")
Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 6
Ruxolitinib: a targeted treatment option for patients with polycythemia vera
Authors Vaddi K, Verstovsek S, Kiladjian J, Iesha L
Received 24 November 2015
Accepted for publication 24 February 2016
Published 12 May 2016 Volume 2016:6 Pages 7—19
DOI https://doi.org/10.2147/BLCTT.S101185
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor David Dingli
Kris Vaddi,1 Srdan Verstovsek,2 Jean-Jacques Kiladjian3
1Drug Discovery, Incyte Corporation, Wilmington, DE, 2Department of Leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, USA; 3Clinical Investigations Center, Hôpital Saint-Louis et Université Paris Diderot, Paris, France
Abstract: Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by erythrocytosis and the presence of Janus kinase (JAK) 2V617F or similar mutations. This review summarizes the pathophysiology of PV, the challenges associated with traditional treatment options, and the scientific rationale and supportive clinical evidence for targeted therapy with ruxolitinib. Accumulating evidence indicates that activating mutations in JAK2 drive the PV disease state. Traditional PV treatment strategies, including aspirin, phlebotomy, and cytoreductive agents such as hydroxyurea, provide clinical benefits for some but not all patients and may not adequately treat PV-related symptoms. Furthermore, traditional treatment approaches are associated with potential side effects that may limit their usage and lead some patients to discontinue the treatment. Ruxolitinib is an orally available small-molecule tyrosine kinase inhibitor that is a potent and selective inhibitor of JAK1/JAK2. Ruxolitinib is approved in the US for patients with PV with an inadequate response or intolerance to hydroxyurea and in Europe for adults with PV who are resistant to or intolerant of hydroxyurea. In the Phase III RESPONSE registration trial, ruxolitinib was superior to the best available therapy in patients with PV who were resistant to or intolerant of hydroxyurea in controlling hematocrit levels, reducing spleen volume, and improving PV-related symptoms and quality-of-life measures. The most common nonhematologic adverse events in ruxolitinib-treated patients were headache, diarrhea, pruritus, and fatigue in the RESPONSE trial; hematologic adverse events were primarily grade 1 or 2. In the Phase IIIb nonregistration RELIEF trial, there were nonsignificant trends toward an improved symptom control in patients with PV on a stable hydroxyurea dose who were generally well controlled but reported disease-associated symptoms and switched to ruxolitinib vs those who continued hydroxyurea therapy. Updated treatment guidelines will be important for educating physicians about the role of ruxolitinib in the treatment of patients with PV.
Keywords: myeloproliferative disorder, polycythemia vera, Janus kinase inhibitor
Introduction
Polycythemia vera (PV) is a disorder predominantly characterized by erythrocytosis.1 As opposed to secondary erythrocytoses, PV and primary familial congenital polycythemia are categorized as primary erythrocytoses, which result from enhanced responses to erythropoietin (EPO).2 In 1951, PV and three other disorders with similar pathophysiologic characteristics (myelofibrosis [MF], essential thrombocythemia [ET], and chronic myeloid leukemia) were characterized as “myeloproliferative disorders” by Dr William Dameshek.3 Subsequent cytogenetic analyses and clonality studies demonstrated that PV is a clonal malignancy acquired through one or more somatic mutations in a pluripotent hematopoietic stem cell, leading to increased myeloid proliferation.4,5 However, a molecular target responsible for PV was not yet identified at that time.6
Early hypotheses regarding the pathologic basis of PV included potential abnormal growth factor signaling pathways,4 transcriptional dysregulation,5 and constitutive activation of signal transducer and activator of transcription (STAT) proteins.5 Polycythemia rubra vera-1 (PRV-1) was once hypothesized as a PV-specific marker that could be important for elucidating the molecular mechanism of the disorder.5,7 The PV diagnostic strategy drastically changed after the discovery of activating mutations in the Janus kinase 2 (JAK2) tyrosine kinase.8–11 Following this discovery, the World Health Organization characterized PV as a chronic myeloproliferative neoplasm (MPN) primarily defined by erythrocytosis and the presence of JAK2V617F or similar mutations.1
Patients diagnosed with PV may have a marked disease burden and a higher mortality risk when compared with the general population, primarily driven by cardiovascular/thrombotic events, disease transformation to MF or acute myeloid leukemia (AML), and solid malignancies.12–15 Traditional PV treatment strategies include aspirin, phlebotomy, hydroxyurea, and other cytoreductive treatments.16 Such options provide clinical benefits for some patients, including a reduced risk of cardiovascular/thrombotic events.17–20 However, traditional treatment options do not provide adequate benefit for some patients21,22 and may not alleviate PV-related symptoms.23,24 As such, there remains an unmet need for improved clinical outcomes, symptom alleviation, and enhancement of quality-of-life (QoL).
The objective of this review is to provide a summary of the pathobiology of PV, including the evolving understanding of the molecular etiology of PV, the challenges associated with traditional PV treatment options, and the scientific rationale and clinical data for ruxolitinib (Jakafi®, Incyte Corporation, Wilmington, DE, USA; Jakavi®, Novartis AG, Horsham, West Sussex, UK) supporting its role as a new targeted treatment option for patients with PV.
Biology of PV: pre- and post-JAK2V617F era
Pre-JAK2V617F molecular understanding of the etiology of PV
Early studies of erythropoiesis provided important information about the hematopoiesis process in the PV setting.4 In vitro analyses demonstrated that bone marrow progenitor cells isolated from patients with PV (but not control bone marrow samples) were able to form EPO-independent endogenous erythroid colonies (EECs).4 In 1987, a review of nine studies reported that 97% of patients with PV had EECs in the bone marrow and/or peripheral blood.25 Although EECs were also observed in some patients with ET,26 their presence is a hallmark of PV and was used as a clinical diagnostic tool.4
The observed increased proliferative responsiveness of PV progenitor cells to EPO, IGF-1, and other growth factors (eg, interleukin [IL]-3, granulocyte-macrophage colony-stimulating factor [GM-CSF], thrombopoietin, and stem cell factor) implicated abnormal cytokine signaling pathways in the molecular underpinnings of PV.4,5,26,27 However, studies that examined mutations related to cytokine signaling targets (eg, the EPO receptor, IGF-1 receptor, IGF-1–binding proteins, and tyrosine phosphatases) were unsuccessful in elucidating the pathogenesis of PV.4 It was also hypothesized that abnormal cytokine signaling was not necessarily related to a limited number of specific mutations but rather to more general defects in transcriptional regulation that could affect a variety of metabolic pathways that play a role in the pathogenesis of PV.4 The transcriptional dysregulation hypothesis was supported by studies of cells from patients with PV, which reported decreased levels of the thrombopoietin receptor c-MPL in platelets28 and an increased proportion of erythroid progenitors expressing BCL-x (an antiapoptotic protein).29 Downstream signal transduction molecules important to cytokine receptor signaling, including EPO-mediated pathways, were further studied to identify the potential PV candidate genes.5 It was hypothesized that EPO-mediated activation of the JAK/STAT pathway induced BCL-x expression, which inhibited apoptosis.6 However, data suggested that constitutive STAT3 activation alone was not the primary molecular cause of PV.30 Therefore, it remained uncertain which one of the signal transducers in this pathway accounted for the increased erythropoiesis observed in patients with PV.6
Collectively, available data up to this point suggested that the JAK/STAT transduction pathway might play a critical role in preventing the apoptosis of erythroid progenitors but not necessarily in the proliferation of erythroid cells.6 The search for the molecular cause of PV continued. Using subtractive hybridization techniques, overexpression of PRV-1 mRNA was observed in the granulocytes of patients with PV but not in normal controls.31 However, the specificity of PRV-1 mRNA overexpression to the PV setting31 was later contradicted by data indicating no consistent differences in PRV-1 protein levels between granulocytes from patients with PV and those from normal controls.32 Furthermore, the sensitivity of PRV-1 expression to GM-CSF exposure31 suggested that alterations in cytokine levels (not the PV disease state itself) explained the variability of PRV-1 levels. As such, the technically demanding and time-consuming EEC assay continued to be the most reliable test for the diagnosis of PV,33 and the molecular cause of PV remained elusive.
Discovery of JAK2V617F and the importance of JAK2 in the pathogenesis of PV
In 2005, four separate groups identified the somatic gain-of-function JAK2V617F mutation,8-11 a constitutively active allele associated with activation of downstream signaling components, including STAT3 and STAT5.8,10,34 In murine models, JAK2V617F bone marrow cells are associated with PV disease features, including erythrocytosis, leukocytosis, and enlarged spleen.10,35-38 Nearly all patients with PV have an activating mutation in JAK2, most often JAK2V617F (95% of patients) or an activating mutation in exon 12 (4% of patients).37-39 Furthermore, patients with elevated JAK2 allele burden (percentage of mutant allele relative to the total [wild type + mutant]) are at increased risk of developing post-PV MF.39
The discovery of the JAK2V617F mutation was critical for understanding the pathobiology of PV and dovetailed with other preclinical data concerning the JAK/STAT signaling pathway. For example, GM-CSF activates JAK140 and drives granulopoiesis in a pathway that includes JAK2 in cell culture systems,41 and IL-12–driven T-cell proliferation requires JAK2 activity.42 EPO signaling through the EPO receptor activates JAK2,43 which in turn activates STAT1, STAT3, and possibly STAT5 in cell culture systems.44,45 Mice lacking JAK2 are embryonic lethal because of extreme anemia.46,47 Unlike wild-type mice, hematopoietic progenitor cells from JAK2−/− mice do not proliferate and form megakaryocytic colonies in response to thrombopoietin.47 In addition, conditional knockout of JAK2 in adult hematopoietic progenitor cells is associated with reduced viability and lowered platelet counts.48 GM-CSF antiapoptotic activity in human eosinophils requires JAK2 activation.49 Finally, JAK2 is activated in response to thrombopoietin binding to the Mpl receptor and is required for the downstream activation of STAT3.50 With the knowledge that activating mutations in JAK2 were driving the PV disease state, researchers and clinicians had the rationale for the development of new, targeted treatment options, which was the impetus for testing ruxolitinib, a potent JAK1/JAK2 inhibitor, in patients with PV.
Clinical presentation of PV
Elevated blood counts
Elevated blood counts are one of the key diagnostic criteria associated with PV. In a large international study evaluating prognosis and survival among 1,545 patients with PV, many patients presented with elevated hemoglobin (median, 18.4 g/dL) and hematocrit values (median, 55%) at diagnosis.14 White blood cell (WBC) and platelet counts were also elevated (median, 10.4×109/L and 466×109/L, respectively).14 In addition, approximately one half of the patients presented with leukocytosis (WBC count >10.5×109/L; 49%) and thrombocytosis (platelet count ≥450×109/L; 53%).14
Patients with PV who have a high JAK2 allele burden may experience further elevated blood counts compared with those who have less allele burden. A prospective study including 173 patients with PV demonstrated that there was a correlation between the JAK2V617F allele burden and the increased risk of erythropoiesis and myelopoiesis.51 Patients with higher JAK2V617F burden at diagnosis also presented with higher hematocrit levels (regression coefficient [r] =0.67; P<0.001) and higher WBC counts (r=0.54; P<0.001).51
Thromboembolic and mortality risk
Although elevated blood counts are important diagnostic markers of PV, thromboembolic events are often responsible for the initial clinical presentation of the disease. Patients with PV are at an increased risk of cardiovascular/thromboembolic events and mortality compared with the general population.12,14,15,52 Results from a large Swedish registry database study (n=11,155 patients with PV and n=44,620 matched controls) indicated that the risk of arterial and venous thrombosis in patients with PV was five- and ninefold higher, respectively, in patients with PV compared with the general population.53
In a large, randomized, controlled trial testing cytoreductive therapy in patients with PV (CYTO-PV), elevated hematocrit levels of 45%–50% (n=183) were associated with a fourfold increase in cardiovascular complications and a significantly higher rate of cardiovascular death compared with those who had hematocrit level of <45% (n=182).20 Data from this trial also indicated that patients with a WBC count of ≥11×109/L were nearly four times more likely to experience major thrombosis when compared with those who had a WBC count of <7×109/L.54 However, available data do not suggest that thrombocytosis is a significant risk factor for cardiovascular/thromboembolic events in patients with PV. The European Collaboration on Low-Dose Aspirin in Polycythemia Vera prospective trial demonstrated that the rates of thromboembolic events and mortality were not significantly different between patients with baseline platelet counts >400×109/L vs those with lower platelet counts.55 The molecular pathway responsible for the increased incidence of thromboembolic events in patients with PV is unclear. However, data suggest that constitutive JAK2 signaling may contribute to several PV features associated with the development of thromboembolic events, including erythrocytosis,10,20 increased adhesive qualities of JAK2V617F erythrocytes,56 leukocytosis,36,54 systemic inflammation,57 and activation of blood cell types.58-65
Signs and symptoms
Patients with PV may experience a variety of disease-related symptoms, reduced QoL,52 and reduced work productivity.66 Fatigue, pruritus, difficulty in sleeping, day or night sweats, and dizziness are among the most frequently reported symptoms of PV.66 Patients with PV may also develop splenomegaly,14 which can be uncomfortable or painful in some patients.67
The specific pathways underlying PV-related symptoms are unknown, but some evidence suggests that cytokine signaling plays an important role. Notably, serum levels of inflammatory cytokines, which signal through JAK1 and/or JAK2,68 are elevated in patients with PV.69 A cytokine profiling study in patients with MF found several clinical correlations between cytokine levels, constitutional symptoms, and splenomegaly.70
Some symptoms may stem from increased blood counts caused by the constitutive activation of JAK2. The JAK2V617F mutation is associated with elevated hematocrit levels in murine models10,35,36 and in patients with PV,51 which may be associated with increased blood viscosity and symptoms that result from an impaired cerebral blood flow, including headache and dizziness.71 Patients with PV who have higher JAK2V617F allele burden may be at increased risk of developing splenomegaly and pruritus and are more frequently in need of cytoreductive therapy.39,51
The effects of PV on symptoms and QoL, as commonly measured by the MPN Symptom Assessment Form (MPN-SAF) and the European Organisation for Research and Treatment of Cancer Quality-of-Life Questionnaire Core 30, are similar to those of other MPNs.13 Some patients report that burdensome PV-related symptoms have negative effects on their QoL, productivity, and activities of daily living.66 In a recent US survey of 380 patients with PV, 66% experienced symptoms that reduced their QoL, 22% were sick from work for ≥1 day in the preceding 30 days, and 48% experienced PV-related interference with daily activities.66
Limitations of traditional treatment options
Treatment overview
Traditional treatment options are effective for many patients with PV but do not target the molecular underpinnings of the disease and are associated with several limitations, suggesting that improved treatment options are warranted. The goals of treatment in PV are to reduce the risk of cardiovascular/thrombotic events and control disease-related symptoms.16 Maintaining a hematocrit level of <45% is associated with a reduced risk of cardiovascular/thrombotic events and related death.20 For patients with splenomegaly, treatment should also aim to reduce spleen size.72 Aspirin, phlebotomy, and cytoreduction are the three main elements of traditional PV treatment strategies.16,72
Aspirin
The European Collaboration on Low-Dose Aspirin in Polycythemia Vera clinical trial demonstrated that treatment with low-dose aspirin was associated with a reduced risk of cardiovascular/thrombotic events.19 Subsequently, a Cochrane meta-analysis reported that low-dose aspirin was associated with a nonsignificant reduction in the risk of fatal thrombotic events.73 However, patients receiving regular aspirin treatment (≥2×325 mg/wk) may be at a dose-dependent increased risk of gastrointestinal bleeding.74 Aspirin is contraindicated in patients with acquired von Willebrand syndrome and/or platelet count of >1,000×109/L, which are associated with an increased risk of bleeding.75,76
Phlebotomy
Phlebotomy with or without cytoreductive treatment to maintain a hematocrit level of <45% was associated with reduced cardiovascular death and major thrombotic events in patients with PV.20 However, hematocrit maintenance at <45% with phlebotomy and other traditional treatment options can be difficult. The CYTO-PV study required hematocrit control within predefined limits with phlebotomy and/or other traditional treatments (eg, aspirin and cytoreductive therapy); however, >25% of the patients did not maintain hematocrit levels within their target range.77
Phlebotomy procedures may be poorly tolerated by some patients, as evidenced by the observation that 28% of patients in the high-hematocrit arm of the CYTO-PV trial discontinued phlebotomy treatment.20 In a large study of phlebotomy patients undergoing venipuncture (N=3,315), 15% of patients reported that they feared the phlebotomy procedure and 3% reported that they avoided the procedure because of that fear.78 In addition, frequent phlebotomies can cause iron deficiency, which may be associated with restless leg syndrome and impairments in cognitive functioning and mental health in some patients.79-81
Cytoreductive agents
Cytoreductive treatment with hydroxyurea is associated with increased survival and may be associated with reduced thromboembolic risk in patients with PV.17,18 However, one retrospective study indicated that 11% and 13% of patients treated with hydroxyurea become resistant and intolerant, respectively.21 In a multivariate analysis, resistance to hydroxyurea was associated with a 6.8-fold higher risk of transformation to AML or MF and a 5.6-fold higher risk of death.21 Hydroxyurea may also be associated with several hematologic and nonhematologic side effects that could limit treatment, including leg ulcers and other mucocutaneous manifestations, gastrointestinal toxicity, and fever.21
Some data suggest that hydroxyurea may increase the risk of transformation to leukemia, especially in younger patients, although further analyses are required to confirm this hypothesis.82,83 The PV Study Group-08 trial reported that the incidence of AML at a median follow-up of 8.6 years was 5.9% in 51 hydroxyurea-treated patients compared with 1.5% in a historical control group of 134 patients from the phlebotomy-only arm of an earlier study; however, the difference between treatment groups was not statistically significant.18 In an analysis of 1,638 patients with PV enrolled in a prospective observational cohort study with a median follow-up of 2.8 years, the incidence of AML/myelodysplastic syndrome (MDS) in patients treated with hydroxyurea was not significantly different from that of patients receiving no treatment, phlebotomy, or interferon.84 The French Polycythemia Study Group was a large, randomized trial with a median follow-up of 16.3 years that compared hydroxyurea with pipobroman as first-line treatment of PV in 285 patients <65 years of age.17 The cumulative incidences of progression to AML/MDS at 10 years, 15 years, and 20 years were 6.6%, 16.5%, and 24.2%, respectively, in the hydroxyurea group and 13.1%, 34.1%, and 52.1%, respectively, in the pipobroman group. The authors acknowledged that the incidence of transformation to AML/MDS in hydroxyurea-treated patients was higher than previously reported. However, they emphasized that the natural evolution of PV should be considered when interpreting the study results. The leukemogenic risk of hydroxyurea appears to be increased when treatment is preceded or followed by alkylating agents (eg, busulfan).83,85 Because hydroxyurea may modestly increase the risk of AML development, it has been suggested that this drug should be used with caution in younger patients (<40 years of age) with MPNs.82
Interferon-α (IFN-α) and pegylated (PEG)-IFN-α variants are not currently indicated by the US Food and Drug Administration (FDA) or the European Medicines Agency for the treatment of patients with PV. However, IFN-α was recommended by the European LeukemiaNet in 2011 as second-line cytoreductive therapy for patients with PV who become resistant to or intolerant of hydroxyurea.86 Treatment with IFN-α and PEG-IFN-α variants has been associated with hematocrit control without phlebotomy, normalization of blood cell counts, and reductions in enlarged spleen size.87-89 In a review article that summarized clinical trial experience with different IFN-α variants in patients with PV published between 1991 and 2008, 60% (182/303) of patients achieved freedom from phlebotomies.90 An objective response was observed in ~80% of patients, although response criteria were heterogenous among these studies.90 In addition, some data suggest that IFN-α treatment may be associated with improvements in some PV-related signs and symptoms, including pruritus91,92 and splenomegaly.87,88,93,94 However, the route of administration (injection) and safety concerns about recombinant IFN-α formulations suggest that IFN-α may not be an ideal treatment option for some patients. A multicenter observational study reported that nonadherence to IFN-α primarily resulted from patients forgetting to administer the injection (50.2%) and other injection-related reasons (32.0%).95 Side effects of IFN-α, including influenza-like symptoms,96 autoimmune disorders, depression, cardiovascular disease, and ocular disease,16 may lead some patients to discontinue the treatment.97 These safety concerns are primarily associated with recombinant IFN-α variants, and it is important to note that PEG-IFN-α variants are associated with fewer toxicity-related treatment discontinuations.97 PEG-IFN-α2a has also demonstrated decreases in JAK2V617F allele burden over time and is associated with preferable hematologic responses in patients with PV.97,98 In two Phase II clinical trials evaluating PEG-IFN-α in patients with PV, complete hematologic response was observed in 70% and 94.6% of patients (median follow-up time, 21 months and 31.4 months, respectively), and complete molecular response (undetectable JAK2V617F) was observed in 14% and 24.1% of evaluable patients, respectively.89,97 Discontinuations because of PEG-IFN-α-related adverse events were 10% and 24.3% in these studies.89,97 Further analyses from ongoing Phase III trials (eg, ClinicalTrials.gov identifiers: NCT01259856, NCT02218047, NCT02523638, NCT01949805, NCT01387763) will be important for determining the role of PEG-IFN-α in treating patients with PV.
Challenges of PV-related symptom alleviation
Traditional treatment options may not alleviate the PV-related symptom burden.23,24 In a survey-based study, patients with MPNs treated with hydroxyurea, IFN-α, busulfan, or 32P reported similar MPN-SAF symptom severity scores as patients who did not receive myelosuppressive agents.23 Furthermore, a prospective analysis of patients with PV reported that treatment with hydroxyurea, aspirin, IFN-α, or phlebotomy was not associated with significant improvements in the MPN-SAF total symptom score (TSS).24 Collectively, available data suggest that a targeted treatment approach aimed at the molecular pathway associated with the pathogenesis of PV could be more effective than traditional treatment options for ameliorating PV-related symptoms.
Ruxolitinib: mode of action, pharmacodynamics, and pharmacokinetics
Mode of action and pharmacodynamics
Ruxolitinib phosphate – (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile phosphate – is a small-molecule tyrosine kinase inhibitor approved by the FDA for the treatment of patients with PV who have had an inadequate response to or are intolerant of hydroxyurea.99 In Europe, ruxolitinib is indicated for the treatment of adult patients with PV who are resistant to or intolerant of hydroxyurea.100 In the US, ruxolitinib is also indicated for the treatment of patients with intermediate- or high-risk MF, including primary MF, post-PV MF, and post-ET MF;99,100 ruxolitinib is approved for a similar population of patients with MF in Europe.100
In preclinical models, ruxolitinib effectively inhibits the JAK/STAT signaling pathway as a potent and selective inhibitor of JAK1 (mean [SD] 50% inhibitory concentration [IC50], 3.3 [1.2] nM) and JAK2 (mean [SD] IC50, 2.8 [1.2] nM).101 Additionally, ruxolitinib has modest selectivity against Tyk2 (mean [SD] IC50, 19 [3.2] nM) and marked selectivity against JAK3 (mean [SD] IC50, 428 [243] nM).101 Ruxolitinib showed no significant inhibition against 26 additional kinases at concentrations 100-fold the IC50 of JAK1 and JAK2, suggesting high specificity for JAK1 and JAK2.101 The effectiveness of ruxolitinib in this pathway has also been demonstrated in cytokine-stimulated whole-blood assays in which preincubation with ruxolitinib inhibited IL-6– and thrombopoietin-mediated STAT3 phosphorylation (mean [SD] IC50, 282 [54] nM and 281 [62] nM, respectively).101
Several preclinical experiments demonstrated that ruxolitinib inhibits JAK pathway signaling of both wild-type and mutant JAK2.101 In cell lines expressing a JAK2V617F mutation, ruxolitinib effectively inhibited the phosphorylation of JAK2 downstream targets (eg, STAT3, STAT5, and ERK1/2) and induced apoptosis in a dose-dependent fashion.101 Ruxolitinib also inhibited erythroid and myeloid progenitor cell proliferation in primary cultures from healthy individuals (burst-forming unit-erythroid IC50, 407 nM; colony-forming unit-erythroid IC50, 551 nM) and patients with PV carrying the JAK2V617F mutation (burst-forming unit-erythroid IC50, 223 nM; colony-forming unit-erythroid IC50, 444 nM).101 In a murine model, ruxolitinib prolonged the survival, reduced the JAK2V617F allele burden, ameliorated splenomegaly, and normalized the elevated levels of circulating proinflammatory cytokines associated with debilitating constitutional symptoms of PV (eg, IL-6 and tumor necrosis factor-α).101
Pharmacokinetics
Ruxolitinib pharmacokinetics supports an oral route of administration.102 Ruxolitinib has a dose-proportional systemic exposure profile, with minimal accumulation following multiple doses.102 In addition, ruxolitinib is rapidly absorbed, with 95% oral bioavailability.103,104 The maximum tolerated dose of ruxolitinib was established at 25 mg twice daily and 100 mg once daily,102 and the average terminal half-life was ~3 hours.102
Ruxolitinib is metabolized primarily by the cytochrome P450 3A4 (CYP3A4) enzyme.103 In healthy human subjects, >99% of ruxolitinib doses are metabolized in a pathway that includes oxidation to single and multiple hydroxylated products, some of which then undergo O-glucuronidation.104 Unmetabolized ruxolitinib is the predominant plasma drug entity until 6 hours postdose (58%).104 Most (96%) of the total dose is excreted within 24 hours postdose, primarily via urine (74%) and feces (22%).104 Patients who receive concomitant potent CYP3A4 inhibitors may require a 50% reduction in the ruxolitinib starting dose; however, data suggest that no dosage change is required when ruxolitinib is coadministered with inducers or mild to moderate inhibitors of CYP3A4.103
Ruxolitinib dose reduction is recommended in patients with hepatic or renal impairment.99 In patients with mild and severe hepatic impairment, exposure to ruxolitinib (area under the curve) was increased because of the reduced clearance, although the magnitude of increase in ruxolitinib exposure was not correlated with the degree of hepatic impairment.105 In contrast, an increasing severity of renal impairment was associated with increased exposure to ruxolitinib’s active metabolites and consequently with increased ruxolitinib pharmacologic activity.105
Ruxolitinib: clinical outcomes
Efficacy
The randomized, open-label Phase III RESPONSE registration trial demonstrated that ruxolitinib was superior to the best available therapy in adult patients with PV who were phlebotomy dependent, had splenomegaly, and were resistant to or intolerant of hydroxyurea.22 Patients were randomized to receive ruxolitinib (n=110) or the best available therapy (n=112). Pharmacological treatment in the best available therapy was chosen at the discretion of the treating physician, primarily hydroxyurea (58.9%) and IFN (11.6%); 15.2% of patients received no medication (except aspirin).22 Several limitations of the RESPONSE trial, including the open-label design and the nonstandardized determination of treatment in the best available therapy arm, may have influenced the study treatment compliance and precluded the study from comparing specific treatments with ruxolitinib. However, the study was not powered for such comparisons, and allowing treating physicians to determine the course of treatment was representative of real-world clinical settings. It is not uncommon for patients to continue treatment with hydroxyurea despite the evidence of resistance or intolerance.
In the primary analysis, a higher proportion of patients receiving ruxolitinib vs the best available therapy achieved the composite primary endpoint of hematocrit control and ≥35% reduction in spleen volume from baseline at Week 32 (20.9% vs 0.9%, P<0.001).22 Higher proportions of patients receiving ruxolitinib also achieved individual components of the primary endpoint (hematocrit control, 60.0% vs 19.6%; ≥35% reduction in spleen volume from baseline, 38.2% vs 0.9%)22 and complete hematologic response (23.6% vs 8.9%, P=0.003)22 at Week 32 (Table 1). A post hoc analysis demonstrated that the degree of splenomegaly at baseline did not influence the achievement of hematocrit control or spleen size reduction with ruxolitinib treatment.106 Data also suggest that ruxolitinib conferred benefits in patients who did not meet the primary study endpoint. In patients who did not achieve hematocrit control at Week 32, ruxolitinib prolonged the median time to subsequent phlebotomy eligibility compared with the best available therapy (52 weeks vs 21 weeks, respectively).107
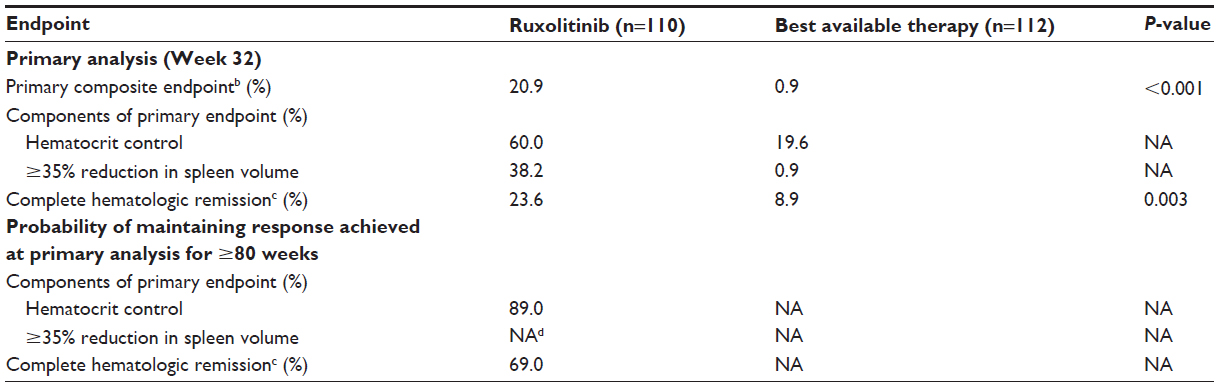 | Table 1 RESPONSE Phase III trial efficacy resultsa |
A total of 96 patients (85.7%) initially randomized to receive the best available therapy crossed over to the ruxolitinib arm because of lack of efficacy; most did so at or soon after the Week 32 visit.22 After cross over to ruxolitinib, a greater proportion of patients achieved ≥35% reduction in spleen size (crossover, 38.5%; best available therapy during randomized treatment, 1.8%), and phlebotomy treatment rates were lower (crossover, 38.5/100 patient-years; best available therapy during randomized treatment, 196.8/100 patient-years).108
A preplanned analysis of RESPONSE conducted 80 weeks after the last patient received his/her first dose demonstrated a durable response with ruxolitinib. This analysis indicated that of the 23 patients randomized to ruxolitinib who achieved the primary endpoint response at Week 32, only one patient lost the response.109 The primary endpoint components were also durable; the probability of maintaining hematocrit control without phlebotomy was 89%, and no patients who achieved ≥35% spleen volume reduction at Week 32 lost their response.109 Furthermore, patients who achieved a complete hematologic response in the primary analysis had a 69% probability of maintaining this response for ≥80 weeks after their initial response.109
Ruxolitinib treatment has also been associated with a decrease in JAK2V617F allele burden in patients with PV. In RESPONSE, the mean JAK2V617F allele burden changes from baseline in the ruxolitinib arm were −12.2% and −34.7% at Weeks 32 and 112, respectively.22 In contrast, patients randomized to receive the best available therapy had a mean 1.2% increase in JAK2V617F allele burden at Week 32.22 A single-center study of 22 patients with PV (n=11) or ET (n=11) reported mean changes in JAK2V617F allele burden of −19% and −28% after 36 months and 60 months of treatment with ruxolitinib, respectively.110
Patient-reported outcomes
Patient-reported outcomes favored ruxolitinib over the best available therapy in Phase III RESPONSE trial, with consistent improvements in the MPN-SAF, European Organisation for Research and Treatment of Cancer Quality-of-Life Questionnaire Core 30, Pruritus Symptom Impact Scale, and Patient Global Impression of Change instruments, compared with little improvement or worsening observed with the best available therapy (Table 2).22 Notably, even patients who did not achieve hematocrit control with ruxolitinib at Week 32 reported a higher degree of symptom improvement as evaluated by the MPN-SAF compared with patients who received the best available therapy (38% vs 4%, respectively).107
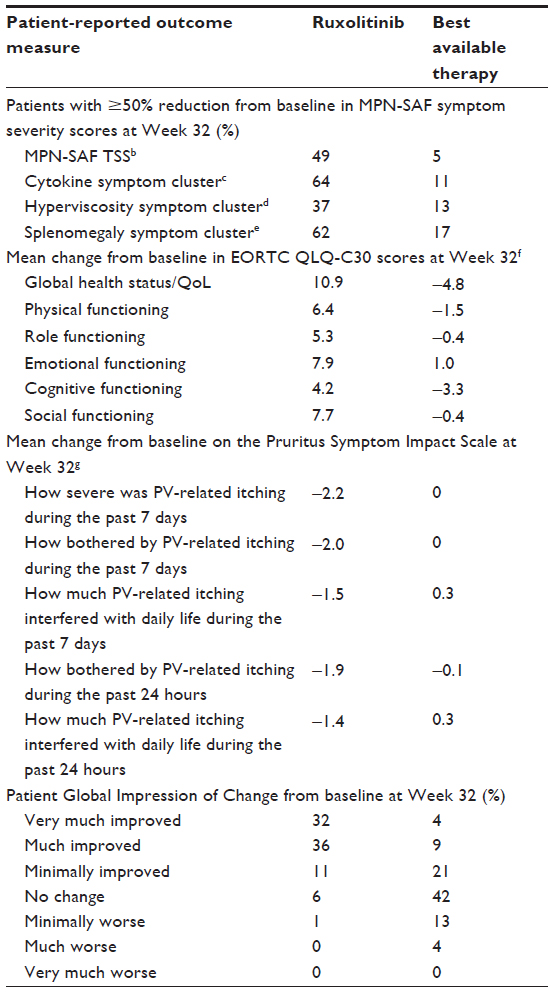 | Table 2 Patient-reported outcomes in the RESPONSE triala |
The randomized, multicenter, double-blind, double-dummy, Phase IIIb RELIEF trial was conducted in patients receiving a stable dose of hydroxyurea who were generally well controlled but reported disease-associated symptoms, comparing the change in PV-related symptom burden in patients continuing their hydroxyurea therapy with those switching to ruxolitinib treatment.111 There was a nonsignificant trend toward a greater improvement in the MPN-SAF cytokine cluster TSS (TSS-C) with ruxolitinib compared with hydroxyurea (proportion of patients achieving ≥50% reduction from baseline in TSS-C at Week 16: 43.4% vs 29.6%, respectively; primary endpoint).111 Additionally, there was a nonsignificant trend toward improvement in individual TSS-C symptoms with ruxolitinib compared with continued hydroxyurea in the RELIEF trial.111
Safety and tolerability
The most common nonhematologic adverse events at Week 32 in patients who received ruxolitinib in the Phase III RESPONSE trial were headache, diarrhea, pruritus, and fatigue (Table 3).22,112 The most frequent grade 3 or 4 nonhematologic adverse events in the ruxolitinib arm were dyspnea, asthenia, abdominal pain, headache, muscle spasms, and pruritus.22 Long-term treatment data indicated that the incidences of the most common nonhematologic adverse events were similar at Week 48 and in the 80-week analysis (headache, 20.9% and 21.8%; diarrhea, 20.0% and 19.1%; pruritus, 20.0% and 17.3%; fatigue, 17.3% and 17.3%, respectively).112
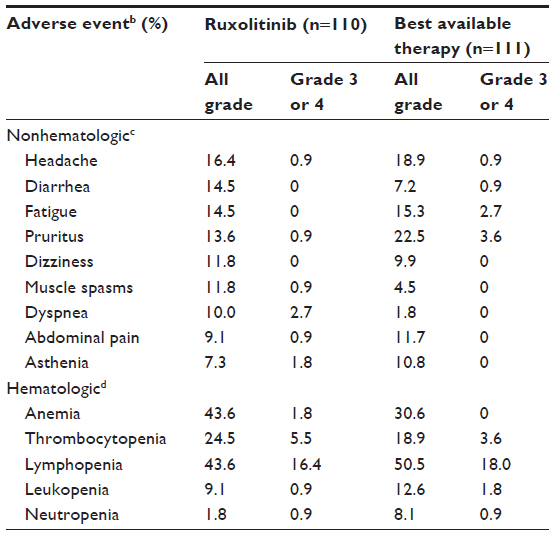 | Table 3 RESPONSE Phase III trial adverse eventsa |
Hematologic adverse events were primarily grade 1 or 2 at both the primary analysis (Table 3) and the 80-week analysis in the RESPONSE trial.22,112 The most common grade 3 or 4 hematologic adverse events in both randomized treatment arms were lymphopenia and thrombocytopenia. The US label for ruxolitinib indicates that thrombocytopenia, anemia, and neutropenia should be managed by dose reduction/interruption or transfusion.99
At Week 32, the rate of any grade 3 or 4 adverse events per 100 patient-years was lower in the ruxolitinib arm (28.8) than in the best available therapy arm (44.0) in the RESPONSE trial.22
Given the risks associated with thromboembolic events and disease transformation in patients with PV,14 it is important to consider such events when evaluating any new treatment option. The incidence of all-grade thromboembolic events at Week 32 was 0.9% in the ruxolitinib arm and 5.4% in the best available therapy arm (grade 3 or 4, 0.9% vs 1.8%, respectively).22 At the time of data cutoff in the primary analysis of RESPONSE, three patients in the ruxolitinib arm and one patient in the best available therapy arm developed MF after randomization; one patient in the ruxolitinib arm also developed AML. Two additional patients randomized to best available therapy experienced MF after cross over to ruxolitinib, one of whom progressed to AML.22
Other important adverse events of interest for patients being treated with ruxolitinib are nonmelanoma skin cancer and infections. At Week 32 in the RESPONSE trial, four patients in the ruxolitinib arm when compared with two patients in the best available therapy arm experienced newly diagnosed nonmelanoma skin cancer. All but one of these patients (best available therapy arm) had a history of precancerous skin lesions or nonmelanoma skin cancer.22 Melanoma skin cancer was diagnosed in zero patients in the ruxolitinib arm and one patient in the best available therapy arm during randomized treatment. The US prescribing information recommends periodic skin examinations for patients treated with ruxolitinib.99 In the RESPONSE trial, the incidence of any infection was similar in the ruxolitinib arm and the best available therapy arm at Week 32 (any grade, 41.8% vs 36.9%, respectively; grade 3 or 4, 3.6% vs 2.7%). However, herpes zoster infection was observed in 6.4% of patients in the ruxolitinib arm (all grade 1 or 2) at Week 32 compared to 0% of patients in the best available therapy arm.22 The US prescribing information states that patients should be advised about the early signs and symptoms of herpes zoster infection and to seek treatment as early as possible if herpes zoster infection is suspected.99
A long-term safety and tolerability evaluation of ruxolitinib was conducted in 241 patients (457 patient-years) who were resistant to or intolerant of hydroxyurea pooled from a Phase II study113 and RESPONSE (randomized and crossover patients).22,114 The most common nonhematologic adverse events were diarrhea, headache, and pruritus.114 Frequent grade 3 or 4 nonhematologic adverse events included dyspnea, herpes zoster, abdominal pain, back pain, headache, fatigue, and pyrexia.114 Hematologic adverse events were primarily grade 1 or 2; grade 3 or 4 anemia and thrombocytopenia were each reported in 3.7% of patients.114 Overall, seven patients had disease transformation to MF, and two patients had disease transformation to AML,114 a rate consistent with prior publications in similar patient populations with PV.21,84,115 The rate of thromboembolic events in the ruxolitinib group of this pooled analysis was 2.2/100 patient-years, whereas the rate in RESPONSE patients during randomized treatment with best available therapy was 8.2/100 patient-years.114
In Phase IIIb RELIEF trial, adverse events in the ruxolitinib arm were primarily grade 1 or 2.111 The most frequent nonhematologic adverse events were fatigue (20.4% [ruxolitinib arm] vs 10.7% [hydroxyurea arm]), headache (16.7% vs 5.4%), and dizziness (13.0% vs 8.9%). Two patients receiving ruxolitinib had grade 3 or 4 neutropenia.111
Place in therapy and ongoing trials
Ruxolitinib is approved by the FDA for the treatment of patients with PV who have had an inadequate response to or are intolerant of hydroxyurea.99 Further research will be important for identifying other populations of patients with PV who may benefit from treatment with ruxolitinib. RESPONSE 2 (ClinicalTrials.gov identifier: NCT02038036) is an ongoing, randomized, open-label, Phase IIIb clinical trial designed to evaluate the efficacy and safety of ruxolitinib compared with the best available therapy in patients with PV without splenomegaly who are resistant to or intolerant of hydroxyurea and require phlebotomy.116 The primary endpoint is achievement of hematocrit control at Week 16 that is maintained through Week 28, together with no phlebotomy eligibility from Weeks 4 to 28.116 Patients who are randomized to the best available therapy and do not meet the primary endpoint are allowed to cross over to ruxolitinib at or after Week 28.116 Safety and durability of response will be evaluated through Week 52.116
Conclusion
Ruxolitinib is the only approved treatment option designed to target the constitutively active JAK/STAT signaling pathway in patients with PV. Ruxolitinib improves hematocrit control without phlebotomy, improves blood cell counts, and reduces the enlarged spleen size.22,107 Accumulating evidence suggests that ruxolitinib may also ameliorate PV-related symptoms in patients who are resistant to and/or intolerant of hydroxyurea. Most adverse events were grade 1 or 2, and 82.7% of patients continued on treatment for ≥80 weeks in Phase III RESPONSE trial. Nonmelanoma skin cancer has been observed with ruxolitinib treatment, and periodic skin examinations should be performed. Herpes zoster infection rates (all grade 1 or 2) were higher with ruxolitinib compared with the best available therapy in the RESPONSE trial,22 and patients receiving ruxolitinib should be advised about the early signs and symptoms of infection and instructed to seek treatment as early as possible if suspected. Rates of disease transformation to MF and AML observed with ruxolitinib22 are similar to those previously published in similar patient populations with PV.84,115
Collectively, clinical trial data indicate that ruxolitinib is an effective treatment option for many patients with PV who are resistant to and/or intolerant of hydroxyurea. Furthermore, the potential benefits of ruxolitinib are unique when compared with the traditional treatment options because ruxolitinib may alleviate the PV-related symptom burden and improve the QoL. Updated treatment guidelines will be important for educating physicians about using ruxolitinib for the treatment of patients with PV.
Acknowledgment
Writing assistance was provided by Phuong Tran, PharmD, MBA (Complete Healthcare Communications, LLC, Chadds Ford, PA, USA, an ICON plc company), whose work was funded by Incyte Corporation, Wilmington, DE, USA.
Disclosure
KV is an employee of Incyte Corporation. SV participated in advisory boards and received research funding from Incyte Corporation. JJK served as a consultant for Incyte Corporation and Novartis and received a travel grant and research funding paid by Novartis to Hôpital Saint-Louis. The authors report no other conflicts of interest in this work.
References
Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14–22. | |
McMullin MF. Diagnosis and management of congenital and idiopathic erythrocytosis. Ther Adv Hematol. 2012;3(6):391–398. | |
Dameshek W. Some speculations on the myeloproliferative syndromes. Blood. 1951;6(4):372–375. | |
Prchal JF, Prchal JT. Molecular basis for polycythemia. Curr Opin Hematol. 1999;6(2):100–109. | |
Pahl HL. Towards a molecular understanding of polycythemia rubra vera. Eur J Biochem. 2000;267(12):3395–3401. | |
Fernandez-Luna JL, Silva M, Richard C, Sanz C, Benito A. Pathogenesis of polycythemia vera. Haematologica. 1998;83(2):150–158. | |
Spivak JL, Barosi G, Tognoni G, et al. Chronic myeloproliferative disorders. ASH Education Program Book. 2003;2003(1):200–224. | |
Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. | |
Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. | |
James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. | |
Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. | |
Hultcrantz M, Kristinsson SY, Andersson TM, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol. 2012;30(24):2995–3001. | |
Stein BL, Moliterno AR, Tiu RV. Polycythemia vera disease burden: contributing factors, impact on quality-of-life, and emerging treatment options. Ann Hematol. 2014;93(12):1965–1976. | |
Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874–1881. | |
Marchioli R, Finazzi G, Landolfi R, et al. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol. 2005;23(10):2224–2232. | |
Vannucchi AM. How I treat polycythemia vera. Blood. 2014; 124(22):3212–3220. | |
Kiladjian JJ, Chevret S, Dosquet C, Chomienne C, Rain JD. Treatment of polycythemia vera with hydroxyurea and pipobroman: final results of a randomized trial initiated in 1980. J Clin Oncol. 2011;29(29):3907–3913. | |
Fruchtman SM, Mack K, Kaplan ME, Peterson P, Berk PD, Wasserman LR. From efficacy to safety: a Polycythemia Vera Study Group report on hydroxyurea in patients with polycythemia vera. Semin Hematol. 1997;34(1):17–23. | |
Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350(2):114–124. | |
Marchioli R, Finazzi G, Specchia G, et al. Cardiovascular events and intensity of treatment in polycythemia vera. N Engl J Med. 2013;368(1):22–33. | |
Alvarez-Larran A, Pereira A, Cervantes F, et al. Assessment and prognostic value of the European LeukemiaNet criteria for clinicohematologic response, resistance, and intolerance to hydroxyurea in polycythemia vera. Blood. 2012;119(6):1363–1369. | |
Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. | |
Johansson P, Mesa R, Scherber R, et al. Association between quality-of-life and clinical parameters in patients with myeloproliferative neoplasms. Leuk Lymphoma. 2012;53(3):441–444. | |
Emanuel R, Dueck AC, Kiladjian JJ, et al. Conventional therapeutic options have limited impact on MPN symptoms: insights from a prospective analysis of the MPN-SAF [abstract 366]. Paper Presented at: European Hematology Association; June 14–17. Amsterdam, the Netherlands: 2012. | |
Reid CD. The significance of endogenous erythroid colonies (EEC) in haematological disorders. Blood Rev. 1987;1(2):133–140. | |
Hinshelwood S, Bench AJ, Green AR. Pathogenesis of polycythaemia vera. Blood Rev. 1997;11(4):224–232. | |
Dai CH, Krantz SB, Dessypris EN, Means RT Jr, Horn ST, Gilbert HS. Polycythemia vera. II. Hypersensitivity of bone marrow erythroid, granulocyte-macrophage, and megakaryocyte progenitor cells to interleukin-3 and granulocyte-macrophage colony-stimulating factor. Blood. 1992;80(4):891–899. | |
Moliterno AR, Hankins WD, Spivak JL. Impaired expression of the thrombopoietin receptor by platelets from patients with polycythemia vera. N Engl J Med. 1998;338(9):572–580. | |
Silva M, Richard C, Benito A, Sanz C, Olalla I, Fernandez-Luna JL. Expression of Bcl-x in erythroid precursors from patients with polycythemia vera. N Engl J Med. 1998;338(9):564–571. | |
Roder S, Steimle C, Meinhardt G, Pahl HL. STAT3 is constitutively active in some patients with polycythemia rubra vera. Exp Hematol. 2001;29(6):694–702. | |
Temerinac S, Klippel S, Strunck E, et al. Cloning of PRV-1, a novel member of the uPAR receptor superfamily, which is overexpressed in polycythemia rubra vera. Blood. 2000;95(8):2569–2576. | |
Klippel S, Strunck E, Busse CE, Behringer D, Pahl HL. Biochemical characterization of PRV-1, a novel hematopoietic cell surface receptor, which is overexpressed in polycythemia rubra vera. Blood. 2002;100(7):2441–2448. | |
Kralovics R, Skoda RC. Molecular pathogenesis of Philadelphia chromosome negative myeloproliferative disorders. Blood Rev. 2005;19(1):1–13. | |
Wolf A, Eulenfeld R, Gabler K, et al. JAK2-V617F-induced MAPK activity is regulated by PI3K and acts synergistically with PI3K on the proliferation of JAK2-V617F-positive cells. JAKSTAT. 2013;2(3):e24574. | |
Bumm TG, Elsea C, Corbin AS, et al. Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Res. 2006;66(23):11156–11165. | |
Tiedt R, Hao-Shen H, Sobas MA, et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood. 2008;111(8):3931–3940. | |
Pasquier F, Cabagnols X, Secardin L, Plo I, Vainchenker W. Myeloproliferative neoplasms: JAK2 signaling pathway as a central target for therapy. Clin Lymphoma Myeloma Leuk. 2014;14(suppl):S23–S35. | |
Milosevic JD, Kralovics R. Genetic and epigenetic alterations of myeloproliferative disorders. Int J Hematol. 2013;97(2):183–197. | |
Passamonti F, Rumi E, Pietra D, et al. A prospective study of 338 patients with polycythemia vera: the impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia. 2010;24(9):1574–1579. | |
Nicholson SE, Oates AC, Harpur AG, Ziemiecki A, Wilks AF, Layton JE. Tyrosine kinase JAK1 is associated with the granulocyte-colony-stimulating factor receptor and both become tyrosine-phosphorylated after receptor activation. Proc Natl Acad Sci U S A. 1994;91(8):2985–2988. | |
Wang L, Xue J, Zadorozny EV, Robinson LJ. G-CSF stimulates Jak2-dependent Gab2 phosphorylation leading to Erk1/2 activation and cell proliferation. Cell Signal. 2008;20(10):1890–1899. | |
Sugimoto N, Nakahira M, Ahn HJ, et al. Differential requirements for JAK2 and TYK2 in T cell proliferation and IFN-gamma production induced by IL-12 alone or together with IL-18. Eur J Immunol. 2003;33(1):243–251. | |
Witthuhn BA, Quelle FW, Silvennoinen O, et al. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74(2):227–236. | |
Penta K, Sawyer ST. Erythropoietin induces the tyrosine phosphorylation, nuclear translocation, and DNA binding of STAT1 and STAT5 in erythroid cells. J Biol Chem. 1995;270(52):31282–31287. | |
Kirito K, Nakajima K, Watanabe T, et al. Identification of the human erythropoietin receptor region required for Stat1 and Stat3 activation. Blood. 2002;99(1):102–110. | |
Grebien F, Kerenyi MA, Kovacic B, et al. Stat5 activation enables erythropoiesis in the absence of EpoR and Jak2. Blood. 2008;111(9):4511–4522. | |
Parganas E, Wang D, Stravopodis D, et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell. 1998;93(3):385–395. | |
Grisouard J, Hao-Shen H, Dirnhofer S, Wagner KU, Skoda RC. Selective deletion of Jak2 in adult mouse hematopoietic cells leads to lethal anemia and thrombocytopenia. Haematologica. 2014;99(4):e52–e54. | |
Simon HU, Yousefi S, Dibbert B, Levi-Schaffer F, Blaser K. Anti-apoptotic signals of granulocyte-macrophage colony-stimulating factor are transduced via Jak2 tyrosine kinase in eosinophils. Eur J Immunol. 1997;27(12):3536–3539. | |
Drachman JG, Millett KM, Kaushansky K. Thrombopoietin signal transduction requires functional JAK2, not TYK2. J Biol Chem. 1999;274(19):13480–13484. | |
Vannucchi AM, Antonioli E, Guglielmelli P, et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia. 2007;21(9):1952–1959. | |
Stein BL, Oh ST, Berenzon D, et al. Polycythemia vera: an appraisal of the biology and management 10 years after the discovery of JAK2 V617F. J Clin Oncol. 2015;33(33):3953–3960. | |
Hultcrantz M, Andersson TM-L, Landgren O, et al. Risk of arterial and venous thrombosis in 11,155 patients with myeloproliferative neoplasms and 44,620 matched controls; a population-based study. Blood. 2014;124:abstract632. (ASH Annual Meeting Abstracts). | |
Barbui T, Masciulli A, Marfisi MR, et al. White blood cell counts and thrombosis in polycythemia vera: a subanalysis of the CYTO-PV study. Blood. 2015;126(4):560–561. | |
Di Nisio M, Barbui T, Di Gennaro L, et al. The haematocrit and platelet target in polycythemia vera. Br J Haematol. 2007;136(2):249–259. | |
De Grandis M, Cambot M, Wautier MP, et al. JAK2V617F activates Lu/BCAM-mediated red cell adhesion in polycythemia vera through an EpoR-independent Rap1/Akt pathway. Blood. 2013;121(4):658–665. | |
Barbui T, Carobbio A, Finazzi G, et al. Inflammation and thrombosis in essential thrombocythemia and polycythemia vera: different role of C-reactive protein and pentraxin 3. Haematologica. 2011;96(2):315–318. | |
Nieswandt B, Pleines I, Bender M. Platelet adhesion and activation mechanisms in arterial thrombosis and ischaemic stroke. J Thromb Haemost. 2011;9(suppl 1):92–104. | |
Harlan JM, Killen PD, Harker LA, Striker GE, Wright DG. Neutrophil-mediated endothelial injury in vitro mechanisms of cell detachment. J Clin Invest. 1981;68(6):1394–1403. | |
Weksler BB, Jaffe EA, Brower MS, Cole OF. Human leukocyte cathepsin G and elastase specifically suppress thrombin-induced prostacyclin production in human endothelial cells. Blood. 1989;74(5):1627–1634. | |
Celi A, Pellegrini G, Lorenzet R, et al. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci U S A. 1994;91(19):8767–8771. | |
Falanga A, Marchetti M, Evangelista V, et al. Neutrophil activation and hemostatic changes in healthy donors receiving granulocyte colony-stimulating factor. Blood. 1999;93(8):2506–2514. | |
Lip GY, Blann A. von Willebrand factor: a marker of endothelial dysfunction in vascular disorders? Cardiovasc Res. 1997;34(2):255–265. | |
Evangelista V, Rajtar G, de Gaetano G, White JG, Cerletti C. Platelet activation by fMLP-stimulated polymorphonuclear leukocytes: the activity of cathepsin G is not prevented by antiproteinases. Blood. 1991;77(11):2379–2388. | |
Hobbs CM, Manning H, Bennett C, et al. JAK2V617F leads to intrinsic changes in platelet formation and reactivity in a knock-in mouse model of essential thrombocythemia. Blood. 2013;122(23):3787–3797. | |
Mesa R, Miller CB, Thyne M, et al. Impact of myeloproliferative neoplasms (MPNs) on patients’ overall health and productivity: results from the MPN LANDMARK SURVEY in the United States [abstract]. Blood. 2014;124:abstract3183. (ASH Annual Meeting Abstracts). | |
Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality-of-life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007;109(1):68–76. | |
Quintas-Cardama A, Kantarjian H, Cortes J, Verstovsek S. Janus kinase inhibitors for the treatment of myeloproliferative neoplasias and beyond. Nat Rev Drug Discov. 2011;10(2):127–140. | |
Pourcelot E, Trocme C, Mondet J, Bailly S, Toussaint B, Mossuz P. Cytokine profiles in polycythemia vera and essential thrombocythemia patients: clinical implications. Exp Hematol. 2014;42(5):360–368. | |
Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011;29(10):1356–1363. | |
Kwaan HC, Wang J. Hyperviscosity in polycythemia vera and other red cell abnormalities. Semin Thromb Hemost. 2003;29(5):451–458. | |
Passamonti F. How I treat polycythemia vera. Blood. 2012;120(2):275–284. | |
Squizzato A, Romualdi E, Passamonti F, Middeldorp S. Antiplatelet drugs for polycythaemia vera and essential thrombocythaemia. Cochrane Database Syst Rev. 2013;4:CD006503. | |
Huang ES, Strate LL, Ho WW, Lee SS, Chan AT. Long-term use of aspirin and the risk of gastrointestinal bleeding. Am J Med. 2011;124(5):426–433. | |
Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013;88(6):507–516. | |
Michiels JJ, Berneman Z, Schroyens W, Finazzi G, Budde U, van Vliet HH. The paradox of platelet activation and impaired function: platelet-von Willebrand factor interactions, and the etiology of thrombotic and hemorrhagic manifestations in essential thrombocythemia and polycythemia vera. Semin Thromb Hemost. 2006;32(6):589–604. | |
Marchioli R, Finazzi G, Specchia G, Masciulli A, Mennitto MR, Barbui T. The CYTO-PV: a large-scale trial testing the intensity of CYTOreductive therapy to prevent cardiovascular events in patients with polycythemia vera. Thrombosis. 2011;2011:794240. | |
Deacon B, Abramowitz J. Fear of needles and vasovagal reactions among phlebotomy patients. J Anxiety Disord. 2006;20(7):946–960. | |
Tobiasson M, Alyass B, Soderlund S, Birgegard G. High prevalence of restless legs syndrome among patients with polycytemia vera treated with venesectio. Med Oncol. 2010;27(1):105–107. | |
Greig AJ, Patterson AJ, Collins CE, Chalmers KA. Iron deficiency, cognition, mental health and fatigue in women of childbearing age: a systematic review. J Nutr Sci. 2013;2:e14. | |
Kim J, Wessling-Resnick M. Iron and mechanisms of emotional behavior. J Nutr Biochem. 2014;25(11):1101–1107. | |
Bjorkholm M, Hultcrantz M, Derolf AR. Leukemic transformation in myeloproliferative neoplasms: therapy-related or unrelated? Best Pract Res Clin Haematol. 2014;27(2):141–153. | |
Barbui T. The leukemia controversy in myeloproliferative disorders: is it a natural progression of disease, a secondary sequela of therapy, or a combination of both? Semin Hematol. 2004;41(2 suppl 3):15–17. | |
Finazzi G, Caruso V, Marchioli R, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005;105(7):2664–2670. | |
Nielsen I, Hasselbalch HC. Acute leukemia and myelodysplasia in patients with a Philadelphia chromosome negative chronic myeloproliferative disorder treated with hydroxyurea alone or with hydroxyurea after busulphan. Am J Hematol. 2003;74(1):26–31. | |
Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761–770. | |
Silver RT. Long-term effects of the treatment of polycythemia vera with recombinant interferon-alpha. Cancer. 2006;107(3):451–458. | |
Sacchi S, Leoni P, Liberati M, et al. A prospective comparison between treatment with phlebotomy alone and with interferon-alpha in patients with polycythemia vera. Ann Hematol. 1994;68(5):247–250. | |
Quintas-Cardama A, Kantarjian H, Manshouri T, et al. Pegylated interferon alfa-2a yields high rates of hematologic and molecular response in patients with advanced essential thrombocythemia and polycythemia vera. J Clin Oncol. 2009;27(32):5418–5424. | |
Kiladjian JJ, Chomienne C, Fenaux P. Interferon-alpha therapy in bcr-abl-negative myeloproliferative neoplasms. Leukemia. 2008; 22(11):1990–1998. | |
Taylor PC, Dolan G, Ng JP, Paul B, Collin R, Reilly JT. Efficacy of recombinant interferon-alpha (rIFN-alpha) in polycythaemia vera: a study of 17 patients and an analysis of published data. Br J Haematol. 1996;92(1):55–59. | |
Muller EW, de Wolf JT, Egger R, et al. Long-term treatment with interferon-alpha 2b for severe pruritus in patients with polycythaemia vera. Br J Haematol. 1995;89(2):313–318. | |
Stasi R, Venditti A, Del Poeta G, et al. Role of human leukocyte interferon-alpha in the treatment of patients with polycythemia vera. Am J Med Sci. 1998;315(4):237–241. | |
Turri D, Mitra ME, Di Trapani R, Lipari MG, Perricone R, Cajozzo A. Alpha-interferon in polycythemia vera and essential thrombocythemia. Haematologica. 1991;76(1):75–77. | |
Devonshire V, Lapierre Y, Macdonell R, et al. The Global Adherence Project (GAP): a multicenter observational study on adherence to disease-modifying therapies in patients with relapsing-remitting multiple sclerosis. Eur J Neurol. 2011;18(1):69–77. | |
Hasselbalch HC. A new era for IFN-alpha in the treatment of Philadelphia-negative chronic myeloproliferative neoplasms. Expert Rev Hematol. 2011;4(6):637–655. | |
Kiladjian JJ, Cassinat B, Chevret S, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood. 2008;112(8):3065–3072. | |
Quintas-Cardama A, Abdel-Wahab O, Manshouri T, et al. Molecular analysis of patients with polycythemia vera or essential thrombocythemia receiving pegylated interferon alpha-2a. Blood. 2013;122(6):893–901. | |
Jakafi® (ruxolitinib). Full Prescribing Information. Wilmington, DE: Incyte Corporation; 2014. | |
Jakavi® (ruxolitinib). Summary of Product Characteristics. Horsham, West Sussex, UK: Novartis AG; 2015. | |
Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15):3109–3117. | |
Shi JG, Chen X, McGee RF, et al. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol. 2011;51(12):1644–1654. | |
Shi JG, Chen X, Emm T, et al. The effect of CYP3A4 inhibition or induction on the pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in healthy volunteers. J Clin Pharmacol. 2012;52(6):809–818. | |
Shilling AD, Nedza FM, Emm T, et al. Metabolism, excretion, and pharmacokinetics of [14C]INCB018424, a selective Janus tyrosine kinase 1/2 inhibitor, in humans. Drug Metab Dispos. 2010;38(11):2023–2031. | |
Chen X, Shi JG, Emm T, et al. Pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in renal and hepatic impairment patients. Clin Pharmacol Drug Dev. 2013;3(1):34–42. | |
Vannucchi A, Verstovsek S, Jones M, et al. Efficacy of ruxolitinib by baseline spleen volume in patients with polycythemia vera resistant to or intolerant of hydroxyurea. Blood. 2014;124(21):abstract1840. (ASH Annual Meeting Abstracts). | |
Verstovsek S, Kiladjian JJ, Mesa R, et al. Ruxolitinib efficacy by hematocrit control in patients with polycythemia vera: an analysis of the RESPONSE trial. Blood. 2014;124(21):abstract3201. (ASH Annual Meeting Abstracts). | |
Kiladjian JJ, Vannucchi A, Griesshammer M, et al. Clinical benefit of ruxolitinib treatment after crossover from best available therapy in patients with polycythemia vera: analysis of the RESPONSE trial. Blood. 2014;124(21):abstract3181. (ASH Annual Meeting Abstracts). | |
Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib in polycythemia vera: follow-up from the RESPONSE trial. J Clin Oncol. 2015;33(No 15_suppl):abstract7087. (ASCO Annual Meeting Abstracts). | |
Pieri L, Pancrazzi A, Pacilli A, et al. JAK2V617F complete molecular remission in polycythemia vera/essential thrombocythemia patients treated with ruxolitinib. Blood. 2015;125(21):3352–3353. | |
Mesa R, Vannucchi AM, Yacoub A, et al. The efficacy and safety of continued hydroxyurea therapy versus switching to ruxolitinib in patients with polycythemia vera: a randomized, double-blind, double-dummy, symptom study (RELIEF). Blood. 2014;124(21):abstract3168. (ASH Annual Meeting Abstracts). | |
Kiladjian JJ, Vannucchi A, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2015;100(s1):abstractS447. (EHA Annual Meeting Abstracts). | |
Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer. 2014;120(4):513–520. | |
Verstovsek S, Harrison CN, Kiladjian J-J, et al. Effect of ruxolitinib on markers of iron deficiency: an analysis of the RESPONSE trial. Haematologica. 2015;100(s1):abstract672. (EHA Annual Meeting Abstracts). | |
Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117(10):755–761. | |
Passamonti F, Saydam G, Lim L, Khan MH, Mounedji N, Griesshammer M. RESPONSE 2: a phase 3b study evaluating the efficacy and safety of ruxolitinib in patients with hydroxyurea (HU)-resistant/intolerant polycythemia vera (PV) versus best available therapy (BAT). J Clin Oncol. 2014;32(5s):abstractTPS7128. (ASCO Annual Meeting Abstracts). | |
Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst. 1993;85(5):365–376. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.