Age and comorbidities, treatments (such as steroids), and environmental exposure may influence the risk of infections, for example, intracellular or extracellular pathogen. For clinical point of view, accurate screening and prompt treatment of infections are mandatory considering the reduction in the levels of inflammatory markers, for example, CRP (drug related) and the possibility of acquiring multidrug-resistant organisms’ infections.65,66 Recently, new definitions and perspectives are recognized regarding sepsis and its high mortality rate.67
More robust data are necessary to answer the question of possible immune derangement of ruxolitinib treatment in MF. In summary, precautions should be implemented to improve adequate screening, prophylaxis, and prompt treatment of infections. Furthermore, the patients should be warned about the possibility of reactivation of infections. Importantly, some authors demonstrated the possibility to cure infections and ultimately treat MF.
Ruxolitinib is the first drug approved in a rare disease of the elderly, MF. At present, it is also approved for the treatment of patients with PV nonresponders or intolerant of hydroxyurea.68
In conclusion, RCT data on this topic are scarce, such as updated data from registry or large case series. At present, other JAK inhibitors are ongoing in phase III studies for the treatment of MPNs. Safety and tolerability of ruxolitinib and JAK inhibitors should be further clarified with the aim of improving responses using the combination of targeted therapies.68
Acknowledgments
The author thanks Dr Nicola Cascavilla, Chief at Hematology with BMT of IRCCS “Casa Sollievo della Sofferenza”, San Giovanni Rotondo (SGR) for his mentorship, and Dr Paola Pauri, Chief at Clinical Pathology Laboratory of Jesi (Ancona) for their discussions that served to strengthen the article. The author is grateful to Dr Ernesto Torres Lopez, Professor and Researcher at Immunovirology Laboratory, Medicine School UANL Postdoctoral and Sabbatical Positions “Harvard University” and Professor Renato Fanin Chief of Hematology with BMT, University of Udine, for their key observations. The author thanks all colleagues from SGR, Jesi for their scientific friendship.
Disclosure
The author reports no conflicts of interest in this work.
References
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. | ||
Barbui T, Thiele J, Vannucchi AM, Tefferi A. Rationale for revision and proposed changes of the WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis. Blood Cancer J. 2015;5:e337. | ||
Bae E, Park CJ, Cho YU, et al. Differential diagnosis of myelofibrosis based on WHO 2008 criteria: acute panmyelosis with myelofibrosis, acute megakaryoblastic leukemia with myelofibrosis, primary myelofibrosis and myelodysplastic syndrome with myelofibrosis. Int J Lab Hematol. 2013;35(6):629–636. | ||
Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1,545 patients with contemporary polychethemia vera: an international study. Leukemia. 2013;27(9):1874–1881. | ||
Passamonti F, Thiele J, Ghordon F, et al. A prognostic model to predict survival in 367 World Health Organization–defined essential thrombocythemia at diagnosis: a study by the International Working Group on Myelofibrosis Research and Treatment. Blood. 2012;120(6):1197–1201. | ||
Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. | ||
Vannucchi A, Guglielminelli P, Rotunno G, et al. Mutation-enhanced international prognostic scoring system (MIPSS) for primary myelofibrosis: an AGIMM & IWG-MRT project. Blood. 2014;124(21):405. | ||
Tefferi A, Guglielminelli P, Finke C, et al. Integration of mutations and karyotype towards a genetics-based prognostic scoring system (GPSS) for primary myelofibrosis. Blood. 2014;124(21):406. | ||
Tefferi A. Myeloproliferative neoplasms: a decade of discoveries and treatment advances. Am J Hematol. 2016;91(1):50–58. | ||
Savona MR. Are we alterating the natural history of primary myelofibrosis? Leuk Res. 2014;38(9):1004–1012. | ||
Pinilla-Ibarz J, Sweet KL, Corrales-Yepez GM, Komrokji RS. Role of tyrosine-kinase inhibitors in myeloproliferative neoplasms. Comparative lesson learned. Onco Targets Ther. 2016;9:4937–4957. | ||
Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. | ||
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. | ||
Gregory SA, Mesa RA, Hoffman R, et al. Clinical and laboratory features of myelofibrosis and limitations of current therapies. Clin Adv Hematol Oncol. 2011;9(9 Suppl 22):1–16. | ||
Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3-year follow-up of COMFORT-I. Haematologica. 2015;100(4):479–488. | ||
Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30(8):1701–1707. | ||
Chen X, Shu JG, Emm T, et al. Pharmacokinetics and pharmacodynamics of orally-administrated ruxolitinib (INCB018424 phosphate) in renal and hepatic impairment patients. Clin Pharmacol Drug Dev. 2014;3(1):34–42. | ||
Talpaz M, Paquette R, Afrin L, et al. Interim analysis of safety and efficacy of ruxolitinib in patients with myelofibrosis and low platelets counts. J Hematol Oncol. 2013;6(1):81. | ||
Quintas-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10(2):127–140. | ||
Tefferi A, Pardanani A. Serious adverse events during ruxolitinib treatment discontinuation in patients with myelofibrosis. Mayo Clin Proc. 2011;86(12):1188–1191. | ||
Theocharides AP, Lundberg P, Lakkaraju AK, et al. Homozygous calreticulin mutations in patients with myelofibrosis lead to acquired myeloperoxidase deficiency. Blood. 2016;127(25):3253–3259. | ||
Heine A, Held SA, Daecke SN, et al. The JAK–inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood. 2013;122(7):1192–1202. | ||
Rudolph J, Heine A, Quast T, et al. The JAK inhibitor ruxolitinib impairs dendritic cell migration via off-target inhibition of ROCK. Leukemia. 2016;30(10):2119–2123. | ||
Schonberg K, Rudolph J, Vonnahme M, et al. JAK inhibition impairs NK cell function in myeloproliferative neoplasms. Cancer Res. 2015;75(11):2187–2199. | ||
Massa M, Rosti V, Campanelli R, et al. Rapid and long-lasting decrease of T-regulatory cells in patients with myelofibrosis treated with ruxolitinib. Leukemia. 2014;28(2):449–451. | ||
Keohane C, Kordasti S, Siedl T, et al. JAK inhibition induces silencing of t helper cytokine secretion and a profound reduction in T regulatory cells. Br J Haematol. 2015;171(1):60–73. | ||
Parampalli Yajnanarayana S, Stübig T, Cornez I, et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br J Haematol. 2015;169(6):824–833. | ||
Al-Ali HK, Griesshammer M, le Coutre P, et al. Safety and efficacy of ruxolitinib in an open-label, multicenter, single-arm phase 3b expanded-access study in patients with myelofibrosis: a snapshot of 1,144 patients in the JUMP trial. Haematologica. 2016;101(9):1065–1073. | ||
Safdar A, Armstrong D. Infections in patients with hematologic neoplasms and hematologic stem cell transplantation: neutropenia, humoral, splenic defects. Clin Infect Dis. 2011;53(8):798–806. | ||
Pea F, Viale P. The antimicrobial therapy puzzle: could pharmacokinetic-pharmacodynamic relationships be helpful in addressing the issue of appropriate pneumonia treatment in critically ill patients? Clin Infect Dis. 2006;42(12):1764–1771. | ||
Levy Hara G, Kanj SS, Pagani L, et al. Ten key points for the appropriate use of antibiotics in hospitalised patients: a consensus from the Antimicrobial Stewardship and Resistance Working Groups of the International Society of Chemotherapy. Int J Antimicrob Agents. 2016;48(3):239–246. | ||
Martí-Carvajal AJ, Anand V, Solà I. Janus kinase-1 and Janus kinase-2 inhibitors for treating myelofibrosis (Review). Cochrane Database Syst Rev. 2015;(4):CD010298. | ||
Deadmond MA, Smith-Gagen JA. Changing Incidence of Myeloproliferative Neoplasms: Trends and Subgroup Risk Profiles in the United States, 1973–2011. J Cancer Res Clin Oncol. 2015;141(12):2131–2138. | ||
Hultcrantz M, Wilkes SR, Kristinsson SY, et al. Risk and cause of death in patients diagnosed with myeloproliferative neoplasms in Sweden Between 1973 and 2005: a population-based study. J Clin Oncol. 2015;33(20):2288–2295. | ||
Landman GW, Arend SM, van Dissel JT. Ruxolitinib can mask symptoms and signs of necrotizing fasciitis. J Infect. 2013;66(3):296–297. | ||
KusanoY, Terui Y, Ueda K, Hatake K. Klebsiella pneumoniae primary liver abscess associated with ruxolitinib. Ann Hematol. 2016;95(9):1561–1562. | ||
Chen YH, Lee CH, Pei SN. Pulmonary tuberculosis reactivation following ruxolitinib treatment in a patient with primary myelofibrosis. Leuk Lymphoma. 2015;56(5):1528–1529. | ||
Abidi MZ, Haque J, Varma P, et al. Reactivation of pulmonary tuberculosis following treatment of myelofibrosis with ruxolitinib. Case Rep Hematol. 2016;2016:2389038. | ||
Shamil E, Cunningham D, Wong BL, Jani P. Ruxolitinib associated tuberculosis presenting as a neck lump. Case Rep Infect Dis. 2015;2015:284168. | ||
Hopman RK, Lawrence SJ, Oh ST. Disseminated tuberculosis associated with ruxolitinib. Leukemia. 2014;28(8):1750–1751. | ||
Colomba C, Rubino R, Siracusa L, et al. Disseminated tuberculosis in a patient treated with a JAK2 selective inhibitor: a case report. BMC Res Notes. 2012;5:552. | ||
Palandri F, Polverelli N, Catani L, Vianelli N. Ruxolitinib-associated tuberculosis: a case of successful ruxolitinib rechallenge. Ann Hematol. 2015;94(3):519–520. | ||
Branco B, Metsu D, Dutertre M, et al. Use of rifampin for treatment of disseminated tuberculosis in a patient with primary myelofibrosis on ruxolitinib. Ann Hematol. 2016;95(7):1207–1209. | ||
Von Hofsten J, Johnsson FM, Zetterberg M. Cytomegalovirus retinitis in a patient who received ruxolitinib. N Engl J Med. 2016;374(3):296–297. | ||
Palmason R, Linden O, Richter J. Case-report: EBV driven lymphoproliferative disorder associated with ruxolitinib. BMC Hematol. 2015;15:10. | ||
Kusano Y, Terui Y, Ueda K, Hatake K. Epstein-Barr virus gastric ulcer associated with ruxolitinib. Ann Hematol. 2016;95(10):1741–1742. | ||
Tong LX, Jackson J, Kerstetter J, Worswick SD. Reactivation of herpes simplex virus infection in a patient undergoing ruxolitinib treatment. J Am Acad Dermatol. 2014;70(3):e59–e60. | ||
Wathers R, Moule S, Milojkovic D. Progressive multifocal leukoencephalopathy associated with ruxolitinib. N Engl J Med. 2013;369(2):197–198. | ||
Coacci G, Murgia F, Podda L, Solinas A, Atzeni S, La Nasa G. Reactivation of hepatitis B virus infection following ruxolitinib treatment in a patient with myelofibrosis. Leukemia. 2014;28(1):225–227. | ||
Shen CH, Hwang CE, Chen YY, Chen CC. Hepatitis B virus reactivation associated with ruxolitinib. Ann Hematol. 2014;93(6):1075–1076. | ||
Yeo W, Johnson PJ. Diagnosis, prevention and management of hepatitis B virus reactivation during anticancer therapy. Hepatology. 2006;43(2):209–220. | ||
Mallet V, van Bommel F, Doerig C, et al. Management of viral hepatitis in patients undergoing haematopoietic stem cell transplantation: recommendations of the 5th European Conference on Infections in Leukemia (ECIL-5). Lancet Infect Dis. 2016;16(5):606–617. | ||
Perrillo RP, Gish R, Falck-Ytter YT. American Gastroenterological Association Institute Technical Review on prevention and treatment of hepatitis B virus reactivation during immunosuppressive drug therapy. Gastroenterology. 2015;148(1):221.e3–244.e3. | ||
Styczynski J, van der Velden W, Fox CP, et al; Sixth European Conference on Infections in Leukemia, a joint venture of the Infectious Diseases Working Party of the European Society of Blood and Marrow Transplantation (EBMT-IDWP), the Infectious Diseases Group of the European Organization for Research and Treatment of Cancer (EORTC-IDG), the International Immunocompromised Host Society (ICHS) and the European Leukemia Net (ELN). Management of Epstein-Barr Virus Infections and post-transplant lymphoproliferative disorders in patients after allogenic hematopoietic stem cell transplantation: sixth European Conference on Infections in Leukemia (ECIL-6) guidelines. Haematologica. 2016;101(7):803–811. | ||
Schmidt-Hieber M, Silling G, Schalk E, et al. CNS infections in patients with hematological disorders (including allogeneic stem-cell transplantation) – Guidelines of the Infectious Diseases Working Party (AGIHO) of the German Society of Hematology and Medical Oncology (DGHO). Ann Oncol. 2016;27(7):1207–1225. | ||
Goldberg RA, Reichel E, Oshry LJ. Bilateral toxoplasmosis retinitis associated with ruxolitinib. N Engl J Med. 2013;369(7):681–683. | ||
Wysham NG, Sullivan DR, Allada G. An opportunistic infection associated with ruxolitinib, a novel janus kinase 1,2 inhibitor. Chest. 2013;143(5):1478–1479. | ||
Chen CC, Chen YY, Huang CE. Criptococcal meningoencephalitis associate with long-term use of ruxolitinib. Ann Hematol. 2016;95(2):361–362. | ||
Lee SC, Feenstra J, Georghiou PR. Pneunomocystis jiroveci pneumonitis complicating ruxolitinib therapy. BMJ Case Rep. 2014;2014. pii: bcr2014204950. | ||
Williams KM, Ahn KW, Chen M, et al. The incidence, mortality and timing of Pneumocystis jiroveci pneumonia after hematopoietic cell transplantation: a CIBMTR. Bone Marrow Transplant. 2016;51(4):573–580. | ||
Green H, Paul M, Vidal L, et al. Prophylaxis of Pneumocystis pneumonia in immunocompromised non-HIV-infected patients: systematic review and meta-analysis of randomized controlled trials. Mayo Clin Proc. 2007;82(9):1052–1059. | ||
Perfect JR, Dismukes WE, Dromer F, et al. Clinical practice guidelines for the management of cryptococcal disease: 2010 update by the infectious diseases society of America. Clin Infect Dis. 2010;50(3):291–322. | ||
Brizendine KD, Baddley JW, Pappas PG. Predictors of mortality and differences in clinical features among patients with cryptococcosis according to immune status. PLoS One. 2013;8(3):e60431. | ||
Baron EJ, Miller M, Weinstein MP, et al. A guide to utilization of the microbiology laboratory for diagnosis of infectious diseases: 2013 recommendations by the infectious diseases society of America (IDSA) and the American Society for Microbiology (ASM). Clin Infect Dis. 2013;57(4):e22–e121. | ||
Marston HD, Dixon DM, Knisely JM, Palmore TN, Fauci AS. Antimicrobial resistance. JAMA. 2016;316(11):1193–1204. | ||
Paczosa MK, Mecsasb J. Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol Mol Biol Rev. 2016;80(3):629–661. | ||
Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. | ||
Bryan JC, Verstovsek S. Overcoming treatment challenges in myelofibrosis and polycythemia vera: the role of ruxolitinib. Cancer Chemother Pharmacol. 2016;77(6):1125–1142. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.