")
Back to Journals » OncoTargets and Therapy » Volume 13
RPL38 Regulates the Proliferation and Apoptosis of Gastric Cancer via miR-374b-5p/VEGF Signal Pathway
Received 1 March 2020
Accepted for publication 28 May 2020
Published 26 June 2020 Volume 2020:13 Pages 6131—6141
DOI https://doi.org/10.2147/OTT.S252045
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjeev K. Srivastava
This paper has been retracted.
Hanshu Ji,1 Xiaoyu Zhang2
1Second Department of General Surgery, Cangzhou Central Hospital, Cangzhou 061000, Hebei Province, People’s Republic of China; 2Third Ward of Tumor Surgery Department, Cangzhou Central Hospital, Cangzhou 061000, Hebei Province, People’s Republic of China
Correspondence: Hanshu Ji
Second Department of General Surgery, Cangzhou Central Hospital Tel +86-15350771923
Email [email protected]
Aim: To explore the role of RPL38 on proliferation and apoptosis of gastric cancer cells by regulating miR-374b-5p/VEGF signal pathway.
Methods: qRT-PCR was used to measure the expression of RPL38. CCK8 assay, Matrigel invasion assay, and flow cytometry were used to detect the role of RPL38in MKN-45 cells. Western blot was used to measure the protein expression of VEGF, p-ERK, ERK, p-AKT, AKT in cells. Dual-luciferase assay was performed to verify the relationship between miR-374b-5p and RPL38, miR-374b-5p and VEGF.
Results: In our research, we found that RPL38 was upregulation in gastric cancer, loss function of RPL38 could inhibit MKN-45 cell proliferation and invasion, accompany with increasing apoptosis. Then, we verified that RPL38 could interact with miR-374b-5p by performed luciferase assay, there was a negative correlation between RPL38 and miR-374b-5p. Furthermore, we observed that VEGF is a potential target of miR-374b-5p, miR-374b-5p negatively regulated the expression of VEGF, and effected ERK/AKT signal pathways. Next, we found that miR-374b-5p inhibitor or overexpression of VEGF could prevent the anti-tumor function of si-RPL38.
Conclusion: Knockdown of RPL38 inhibits the proliferation and apoptosis of gastric cancer via miR-374b-5p/VEGF signal pathway.
Keywords: RPL38, gastric cancer, miRNA, VEGF, proliferation and apoptosis
Introduction
As a common malignant tumor in the world, gastric cancer (GC) ranks the 4th in incidence and the 2nd in mortality.1,2 The early stage of gastric cancer has no specific manifestations and lacks sensitive serum indicators, and most patients are already in the advanced stage when diagnosed with poor prognosis.3,4 Gastric cancer is a complex disease caused by many factors such as heredity and environment. Gene mutation,5,6 loss of tumor suppressor and abnormal regulation of cell proliferation signaling pathway are all related to the occurrence and development of gastric cancer.7,8 Therefore, it is very important for the diagnosis and treatment of gastric cancer to find effective early warning molecules, improve the early diagnosis and find out the molecular mechanism of invasion and metastasis of gastric cancer.
RPL38 belongs to the L38E family of ribosomal proteins. It is located in the cytoplasm. Alternative splice variants have been identified, both encoding the same protein. As is typical for genes encoding ribosomal proteins, there are multiple processed pseudogenes of this gene dispersed through the genome, including one located in the promoter region of the type 1 angiotensin II receptor gene. Sahin et al identified RPL38, predominantly expressed in the pancreatic ductal epithelium. They also demonstrated that RPL38 is highly expressed in a panel of pancreatic cancer cell lines and can potentially be useful in tumor targeting or as a tumor marker.9 Zheng et al identified RPL38 as differentially expressed gene, which may function in the prognosis of pancreatic ductal adenocarcinoma.10 In this study, we explored the role of RPL38 in GC development and progression.
Many studies have confirmed that miRNA is abnormally expressed in various types of tumors. MiRNA can silence or enhance mRNA expression through binding of regulatory genes or mRNA related to GC,11,12 leading to invasion and metastasis of gastric cancer cells. Such as, miR-574-5p, miR-574-3p,13 miR-502-5p14 and miR-198.15
Vascular endothelial growth factor (VEGF) is a dimer glycoprotein,16 which can stimulate the proliferation, growth and migration of endothelial cells and has a high level of expression in a variety of tumor cells.17 It is not only one of the most important pro-angiogenic factors in tumor angiogenesis but also plays an important role in tumor immunity. We observed that RPL38 was positively correlated with VEGF, and regulates the pathways of EKR and AKT to effect the process of gastric cancer.
Materials and Methods
Clinical Samples
Fresh tissue samples and adjacent normal tissue samples were collected from 60 GC patients at Cang zhou Central Hospital. After the collection, the tissues were immediately stored in liquid nitrogen, and then kept at −80°C for further use. Experimental procedures were accomplished in accordance with the guidelines released by the Ethics Committee of Cang zhou Central Hospital. Signed written informed consent was obtained from each patient.
Cell Culture
GES-1, AGS, SGC-7901, BGC-823, MKN-28 and MKN-45 cell lines were purchased from the Science Cell Laboratory. Cells were cultured in PRIM 1640 (GIBCO, United States) supplemented with 10% fetal bovine serum (Cromwell, USA) and 100 μL/mL penicillin and streptomycin (Sigma-Aldrich, USA) and placed at 37°C with 5% CO2.
Cell Transfection
SiRNA was produced by Genomeditech, Co. (Shanghai, China) si-RPL38#1 and si-RPL38#2. A scrambled siRNA was used as the negative control (si-NC). About 5×105 cells per well were seeded in 6 well plates, transfection of siRNA into the cells was performed using lipo2000 (Thermo Fisher Scientific, USA) according to the manufacturer’s recommendations. Cells were transfected with 20 nmol/L siRNA for 48 h, and then siRNA transfection in the best condition was performed.
Nuclear and Cytoplasmic Protein Extraction Kit
Wash with PBS and scrape cells, or treat cells by EDTA, and stroke cells with a pipette. Then, centrifuge for several minutes to collect cells, discard supernatant, and keep cell pellets for use. Do not digest with pancreatin to avoid degradation of proteins. Add 200 μL of CER A (containing PMSF) into per 20 μL of cell pellets. Vortex at maximum speed for 5 s to suspend cell pellets. Incubate on ice for 10–15 min. Add 10 μL of CER B. Vortex at maximum speed for 5 s, and incubate on ice for 1 min. Vortex at maximum speed for 5 s; then centrifuge at 12000–16000 g at 4°C for 5 min. Immediately pipette the supernatant (cytoplasmic proteins) into a pre-cooling tube. Assay the cytoplasmic proteins immediately, or store at −80°Cfor use
qRT-PCR
RNA isolation, reverse transcription and quantitative expression were carried according to manufacturer’s instructions. Total RNA was isolated from tissues or cultured cells using Trizol reagent (Invitrogen, USA), DNA was generated using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR was performed on an ABI7500 FAST real-time PCR System (Applied Bio-systems) for 40 cycles. After the reaction cycles, the threshold cycle (Ct) values were determined, and the relative mRNA levels were calculated based on the Ct values and normalized to the GAPDH/U6 level in each sample. Gene expression was calculated using the 2-ΔΔCt method. The primer sequences were shown as follows:
RPL38-Forward, 5ʹ-CAAAGACAGCCCTCGAAAAG-3ʹ;
RPL38-Reverse, 5ʹ-TTTTC CGTACGCGTTAAAGG-3ʹ.
VEGF-Forward, 5ʹ-GCAGAATCATCACGAAGTGG-3
VEGF-Reverse, 5ʹ-ATCAGGGGCACACAGGAT-3ʹ
GAPDH-Forward,5′-ATGGGGGAAGGTGAAGGTCG-3′;
GAPDH-Reverse, 5′-GGGGTCATTGATGGCAACAATA-3′.
MiR-374b-5p-Forward, 5ʹ-TCTGATGGCTGGGTTGGCTTAG-3ʹ;
MiR-374b-5p-Reverse, 5ʹ-ATCATTCTTTGAAGATGCCTGCTTG-3ʹ.
U6-Forward, 5ʹ-ATTGGAACGATACAGAGAAGATT-3ʹ;
U6-Reverse, 5ʹ-GGAACGCTTCACGAATTTG-3ʹ.
Western Blot
Total protein was collected from cells with RIPA lysis Mix. Briefly, 60 μg protein extraction was loaded via SDS-PAGE and transferred onto nitrocellulose membranes (Absin, China); the membranes were incubated in 5% non-fat milk blocking buffer for 3 h, and then incubated with primary antibodies for 2 h at room temperature and plated at 4°C overnight. After incubation with secondary antibodies, the membranes were scanned using an Odyssey, and data were analyzed with Odyssey software (LI-COR, USA). The primary antibodies were purchased from proteintech, VEGF (19003-1-AP, 1:500), ERK (67170-1-Ig, 1:500), AKT (10176-2-AP,1:1000), p-AKT (66444-1-Ig, 1:500), p-ERK (sc-7383, 1:200) was purchased from SANTA CRUZ biotechnology, and Gapdh (60004-1-Ig, 1:2000) was used as an internal control.
CCK8 Assay
Cells were seeded in 96-well cell plates, and added CCK-8 solution (Vazyme, China) at 0, 24, 48 and 72 h. 2 h later, measure the OD value at 450 nm using an MK3ELISA Reader (Thermo fisher scientific, USA).
Matrigel Invasion Assay
Cells in logarithmic growth phase were adjusted to 2 × 105 cells/well of medium (without serum) and plated into the upper chamber insert pre-coated with 1μg/μL Matrigel. Lower chamber was added with 500 μL of medium (with 10% FBS), and then incubate the chamber at 37°C for 48 h. Then, the invading cells were visualized by the crystal violet and inverted microscope.
In vivo Tumor Growth Assay
Nude mice were purchased from Beijing Charles river. The nude mice were injected with AAV9-si-NC/AAV9-si-RPL38#1, after 1 week, MKN-45 cells (5 × 106) were subcutaneously injected in the right lower limb of the nude mice. Tumor size was measured every 5 days. After another 15 d of injection, the tumor was removed for follow-up study.
Cell Cycle Assay
The cells were digested with 1 mL trypsin for 2–3 min, 5 mL PBS was added, and the bottle wall was blown open to separate the cells into single-cell suspension. After collection, centrifuge at 1000 RPM for 5 min at 4°C. After the supernatant was discarded, 10 mL PBS was added to re-suspension, and the cells were precooled overnight with 70% ethanol and ice (4°C) to fix. The next day, the cell suspension was filtered with a 300-mesh sieve, centrifuged at 1000 RPM at 4°C for 5 min, and the supernatant was discarded. The cells were fixed with 1 mL PI solution and kept at 4°C for 30 min in dark. Flow cytometer was used to detect cell cycle.
Cell Apoptosis Assay
The cells were counted, about 5×105–1×106 cells/mL. Then, 1 mL cells were centrifuged, 1000 rpm, 10 min, 4°C, and the supernatant was thrown away. Cell suspension was added with 1 mL precooled PBS and the supernatant was thrown away, 1000 rpm, 10 min, 4°C. The cells were resuspended in 200 μL Binding Buffer with10 Μl Annexin V-FITC and 10 μL PI solution at room temperature in darkness for 15 min after which 300 μL Binding Buffer was mixed into the resuspension. Flow cytometry was used to detect cell apoptosis within 1 h.
Dual-Luciferase Reporter Assay
HEK293T cells were transfected with 20 mmol/L miRNA mimic or miR-NC together with RPL38-WT or RPL38-mutation/VEGF-WT or VEGF-mutation. Luciferase activity was determined using Dual-Luciferase Reporter Assay Kit (Promega, USA) on luminometer (Promega, USA) 48 h after the transfection.
EdU Assay
EdU incorporation. For EdU labeling, a 1:1,000 dilution of EdU-labeling reagent (Beyotime, China) was added to islet culture medium during the last 18 h of cell culture.
Immunohistochemistry (IHC)
The tumor tissue was cut into 5-μm-thick sections. The sections were dewaxed and deparaffinized in xylene and rehydrated in graded alcohol solutions. Sections were heated in tris-EDTA buffer for 30 min to extract antigens. The samples were incubated with primary antibodies for ki-67 (27309-1-AP, 1:100), VEGF (19003-1-AP, 1:100), p-ERK (sc-7383, 1:20), p-AKT (66444-1-Ig, 1:50), cleaved-caspase3 (ab2302, 1:100, abcam, USA) and HRP-labeled Streptavidin (A0303, 1:200, Beyotime, China) secondary antibodies. Sections were avoided light with hematoxylin, then dehydrated and secured. Images were catched with an Olympus camera.
Tumor Allograft
Thirty nude mice were randomly divided into two groups. AAV9-si-RPL38 # 1 or AAV9-si-NC was injected through the tail vein. One week later, MKN-45 cells were injected subcutaneously into nude mice, and tumors were observed at a time point. After the volume change was measured at the last time point, the nude mice were euthanized to collect tumor tissues of the nude mice for quality detection and immunohistochemistry.
Statistical Analysis
All values are presented as the mean ± S.E.M. Significant differences were determined using GraphPad 6.0 software (GraphPad, Inc.). One-way analysis of variance was used to determine statistical differences between ≥2 groups, followed by t-test. Unpaired or paired t-tests were used to determine statistical differences between two groups. P values of less than 0.05 were considered significant. In order to evaluate the prognostic value of RPL38 in GC samples, we selected the appropriate cut-off value for subdivision. The optimal cut-off was defined by the statistical distribution of RPL38 expression. RPL38 expression was normally distributed in GC samples, and there was a significantly evident distinction along the median value. Therefore, GC samples were divided into RPL38 high and RPL38 low groups by the median RPL38 expression Error bars on all graphs are presented as the SEM of the mean unless otherwise indicated.
Results
RPL38 Was Upregulation in Gastric Cancer and MKN-45 Cells
To explore the role of RPL38 in GC, we collected cancerous and para-cancer normal tissue from 60 cancer patients, qRT-PCR was performed to determine the level of RPL38. We found that the expression of RPL38 was upregulation in tumor tissue compared with the normal tissue (P < 0.001) (Figure 1A). We also detected the level of RPL38 in different human GC cell lines (AGS, SGC-7901, BGC-823, MKN-28, MKN-45), the GES-1 was indicated as control. The results showed that RPL38 was increased in the serious of human GC cell lines (P < 0.05) (Figure 1B). Through follow-up interviews with patients, we found that patients with low RPL38 expression had a higher survival rate (Figure 1C). The results indicated that RPL38 may play a key role in GC progression.
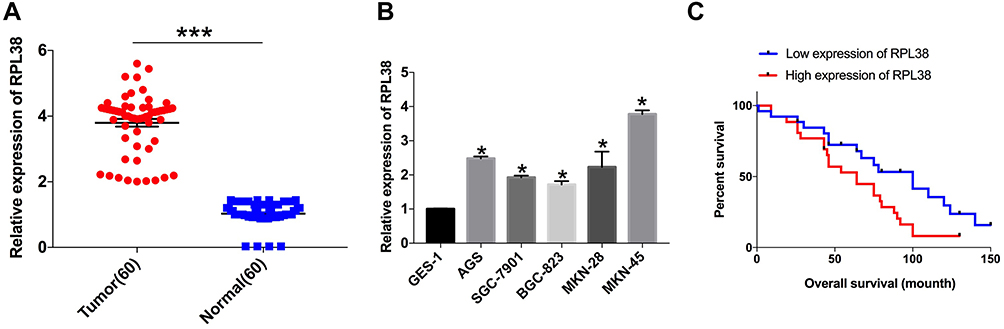 |
Figure 1 RPL38 is upregulation in GCs. (A) qRT-PCR assay was performed to detect the expression of RPL38 in tumor and adjacent tissues. (B) The expression of RPL38 in each cell line. (C) The survival rates of patients with high expression of RPL38 and low expression of RPL38. *P<0.05, ***P<0.001. |
Knockdown of RPL38 Inhibits the GC Cells Proliferation, Invasion, and Metastasis
To further explore the role of RPL38 in GC, the si-RNA was constructed to induce the loss function of RPL38. qRT-PCR was used to detect the knockout efficiency of si-RPL38#1and si-RPL38#2 (P < 0.05) (Figure 2A). Next, we transfected the si-RPL38 or si-NC into MKN-45 cells; CCK8 and EdU assays showed that si-RPL38#1 and #2 both significantly inhibited cell proliferation (P < 0.01) (Figure 2B and C). We found that the inhibition of RPL38 inhibited the invasion of cancer cells (P < 0.01) (Figure 2D). Then, cell cycle distribution in MNK-45 cell line was analyzed by flow cytometry, si-RPL38#1 and #2 siRNA increased cells in the G0/G1 phase and prevented them into the S phase (P < 0.05) (Figure 2E). The early apoptotic signal of MNK-45 cells was detected by flow cytometry. As Figure 2F has shown, si-RPL38 induced apoptosis, and the proportion of living cells was significantly decreased. Taken together, RPL38 may participant the progress of GC (P < 0.01).
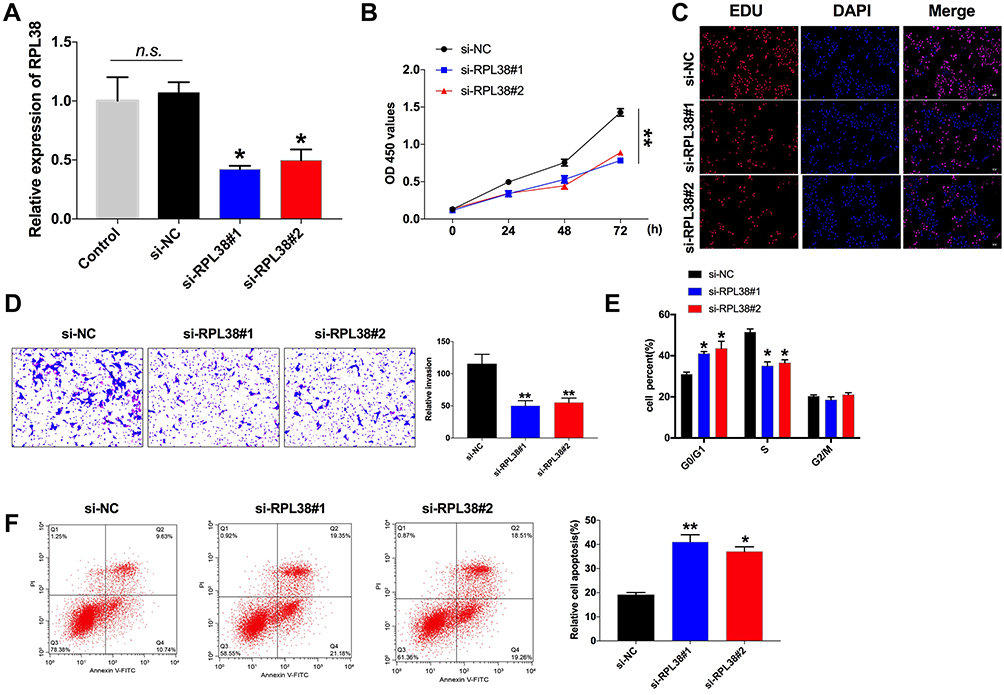 |
Figure 2 Knockdown of RPL38 inhibits proliferation, invasion and promote apoptosis of MKN-45 cells. (A) The knockdown efficiency of si-RPL38#1 and RPL38#2. (B and C) CCK8 and EdU assay were used to detect cell proliferation in each group. (D) Cell invasion ability was detected in each group. (E) The cell cycle was analyzed by flow cytometry. (F) Cell apoptosis was analyzed by flow cytometry. *P<0.05, **P<0.01. |
RPL38 Was Negatively Correlated with miR-374b-5p
By isolating the nuclear and cytoplasm protein, we found that RPL38 was mainly expressed in the cytoplasm (Figure 3A). Then, we predicted that there was the binding site of miR-374b-5p on RPL38 by starbase, the luciferase report verified the supposition that miR-374b-5p could bind with RPL38, RPL38-WT co-transfected with miR-374b-5p performed decreased luciferase activity compared with miR-NC, while, RPL38-mutant did not affect the luciferase activity with miR-374b-5p (P < 0.01) (Figure 3B). After transfecting miR-NC, miR-374b-5p mimics or miR-374b-5p inhibitor, we detected the expression of RPL38 by Western blot, miR-374b-5p mimics inhibited the level of RPL38, while co-transfected with miR-374b-5p inhibitor recovered the level of RPL38 (P < 0.05) (Figure 3C). Next, knockdown RPL38 could increase the expression of miR-374b-5p (P < 0.01) (Figure 3D).
 |
Figure 3 RPL38 could interact with miR-374b-5p. (A) The expression of RPL38 in nuclear and cytoplasm. (B) Targeting prediction results of miR-374b-5p and RPL38 (http://starbase.sysu.edu.cn/index.php). The results of luciferase assay. (C) The protein level of RPL38 after transfecting with miR-NC, miRNA or miRNA inhibitor. (D) The expression of miR-374b-5p was detected after knockdown RPL38. *P<0.05, **P<0.01. |
MiRNA-374-5b Binds with VEGF and Regulates AKT/ERK Signal Pathway
VEGF gene expression was regulated by many factors, among which hypoxia is the main regulation mode. VEGF is the target gene of hypoxia-inducible factor (HIF), and the inhibition of HIF1 obviously reduces the expression level of VEGF and inhibits tumor angiogenesis. The luciferase assay showed that VEGF could interact with miR-374b-5p (P < 0.01) (Figure 4A and B). The expression of VEGF was up-regulated after the transfection of miR-374b-5p mimic (P < 0.01), and down-regulated after the transfection of miR-374b-5p inhibitor (P < 0.05) (Figure 4C and D). Then, we detected the protein level of phosphorylation-ERK, ERK, phosphorylation-AKT and AKT, the results performed that miR-374b-5p inhibitor transfection could induce the phosphorylation of ERK and AKT (P < 0.05) (Figure 4D). The above results showed that miR-374b-5p could regulate the expression of VEGF, and control the ERK, AKT signal pathway.
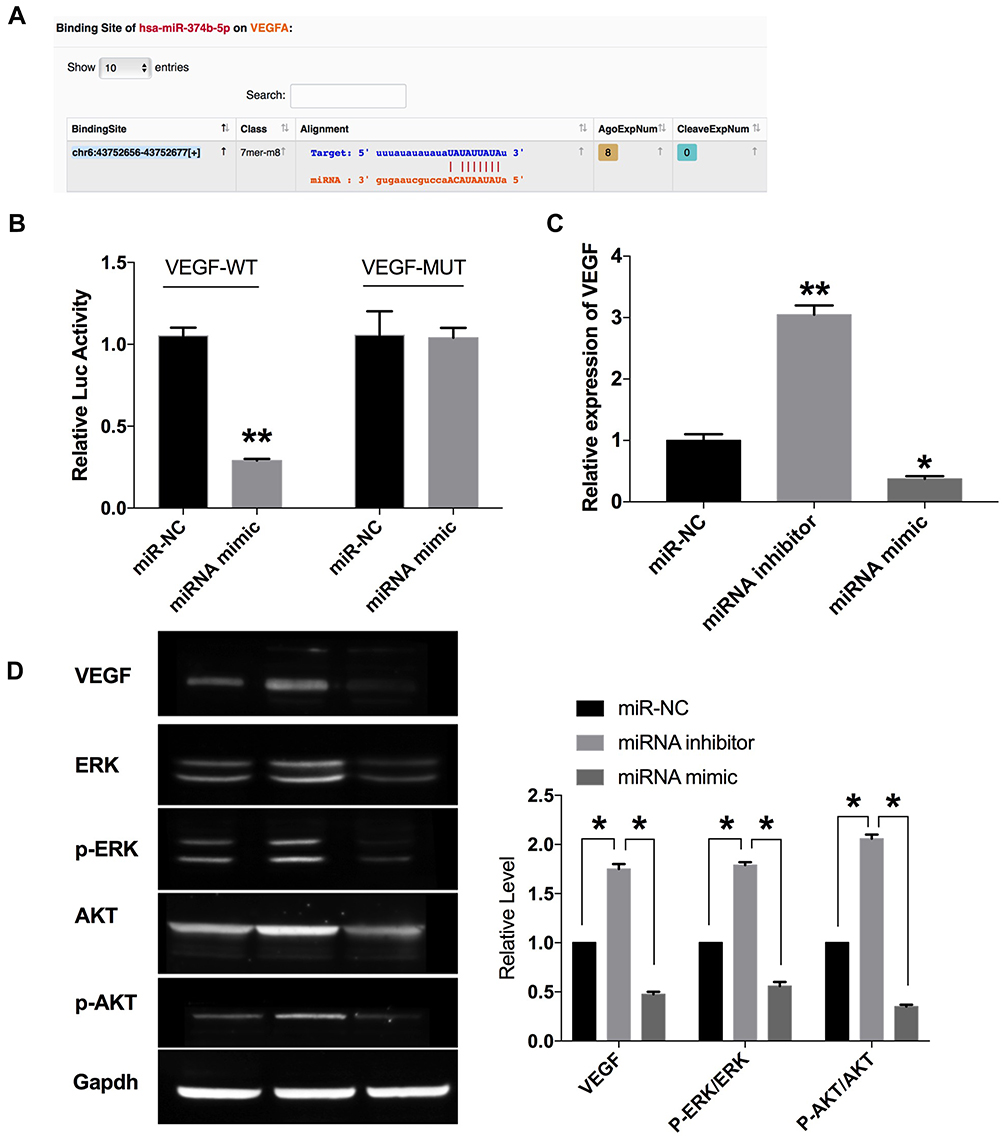 |
Figure 4 VEGF is a target of miR-374b-5p. (A) Targeting prediction results of miR-374b-5p and VEGF (http://starbase.sysu.edu.cn/index.php). (B) The luciferase report was performed to verify the correlation of miR-374b-5p and VEGF. (C) The expression of VEGF in each group. (D) The protein level of VEGF, ERK, p-ERK, AKT, p-AKT were detected by Western blot. *P<0.05, **P<0.01. |
Knockdown of RPL38 Attenuates Gastric Cancer Progress via Regulating miR-374b-5p and VEGF
Co-transfecting si-RPL38#1 with miR-374b-5p, we assessed the level of VEGF via qRT-PCR, si-RPL38#1 induced the downregulation of VEGF, while co-transfection with miR-374b-5p inhibitor recovered the level of VEGF (P < 0.05) (Figure 5A). Furthermore, the decreased level of VEGF induced by si-RPL38#1 was recovered by miR-374b-5p inhibitor, knockdown of RPL38 would activate phosphorylation of ERK and AKT, which were reversal by miR-374b-5p inhibitor (P < 0.05) (Figure 5B). Next, we found that miR-374b-5p inhibitor and VEGF could inhibit the anti-proliferation and anti-invasion (P < 0.01) function of si-RPL38#1 (Figure 5C–E); meanwhile, miR-374b-5p inhibitor and VEGF restored the number of G0/G1 and S cells (P < 0.05) (Figure 5F). Taken together, RPL38 regulated the progression of GC via controlling miR-374b-5p and VEGF.
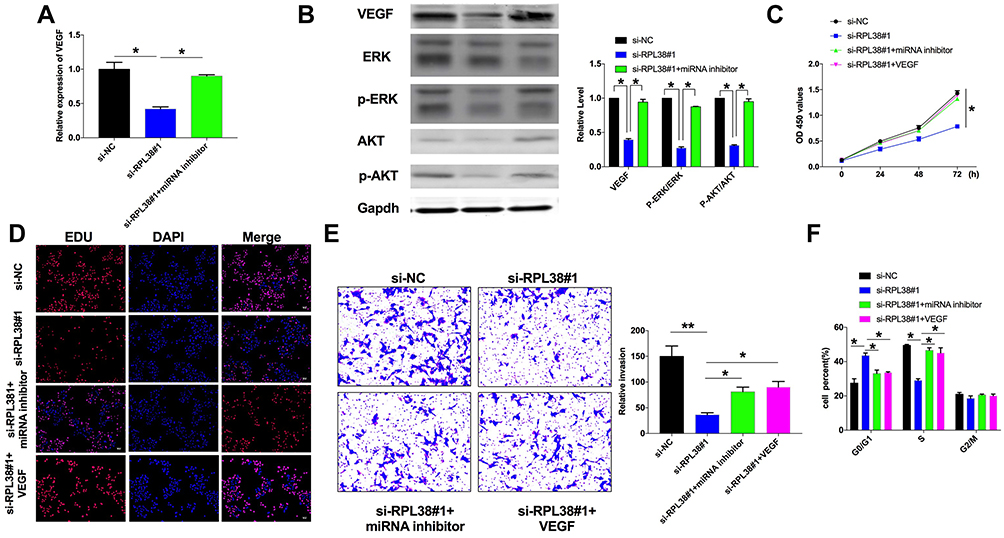 |
Figure 5 Knockdown of RPL38 can inhibit proliferation and invasion in MKN-45 by controlling miR-374b-5p and VEGF. (A) The expression of VEGF in each group. (B) The protein level of VEGF, ERK, p-ERK, AKT, p-AKT were detected by Western blot. (C and D). CCK8 and EdU assay were used to detect cell proliferation in each group. (E) Cell invasion ability was detected in each group. (F) The cell cycle was analyzed by flow cytometry. *P<0.05, **P<0.01. |
Knockdown of RPL38 Prevents Tumor Growth in vivo
To further explore the function of RPL38 in GC tumor, we set up a xenograft nude mice model. Thirty mice were divided into two groups randomly, AAV9-si-RPL38#1 or AAV9-si-NC were injected via the tail vein; after 1 week, MKN-45 cells were subcutaneously injected into nude mice, and we measured tumor volume. AAV9-si-RPL38#1 significantly reduced tumor volume (P < 0.05) and weight (P < 0.01) (Figure 6A–C). The tumor tissue was sectioned for immunohistochemical staining (Figure 6D). The Ki-67 staining performed that AAV9-si-RPL38#1 significantly inhibited the proliferation of tumor, which was consistent with the results in vitro. AAV9-si-RPL38#1 decreased the level of VEGF, p-ERK and p-AKT, accompanied with increased the expression of cleaved-Caspase3.
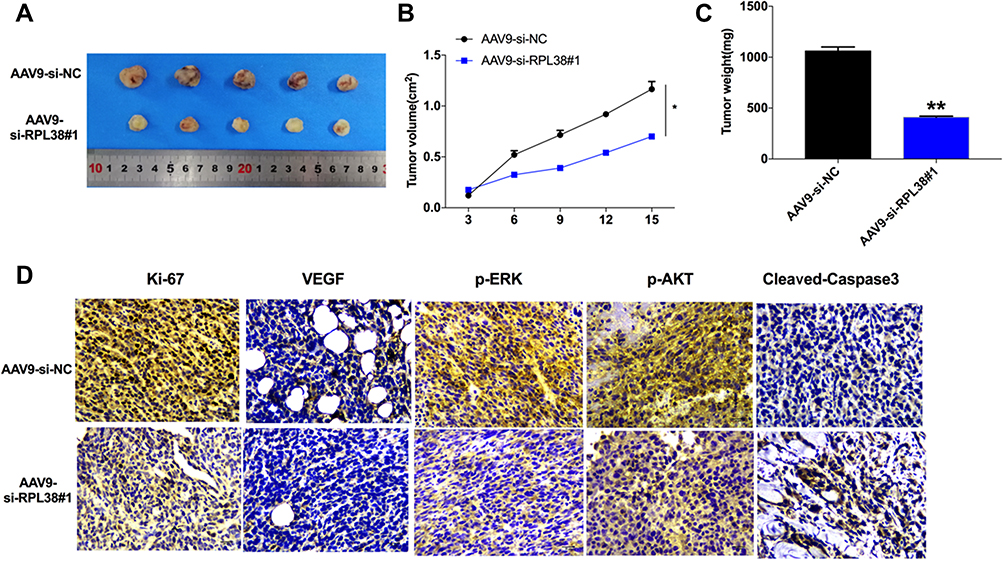 |
Figure 6 RPL38 regulates tumorigenesis in vivo. (A) Effects of RPL38 knockdown on the size of MKN-45 xenograft tumors in nude mice. (B and C) The significantly smaller average tumor volume and weights of the AAV9-si-RPL38 group are compared to AAV9-si-NC group. (D) Representative images of the IHC staining of ki-67, VEGF, p-ERK, p-AKT and cleaved-caspase3. **P<0.01. |
Discussion
At present, the diagnosis of gastric cancer depends on the examination of gastroscope, CEA, CA19-9, pathology and cancer tissue,18 and the diagnosis is clear but traumatic. The early detection, diagnosis and treatment of gastric cancer have always been the focus of gastric cancer research.19 Searching for effective markers is an important factor to improve the survival and prognosis of gastric cancer patients.20 Tumor cell metastasis is a very complicated process, including the transformation, proliferation, angiogenesis, invasion, transmission and survival of tumor cells in circulation, and subsequent adhesion and colonization of secondary organs or tissues.21 Therefore, we need to further explore the mechanism of gastric cancer, especially look for new gene variants associated with gastric cancer, which could provide a new index for early diagnosis of gastric cancer.
Ribosomes, the organelles that catalyze protein synthesis, consist of a small 40S subunit and a large 60S subunit. Together these subunits are composed of 4 RNA species and approximately 80 structurally distinct proteins. This gene encodes a ribosomal protein that is a component of the 60S subunit. Ribosome biogenesis is one of the most multifaceted and energy-demanding processes in biology, involving a large number of assembly and maturation factors.22,23 Ribosomal protein regulates the redistribution of cell cycle by regulating ribosomal protein, which blocks the cell cycle of tumor in G0~G1 phase and reduces the proportion of cells in G2/M phase. RNA polymerase I protease involved in ribosomal protein synthesis was used as a target to indirectly regulate ribosomal protein therapy, which showed significant anti-tumor activity in animal models. In patients with prostate cancer and ERB2-positive breast cancer, RPL19 levels were significantly increased.24 In early hepatocellular carcinoma, the overall survival rate of patients with high expression of RPL36 is higher, which suggests that RPL36 can be used as an index to evaluate the postoperative prognosis of patients with hepatocellular carcinoma.25 It is speculated that RP is involved in the regulation of tumor cell growth, proliferation and malignant transformation. RPL3 regulates cell cycle progression by regulating the expression of P21, which is a cyclin-dependent kinase inhibitor and an inhibitory protein that regulates cell cycle.26
RPL38 was cloned as one of the ribosomal proteins overexpressed in a colon cancer cell line, HF-29.27 Previous study shows that RPL38 is predominantly expressed in the normal ductal epithelium of the pancreas and each of the pancreatic cancer cell lines examined.9 In this study, we found RPL38 was mainly located in the cytoplasm, and knockdown of RPL38 prevented the proliferation and invasion of GC cells. Meanwhile, RPL38 interacted with miR-374b-5p. The main role of miRNA is to participate in the regulation of central genes, which can regulate the expression of protein and a variety of signal pathways affecting tumor growth, so its value as a target molecule for gastric cancer treatment is significantly higher than that of gene coding. Extracting peripheral venous blood from patients for miRNA expression level detection can be used as an important indicator for the diagnosis and prognosis of gastric cancer.
Angiogenesis in tumor growth and metastasis of the tumor has a leading role in the process, provides conditions for the rapid growth of cells, and further accelerate the occurrence of solid tumors, and turn to move. VEGF with endothelial cell surface-specific transmembrane receptor stimulates endothelial cell division and proliferation, thus promotes the growth of new blood vessels and promote intravascular material to the blood vessels outside exchange, basic conditions for the transfer of tumor cells. In this study, we found that VEGF is a potential target of miR-374b-5p, and positively correlated with RPL38 expression level. Meanwhile, RPL38 regulated the GC progression by regulating AKT/ERK signal pathway. The pathogenesis of gastric cancer was further revealed.
Conclusion
In conclusion, our results showed that RPL38 acted as a novel factor in the regulation of GC cell proliferation and invasion via miR-374b-5p/VEGF and controlling ERK/AKT signal pathway. In addition, there should be a more clinical experimental basis for the clinical detection effect of RPL38 on gastric cancer. On this basis, more prospective studies should be carried out to promote diagnosis and treatment, the prediction and the prognosis of gastric cancer, so as to benefit patients with gastric cancer.
Institutional Review Board Statement
The present study was supported by the Ethics Committee of Cangzhou Central Hospital and also has been carried out in accordance with the World Medical Association Declaration of Helsinki. The subjects had been informed of the objective. Certainly, written consents were signed by every subject in the present study. In addition, this animal study was approved by the Animal Clinical Committee of Cangzhou Central Hospital. Nude mice were euthanized and operated strictly in accordance with the Guidelines for the Care and Use of Laboratory Animals of the National Institutes of Health.
Disclosure
The authors declare that there are no conflicts of interest.
References
1. Gantuya B, Oyuntsetseg K, Bolor D, et al. Evaluation of serum markers for gastric cancer and its precursor diseases among high incidence and mortality rate of gastric cancer area. Gastric Cancer. 2019;22(1):104–112. doi:10.1007/s10120-018-0844-8
2. Kinoshita H, Hayakawa Y, Koike K. Metaplasia in the stomach-precursor of gastric cancer? Int J Mol Sci. 2017;18(10):2063. doi:10.3390/ijms18102063
3. Figueiredo C, Camargo MC, Leite M, Fuentes-Panana EM, Rabkin CS, Machado JC. Pathogenesis of gastric cancer: genetics and molecular classification. Curr Top Microbiol Immunol. 2017;400:
4. Francis R, Guo H, Streutker C, et al. Gastrointestinal transcription factors drive lineage-specific developmental programs in organ specification and cancer. Sci Adv. 2019;5(12):eaax8898. doi:10.1126/sciadv.aax8898
5. Takeda H, Rust AG, Ward JM, Yew CC, Jenkins NA, Copeland NG. Sleeping beauty transposon mutagenesis identifies genes that cooperate with mutant Smad4 in gastric cancer development. Proc Natl Acad Sci U S A. 2016;113(14):E2057–2065. doi:10.1073/pnas.1603223113
6. Zong L, Hattori N, Yoda Y, et al. Establishment of a DNA methylation marker to evaluate cancer cell fraction in gastric cancer. Gastric Cancer. 2016;19(2):361–369. doi:10.1007/s10120-015-0475-2
7. Xu H, Peng L, Shen M, Xia Y, Li Z, He N. Shiga-like toxin I exerts specific and potent anti-tumour efficacy against gastric cancer cell proliferation when driven by tumour-preferential Frizzled-7 promoter. Cell Prolif. 2019;52(3):e12607. doi:10.1111/cpr.12607
8. Park KJ, Cho SB, Park YL, et al. Prospero homeobox 1 mediates the progression of gastric cancer by inducing tumor cell proliferation and lymphangiogenesis. Gastric Cancer. 2017;20(1):104–115. doi:10.1007/s10120-015-0592-y
9. Sahin F, Qiu W, Wilentz RE, Iacobuzio-Donahue CA, Grosmark A, Su GH. RPL38, FOSL1, and UPP1 are predominantly expressed in the pancreatic ductal epithelium. Pancreas. 2005;30(2):158–167. doi:10.1097/01.mpa.0000151581.45156.e4
10. Zheng B, Peng J, Mollayup A, et al. Construction of a prognostic prediction system for pancreatic ductal adenocarcinoma to investigate the key prognostic genes. Mol Med Rep. 2018;17(1):216–224. doi:10.3892/mmr.2017.7850
11. Shin VY, Chu KM. MiRNA as potential biomarkers and therapeutic targets for gastric cancer. World J Gastroenterol. 2014;20(30):10432–10439. doi:10.3748/wjg.v20.i30.10432
12. Alessandrini L, Manchi M, De Re V, Dolcetti R, Canzonieri V. Proposed molecular and miRNA classification of gastric cancer. Int J Mol Sci. 2018;19(6):1683. doi:10.3390/ijms19061683
13. Vo JN, Cieslik M, Zhang Y, et al. The Landscape of Circular RNA in Cancer. Cell. 2019;176(4):869–881.e813.
14. Zhang J, Hou L, Liang R, et al. CircDLST promotes the tumorigenesis and metastasis of gastric cancer by sponging miR-502-5p and activating the NRAS/MEK1/ERK1/2 signaling. Mol Cancer. 2019;18(1):80. doi:10.1186/s12943-019-1015-1
15. Huang X, Li Z, Zhang Q, et al. Circular RNA AKT3 upregulates PIK3R1 to enhance cisplatin resistance in gastric cancer via miR-198 suppression. Mol Cancer. 2019;18(1):71. doi:10.1186/s12943-019-0969-3
16. Frezzetti D, Gallo M, Maiello MR, et al. VEGF as a potential target in lung cancer. Expert Opin Ther Targets. 2017;21(10):959–966. doi:10.1080/14728222.2017.1371137
17. Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to anti-angiogenic therapy in cancer-alterations to anti-VEGF pathway. Int J Mol Sci. 2018;19(4):1232. doi:10.3390/ijms19041232
18. Tan Z. Recent advances in the surgical treatment of advanced gastric cancer: a review. Med Sci Monit. 2019;25:
19. Yu G, Torres J, Hu N, et al. Molecular characterization of the human stomach microbiota in gastric cancer patients. Front Cell Infect Microbiol. 2017;7:302. doi:10.3389/fcimb.2017.00302
20. Ohi M, Toiyama Y, Omura Y, et al. Possibility of limited gastrectomy for early gastric cancer located in the upper third of the stomach, based on the distribution of sentinel node basins. Surg Today. 2019;49(6):529–535. doi:10.1007/s00595-019-1768-6
21. Lin C, He H, Liu H, et al. Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut. 2019;68(10):1764–1773. doi:10.1136/gutjnl-2018-316324
22. Derenzini M, Montanaro L, Trere D. Ribosome biogenesis and cancer. Acta Histochem. 2017;119(3):190–197. doi:10.1016/j.acthis.2017.01.009
23. Pelletier J, Thomas G, Volarevic S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat Rev Cancer. 2018;18(1):51–63. doi:10.1038/nrc.2017.104
24. Deisenroth C, Franklin DA, Zhang Y. The evolution of the ribosomal protein-MDM2-p53 pathway. Cold Spring Harb Perspect Med. 2016;6(12):a026138. doi:10.1101/cshperspect.a026138
25. Shenoy N, Kessel R, Bhagat TD, et al. Alterations in the ribosomal machinery in cancer and hematologic disorders. J Hematol Oncol. 2012;5:32.
26. Russo A, Russo G. Ribosomal proteins control or bypass p53 during nucleolar stress. Int J Mol Sci. 2017;18(1):140. doi:10.3390/ijms18010140
27. Espinosa L, Martín M, Nicolas A, Fabre M, Navarro E. Primary sequence of the human, lysine-rich, ribosomal protein RPL38 and detection of an unusual RPL38 processed pseudogene in the promoter region of the type-1 angiotensin II receptor gene. Biochimica Et Biophysica Acta. 1997;1354(1):58–64. doi:10.1016/S0167-4781(97)00124-3
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.