")
Back to Archived Journals » Journal of Neurorestoratology » Volume 2
Rotenone-induced nigrostriatal toxicity is reduced by methylene blue
Authors Abdel-Salam O , Omara E, Youness E, Khadrawy Y, Mohammed N, Sleem A
Received 4 December 2013
Accepted for publication 5 February 2014
Published 8 May 2014 Volume 2014:2 Pages 65—80
DOI https://doi.org/10.2147/JN.S49207
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Omar ME Abdel-Salam,1 Enayat A Omara,2 Eman R Youness,3 Yasser A Khadrawy,4 Nadia A Mohammed,3 Amany A Sleem5
1Department of Toxicology and Narcotics, 2Department of Pathology, 3Department of Medical Biochemistry, 4Department of Medical Physiology, 5Department of Pharmacology, National Research Centre, Cairo, Egypt
Abstract: This study investigated the effect of subcutaneously injected methylene blue on oxidative stress and nigrostriatal damage induced in the rat brain by systemic rotenone injection. Rotenone 1.5 mg/kg (injected subcutaneously three times per week) was given alone or in combination with methylene blue (5, 10, or 20 mg/kg subcutaneously daily) for 2 weeks. Brain concentrations of malondialdehyde, reduced glutathione, nitric oxide (nitrite), and acetylcholinesterase, paraoxonase 1 activity, and the antiapoptotic marker Bcl-2 (B-cell lymphoma 2) were determined. Histopathology, tyrosine hydroxylase immunoreactivity, tumor necrosis factor-alpha (TNF-α), and caspase-3 immunohistochemistry were also performed. Injection of rotenone resulted in increased oxidative stress in different regions of the brain. Substantial increases in malondialdehyde (by 59.6%–77.3%) and nitric oxide (by 43.7%–58.7%) were observed in the cortex, striatum, hippocampus, and medulla of rotenone-treated rats. Meanwhile, glutathione decreased by 24.3%–59.8% in these brain regions. Rotenone administration was associated with a significant decrease in paraoxonase 1 activity in the cerebral cortex, striatum, hippocampus, medulla, midbrain, and cerebellum (by 66%–73.6%). Further, acetylcholinesterase activity decreased by 44.4% in the cortex and Bcl-2 decreased in the striatum by 33.9% after rotenone injection. Rotenone induced a marked decrease in tyrosine hydroxylase immunoreactivity and increased both TNF-α and caspase-3 immunoreactivity in the striatum. These effects of rotenone were substantially reduced by coadministration of methylene blue, which resulted in significantly decreased malondialdehyde and nitrite and increased glutathione in different regions of the brain. In addition, there was increased paraoxonase 1 and acetylcholinesterase activity in response to treatment with methylene blue. Bcl-2 levels in the striatum increased significantly after the highest dose of methylene blue. Methylene blue also significantly decreased striatal expression of TNF-α and caspase-3 (apoptosis), and the degree of neuronal degeneration and gliosis in the striatum, substantia nigra, cortex, and hippocampus. Treatment with methylene blue resulted in an increased number of tyrosine hydroxylase-positive cells in the striatum, substantia nigra, and cerebral cortex, compared with the rotenone control group. The present findings suggest that treatment with methylene blue could prevent neuronal degeneration induced by rotenone injection in the rat brain. This effect may be mediated by inhibition of oxidative stress and apoptotic markers, and by enhancement of apoptotic markers and acetylcholinesterase activity.
Keywords: methylene blue, nigrostriatal, oxidative stress, rotenone
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that affects about 1% of the population over the age of 65 years.1,2 In PD, there is a selective degeneration and loss of pigmented neurons of the substantia nigra pars compacta of the midbrain.3 This results in motor impairment, such as rest tremor, bradykinesia, rigidity, and postural instability.4 The disease is largely sporadic, and only 5% of cases have an identified genetic origin.5,6 The exact cause of sporadic PD is not yet known, but a combination of genetic and environmental factors are likely to contribute to development of the disease.7,8 In this context, there is evidence to suggest the involvement of pesticides, such as rotenone, paraquat, or maneb, in contributing to the higher incidence of sporadic parkinsonism in rural populations.9,10
Oxidative stress has been strongly implicated in the pathophysiology of PD.11,12 Under physiological conditions, the body is continuously exposed to reactive oxygen metabolites and nitrogen species derived from such sources as the mitochondrial electron transport, xanthine oxidase, nicotinamide adenine dinucleotide phosphate oxidases, activated phagocytes, and nitric oxide synthases. Being highly unstable molecules with unpaired electrons, reactive oxygen metabolites (eg, the superoxide radical, hydrogen peroxide, and hydroxyl radical) can attack cellular macromolecules (membrane lipids, enzyme proteins, DNA) leading to cellular perturbations. The production of reactive oxygen metabolites and nitrogen species is counterbalanced by a number of enzymatic (eg, catalase, glutathione peroxidase, and superoxide dismutase) and nonenzymatic (eg, glutathione, ascorbic acid, carotenoids, and vitamin E) antioxidant mechanisms.13–15 Oxidative stress occurs when these antioxidant mechanisms are overwhelmed by free radicals which could result in potential cellular and tissue damage.15 Several studies have indicated the existence of elevated levels of oxidative stress in postmortem brain tissue from patients with Parkinson’s disease, eg, increased cholesterol lipid hydroperoxides in the substantia nigra16 and increased carbonyl levels in the substantia nigra, caudate nucleus, and putamen, as well as in other areas of the brain,17 intense cytoplasmic 8-hydroxyguanosine immunoreactivity in neurons of the substantia nigra,18 increased oxidative damage to several brain mitochondrial complex I proteins,19,20 and decreased glutathione levels in the substantia nigra.21
Methylthioninium chloride, also known as methylene blue, is used in humans in the treatment of methemoglobinemia and in cyanide poisoning.22,23 Methylene blue is a reduction-oxidation agent with potent antioxidant properties, that prevents formation of mitochondrial oxygen free radicals.24–26 The dye also acts as an enhancer of the electron transport chain, thereby promoting oxygen consumption.27,28 Methylene blue readily crosses the blood–brain barrier, resulting in high concentrations in the central nervous system.29 This dye has recently been a focus of interest in view of its possible role in the treatment of neurodegenerative disorders. Thus, methylene blue has been shown to increase neuronal survival and to decrease neurodegeneration in both in vitro and in vivo models of Huntington’s disease,30 and to inhibit Aβ oligomerization,31 reduce Aβ levels, and improve learning and memory deficits in a genetic mouse model of Alzheimer’s disease.32 It has also been reported to prevent retinal neurodegeneration induced in mice by intravitreal rotenone injection33 and to decrease spinal cord injury due to ischemia-reperfusion.34
The aim of the present study was therefore to investigate the effect of systemic administration of methylene blue on several oxidative stress and apoptotic markers and on acetylcholine esterase (AChE) activity in a model of Parkinson’s disease induced by rotenone in the rat.
Materials and methods
Animals
Male Sprague Dawley rats weighing 130–140 g were obtained from the animal house colony of the National Research Centre, Cairo, Egypt. Standard laboratory food and water were provided ad libitum. Animal procedures were performed in accordance with the ethics committee of the National Research Centre and followed the recommendations of the National Institutes of Health Guide for Care and Use of Laboratory Animals (Publication No 85-23, revised 1985). Equal groups containing ten rats each were used in all experiments. The doses of methylene blue used were based on the relevant literature.35–37
Drugs and chemicals
Rotenone and methylene blue were obtained from Sigma-Aldrich (St Louis, MO, USA). Rotenone was dissolved in 100% dimethyl sulfoxide. Methylene blue was dissolved in saline to obtain the necessary doses. Anti-tyrosine hydroxylase antibody, anti-caspase-3 antibody, and anti-tumor necrosis factor-alpha (TNF-α) antibody kits (Abcam, Cambridge, UK), an avidin-biotin complex kit (Vector Laboratories, Burlingame, CA, USA), a biotinylated peroxidase-based universal kit (Dako, Carpinteria, CA, USA), and 3,3′-diaminobenzidine (Sigma-Aldrich) were used. All other chemicals used were obtained from Sigma-Aldrich.
Experimental design
Two cohorts of rats were used; the first cohort for biochemical assays and the second for histopathology and immunohistochemistry. For the biochemical assays, the rats were randomly divided into five groups, with six rats in each group. Group 1 received the vehicle (dimethyl sulfoxide) daily; group 2 received a subcutaneous injection of rotenone 1.5 mg/kg three times per week; groups 3, 4, and 5 received rotenone 1.5 mg/kg subcutaneously three times per week in combination with methylene blue (5, 10, or 20 mg/kg subcutaneously daily, respectively). The injection volume was 0.2 mL per rat. All animals received the treatments for 2 weeks and were then euthanized under ether anesthesia for tissue collection. The brains were quickly dissected out on an ice-cold plate into different areas (cerebral cortex, striatum, hippocampus, medulla, midbrain, and cerebellum), washed with ice-cold phosphate-buffered saline (pH 7.4), weighed, and stored at −80°C until the biochemical analyses. The tissues were homogenized with 0.1 M phosphate-buffered saline (pH 7.4) to give a final concentration of 10% weight/volume (w/v) for the biochemical assays. A separate cohort of rats were administered dimethyl sulfoxide, rotenone, or rotenone and methylene blue (n=6 per group), and were euthanized 2 weeks after starting treatment for histopathology and immunohistochemistry.
Biochemical analysis
Determination of lipid peroxidation
Lipid peroxidation was assayed by measuring the level of malondialdehyde in brain tissues. Malondialdehyde was determined by measuring thiobarbituric acid-reactive species according to the method devised by Ruiz-Larrea et al38 in which thiobarbituric acid-reactive substances react with thiobarbituric acid to produce a red-colored complex having peak absorbance at 532 nm. In brief, 2.25 mL of working reagent (one volume of 0.8 g thiobarbituric acid dissolved in 100 mL of 10% perchloric acid and three volumes of 20% trichloroacetic acid) were added to 0.25 mL of sample, incubated for 20 minutes in a boiling water bath, and then left to cool at room temperature before centrifugation at 3,000 rpm for 5 minutes at 0°C. The pink color was measured using a ultraviolet (UV)-VI8 recording spectrophotometer (Shimadzu Corporation, Rydalmere, Australia) at a 532 nm wavelength against the blank solution, which was prepared by addition of 0.25 mL of distilled water to 2.25 mL of working reagent.
Determination of reduced glutathione
Reduced glutathione was determined in the supernatants by Ellman’s method.39 This procedure is based on the reduction of Ellman’s reagent by –SH groups of glutathione to form 2-nitro-s-mercaptobenzoic acid; the nitromercaptobenzoic acid anion has an intense yellow color which can be determined spectrophotometrically using a UV-VI8 recording spectrophotometer. The reduced glutathione concentration was calculated by comparison with a standard curve.
Determination of nitric oxide
Nitric oxide measured as the nitrite was determined using Griess reagent, according to the method devised by Moshage et al,40 which is based on measurement of endogenous nitrite concentration as an indicator of nitric oxide production. It depends on the addition of Griess reagent which converts nitrite into a deep purple azo compound, the absorbance of which is read at 540 nm.
Determination of paraoxonase activity
The arylesterase activity of paraoxonase was measured spectrophotometrically in the supernatants using phenylacetate as a substrate.41,42 In this assay; arylesterase/paraoxonase catalyzes the cleavage of phenyl acetate, resulting in formation of phenol. The rate of formation of phenol is measured by monitoring the increase in absorbance at 270 nm at 25°C. The working reagent consists of 20 mM Tris/HCl buffer (pH 8.0) containing 1 mM CaCl2 and 4 mM phenyl acetate as the substrate. Samples diluted 1:3 in buffer were added and the change in absorbance was recorded following a 20-second lag time. Absorbance at 270 nm was taken every 15 seconds for 120 seconds. One unit of arylesterase activity is equal to 1 μM of phenol formed per minute. The activity is expressed in kU/L, based on the extinction coefficient of phenol of 1,310 M−1 cm−1 at 270 nm, pH 8.0, and 25°C. Blank samples containing water are used to correct for the spontaneous hydrolysis of phenylacetate.
Determination of Bcl-2
The level of human B-cell leukemia/lymphoma 2 (Bcl-2) in striatal tissue was determined by double-antibody sandwich enzyme-linked immunosorbent assay kit according to the manufacturer’s instructions (Glory Science Co, Ltd, Del Rio, TX, USA).
Determination of acetylcholinesterase activity
The procedure used to determine acetylcholinesterase activity was a modification of the method of Ellman et al43 as described by Gorun et al.44 The principle of the method involves measurement of the thiocholine produced as acetylthiocholine is hydrolyzed. The color was read immediately at 412 nm. The following reagents were pipetted in a cuvette: 0.14 mL of phosphate buffer 20 mM (pH 7.6), 0.05 mL of 5 mM acetylthiocholine iodide, and 0.01 mL of tissue homogenate. After 10 minutes of incubation at 38°C, the reaction was stopped with 1.8 mL of 5,5′-dithiobis-2-nitrobenzoic acid (DTNB)-phosphate ethanol reagent. The DTNB-phosphate ethanol reagent was prepared by dissolving 12.4 mg of DTNB in 120 mL of 96% ethanol, 80 mL of distilled water, and 50 mL of 0.1 mM phosphate buffer (pH 7.6). Glutathione 2.5 mM was used as the standard.
Histopathology
Brain sections were fixed in freshly prepared 10% neutral buffered formalin, processed routinely, and embedded in paraffin. Paraffin sections 4 μm thick were prepared and stained with hematoxylin and eosin for histopathological examination. Sections were examined using a light microscope.
Histomorphometric quantification
Further histopathological evaluation was done using quantitative morphometric analysis of the pathological changes. The amount of brain tissue affected by damaged neurons with included nuclei that were rather intensely stained, cytoplasmic vacuolation scarcely separated from surrounding cytoplasm, shrunken and neuronal atrophy (damaged area) were determined using a computer-assisted automated image analyzer. A Leica QWin image processing and analysis system (Cambridge, UK, Image Analyzer Unit; Pathology Department, National Research Center, Cairo, Egypt) was used for interactive automatic measurement of the percentage of damaged areas on slides stained with hematoxylin and eosin by analyzing ten random fields per slide.
Immunohistochemistry for tyrosine hydroxylase
Sections were immunostained for tyrosine hydroxylase at room temperature with a biotinylated peroxidase-based kit. The same sets of tissue were used for hematoxylin and eosin staining and for tyrosine hydroxylase (n=6 per group). Deparaffinized tissue sections were rinsed in phosphate-buffered saline for 2.5 minutes and then placed in 0.3% H2O2 for 30 minutes to reduce endogenous peroxidase activity. The tissue was then rinsed in phosphate-buffered saline for 2.5 minutes and blocked in a solution of phosphate-buffered saline containing 3% horse serum and 0.1% Triton for 60 minutes. Without rinsing, the tissue sections were incubated in blocking solution that also contained a 1:20,000 dilution of monoclonal mouse tyrosine hydroxylase antibody for 60 minutes. Tissue was rinsed for 2.5 minutes with phosphate-buffered saline and incubated with the blocking solution that included a 1:300 dilution of biotinylated anti-mouse antibody made in a horse. After rinsing in phosphate-buffered saline for 2.5 minutes, slices were incubated with avidin-peroxidase reagent diluted as instructed by the manufacturer. Sections were transferred to a 50 mM Tris-buffered 0.9% saline solution, rinsed, treated with 3,3′-diaminobenzidine and a final rinse in phosphate-buffered saline for 2 minutes, and then counterstained with hematoxylin and eosin. Sections were mounted on slides, allowed to dry in air for 24 hours, dehydrated with a series of ethanol rinses, and then cleared with xylene. Sections were cover-slipped after at least 24 hours of drying in air.
Immunohistochemical assessment of TNF-α and caspase-3
Immunohistochemical staining of TNF-α and anti-caspase-3 antibody in the cortex and striatum was performed with 4 μm thick sections that were deparaffinized and incubated with fresh 0.3% H2O2 in methanol for 30 minutes at room temperature. Briefly, deparaffinized brain slides were incubated with the antibodies against TNF-α diluted 1:50 and cleaved caspase-3 diluted 1:100. Positive cells were then determined with streptavidin biotin-peroxidase secondary antibody (Dako). The antibody binding sites were visualized with 3,3′-diaminobenzidine. The sections were then counterstained with hematoxylin, dehydrated using graded alcohols and xylene, and mounted. The immunostaining intensity and cellular localization of TNF-α and cleaved caspase-3 were analyzed by light microscopy.
Image analysis for quantitative immunohistochemistry
Optical density measurements of tyrosine hydroxylase, TNF-α, and caspase-3 immunoreactivity were determined using the computer-assisted image analysis system composed of a high precision illuminator, a digital camera, and a computer with specific image analysis software. The mean optical density of each region was measured bilaterally in selected brain regions using consecutive sections from each rat. The degree of reaction was chosen by the color detect menu, the areas of reactivity were masked by binary color, and area was measured using an objective lens with a magnification of 40× and an eye lens with a magnification of 10× (total magnification 400×). A total of ten measurements were taken per region by an investigator blinded to experimental group allocation. These measurements were averaged to obtain one mean per region for each animal.
Statistical analysis
The data are expressed as the mean ± standard error of the mean. The data were analyzed by one-way analysis of variance followed by Tukey’s test, using SPSS software (SPSS Inc., Chicago, IL, USA). A P-value of less than 0.05 was considered to be statistically significant.
Results
Biochemical results
Malondialdehyde
Following injection of rotenone, there was a significant increase in lipid peroxidation, as assessed by malondialdehyde levels, with significantly (P<0.05) increased levels in the cerebral cortex (68.2%), striatum (59.6%), hippocampus (70.4%), and medulla (77.3%). Administration of methylene blue resulted in a dose-dependent decrease in malondialdehyde in these brain areas. In the cortex and striatum, malondialdehyde decreased by 15.4%–29.2% and by 17.8%–28%, respectively, after administration of 5–20 mg/kg methylene blue. Malondialdehyde levels decreased in the hippocampus by 18.5% and 44.6% after methylene blue doses of 10 mg/kg and 20 mg/kg, respectively. Malondialdehyde levels also decreased in the medulla by 43.3% after methylene blue 20 mg/kg (Table 1).
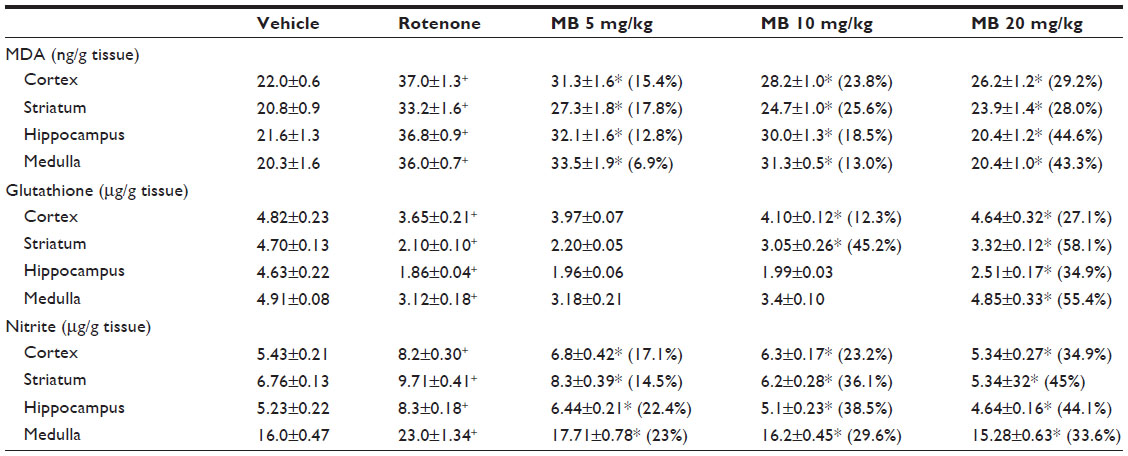 | Table 1 Effect of methylene blue on malondialdehyde, reduced glutathione, and nitric oxide (nitrite) in different brain regions after systemic administration of rotenone |
Reduced glutathione
Injection of rotenone resulted in a significant decrease in glutathione concentration in the cerebral cortex (−24.3%), striatum (−55.3%), hippocampus (−59.8%), and medulla (−36.4%). In rotenone-treated rats, a dose-dependent increase in glutathione concentration was found in the cerebral cortex (12.3%–27.1%) and in the striatum (45.2%–58.1%) after subcutaneous injection of methylene blue 10 mg/kg and 20 mg/kg, respectively. A 43.3% increase in glutathione concentration was observed in the medulla after administration of methylene blue 20 mg/kg (Table 1).
Nitric oxide
In rotenone-treated rats, a significant increase in nitric oxide (measured as nitrite) levels was observed in the cerebral cortex (51%), striatum (43.6%), hippocampus (58.7%), and medulla (43.7%). Following treatment with methylene blue 5–20 mg/kg, a dose-dependent decrease in nitrite concentration was found in the cerebral cortex (17.1%–34.9%), striatum (14.5%–45%), hippocampus (22.4%–44.1%), and medulla (23%–33.6%), (Table 1).
Paraoxonase activity
Rotenone produced a significant decrease in paraoxonase 1 activity in the cerebral cortex (−70%), striatum (−71.6%), hippocampus (−80.7%), medulla (−66%), midbrain (−73.6%), and cerebellum (−67.4%). Methylene blue at 10 mg/kg and 20 mg/kg resulted in increased paraoxonase 1 activity in the cortex (21%–34%), striatum (21.1%–57.7%), hippocampus (72.2%–334.4%), medulla (17.4%–25.8%), midbrain (24%–129.3%), and in cerebellum (21.3%–144%), (Table 2).
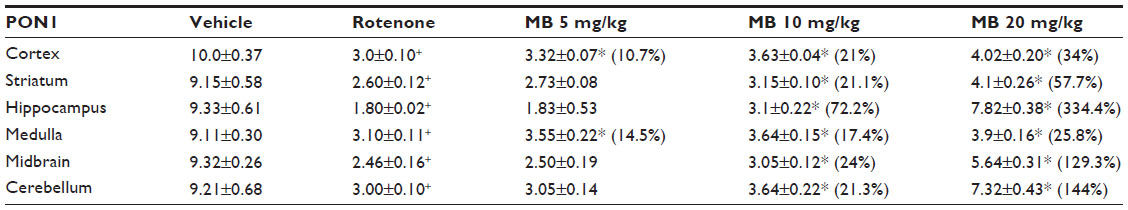 | Table 2 Effect of methylene blue on paraoxonase 1 activity in different brain regions after systemic administration |
Bcl-2
Striatal Bcl-2 concentration showed a significant decrease (−33.9% of control value) after injection of rotenone. Administration of methylene blue 5 mg/kg or 10 mg/kg had no significant effect on the concentration of Bcl-2 in the striatum compared with the rotenone only group. A significant (P<0.05) increase in the Bcl-2 concentration of 17.7% was observed after treatment with methylene blue 20 mg/kg as compared with the rotenone only group. However, Bcl-2 in rats treated with methylene blue 20 mg/kg was markedly and significantly decreased compared with the control group receiving vehicle only (Figure 1).
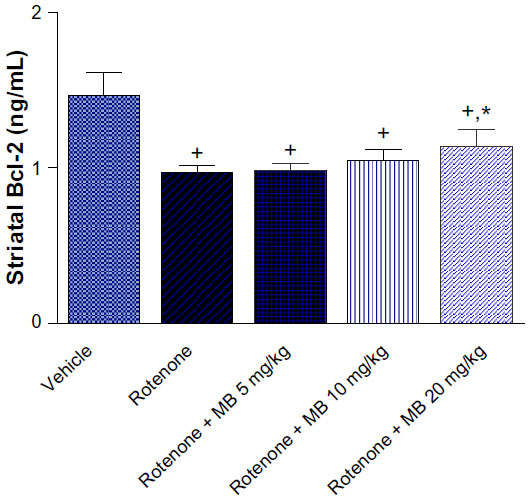 | Figure 1 Effect of methylene blue on Bcl-2 in striatal tissue from rotenone-treated rats. |
Acetylcholinesterase activity
AChE activity in the cortex was significantly decreased by 44.4% after rotenone injection. Treatment with methylene blue 5, 10, and 20 mg/kg resulted in a significant and dose-dependent increase in AChE activity by 47.6%, 73.2%, and 85.4%, respectively, compared with the rotenone only group (Figure 2).
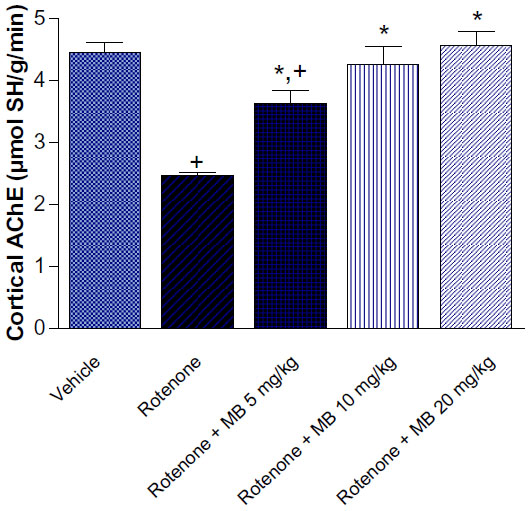 | Figure 2 Effect of methylene blue on AChE activity in the cortex of rotenone-treated rats. |
Histopathology results
Cerebral cortex
Hematoxylin and eosin-stained sections of the cerebral cortex from the control group showed a normal appearance, with neurons having either single or double open-face nuclei with prominent nucleoli surrounded with basophilic cytoplasm. Astrocytes with sharply demarcated nuclei were seen (Figure 3A). Sections from rotenone-treated rats revealed degenerated neurons that appeared shrunken with dark cytoplasm and pyknotic nuclei (Figure 3B). Necrotic astrocytes and gliosis were also seen (Figure 3C). Sections from rotenone-treated rats given methylene blue 20 mg/kg contained almost normally appearing neuronal cells, except for pyknosis in some neurons (Figure 3D).
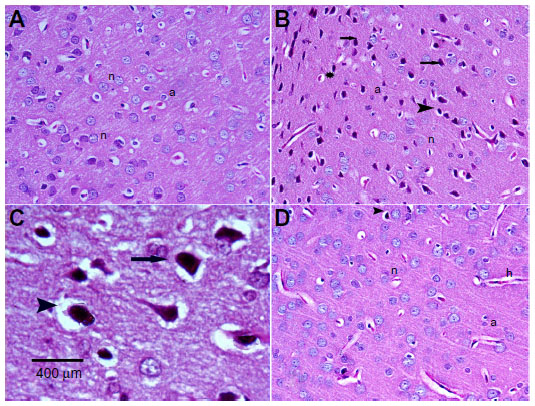 | Figure 3 Representative light microphotographs of hematoxylin and eosin-stained sections from the cortical tissue of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B–C), or rotenone and 20 mg/kg methylene blue (D). |
Striatum
Figure 4A represents hematoxylin and eosin-stained sections of the striatum from control rats and they have a normal appearance. Rotenone injection resulted in neurodegenerative changes with pyknotic and apoptotic nuclei, neuropil vacuolation, and congested vascular channels (Figure 4B and C). Fewer degenerative changes were observed after treatment with methylene blue 20 mg/kg (Figure 4D).
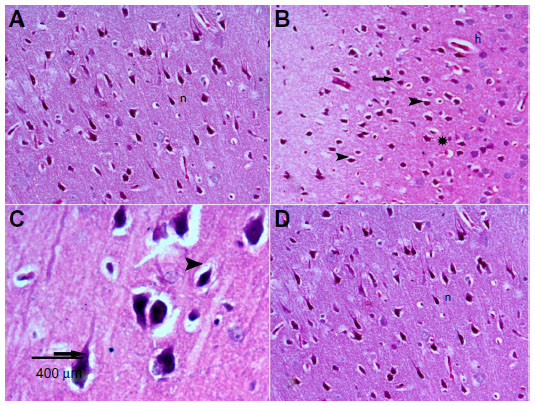 | Figure 4 Representative light microphotographs of hematoxylin and eosin-stained sections from the striatal tissue of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B–C), or rotenone and 20 mg/kg methylene blue (D). |
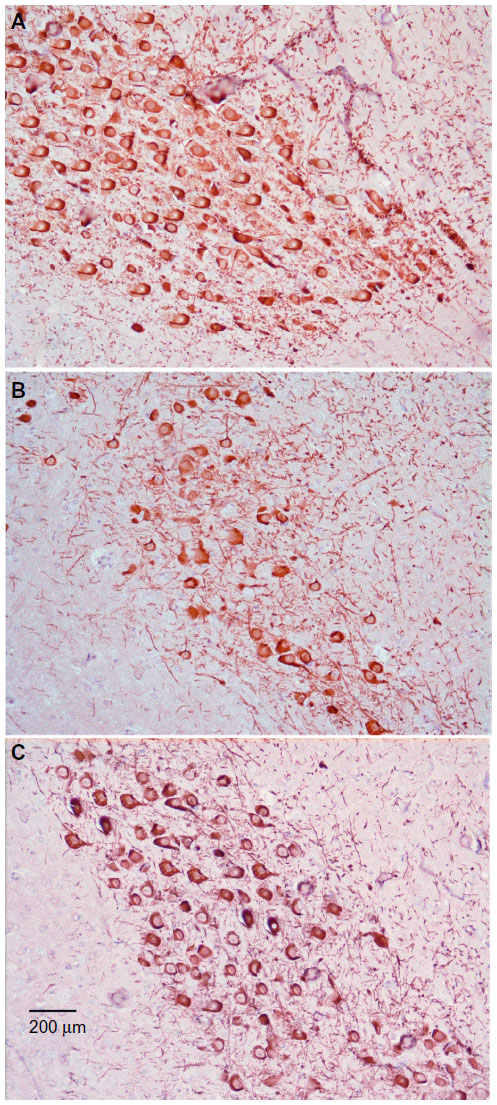
Substantia nigra
Sections from control rats showed normal substantia nigra neurons with normal nuclei. The cytoplasm of these cells was basophilic (Figure 5A). The substantia nigra of rotenone-treated rats showed marked neuronal degeneration. Neurons were reduced in number and stained darkly (Figure 5B and C). Administration of methylene blue 20 mg/kg was associated with a lesser degree of neuronal degeneration, with few cells having pyknotic nuclei (Figure 5D).
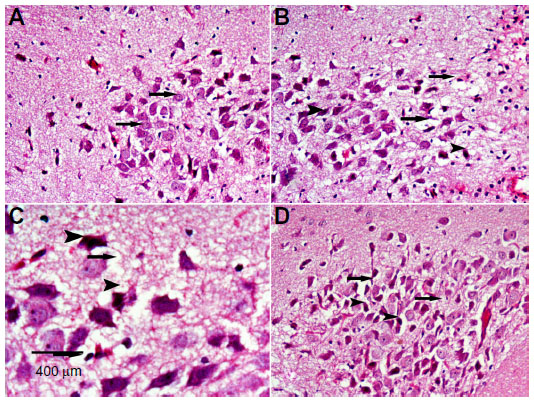 | Figure 5 Representative light microphotographs of hematoxylin and eosin-stained sections from the substantia nigra of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B–C), or rotenone and 20 mg/kg methylene blue (D). |
Hippocampus
The hippocampal region in control rats showed normal cellular composition in all layers (molecular, Purkinje, and granular, Figure 6A). Following rotenone injection, hippocampal neuronal cells showed fewer histopathological changes compared with those seen in the cortex and striatum. The hippocampus showed neuronal cell loss with mild degeneration when compared with controls. Shrunken apoptotic cells were observed, as well as degenerated and necrotic cells. Neuronal cells with dense pyknotic nuclei were also seen (Figure 6B and C). Methylene blue 20 mg/kg decreased this neuronal degeneration, with only small neuronal loss in the hippocampus being seen as compared with the rotenone only group (Figure 6D).
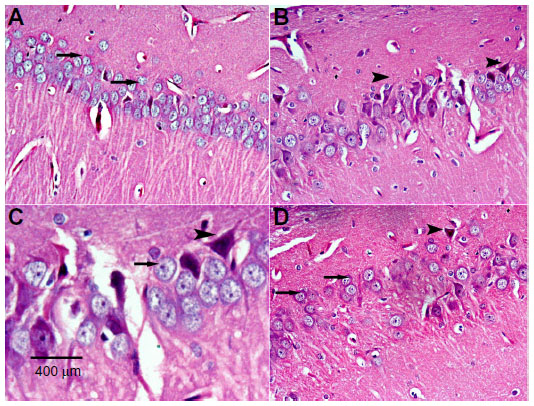 | Figure 6 Representative light microphotographs of hematoxylin and eosin-stained sections from the hippocampus of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B–C), or rotenone and 20 mg/kg methylene blue (D). |
Histomorphometric results
Histomorphometric analysis to determine the extent of damage in the different brain regions (cortex, substantia nigra, striatum, and hippocampus) quantitatively ascertained that the area of damage was significantly greater in the rotenone group than in the control group. Treatment with methylene blue 20 mg/kg resulted in a significant reduction of tissue damage in these regions (Table 3).
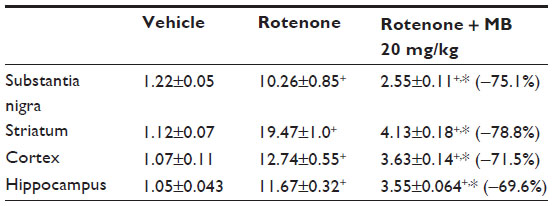 | Table 3 Effect of methylene blue on the area of damage in different brain regions in rats treated with rotenone |
Immunohistochemical results
Tyrosine hydroxylase
Immunostaining for tyrosine hydroxylase indicated that administration of rotenone decreased the number of tyrosine hydroxylase-immunostained neurons in the substantia nigra, striatum, and cortex as compared with controls (Figures 7–12). Optical density measurements of tyrosine hydroxylase-positive cells in the substantia nigra, striatum, and cortex of rats treated with rotenone decreased by −25%, −43.8%, and −46.8%, respectively, compared with their respective control values. Methylene blue 20 mg/kg markedly increased tyrosine hydroxylase immunostaining in these areas as compared with the rotenone only group (Figures 7C, 8D, 9C, 10D, 11C, 12D and Table 4).
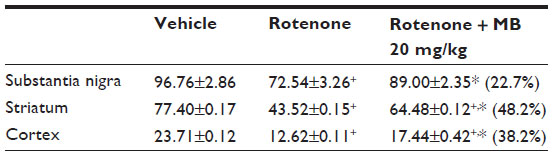 | Table 4 Effect of methylene blue on optical density measurements of tyrosine hydroxylase immunoreactivity in the substantia nigra, striatum, and cortex of rats treated with rotenone |
 | Figure 7 Representative light microphotographs of TH expression in the substantia nigra of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B), or rotenone and 20 mg/kg methylene blue (C). |
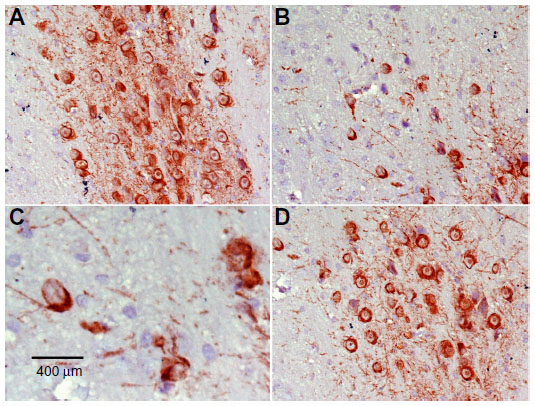 | Figure 8 Representative light microphotographs of tyrosine hydroxylase expression in the substantia nigra of rats treated with (A)vehicle (dimethyl sulfoxide), (B–C) rotenone, or (D) rotenone and 20 mg/kg methylene blue. |
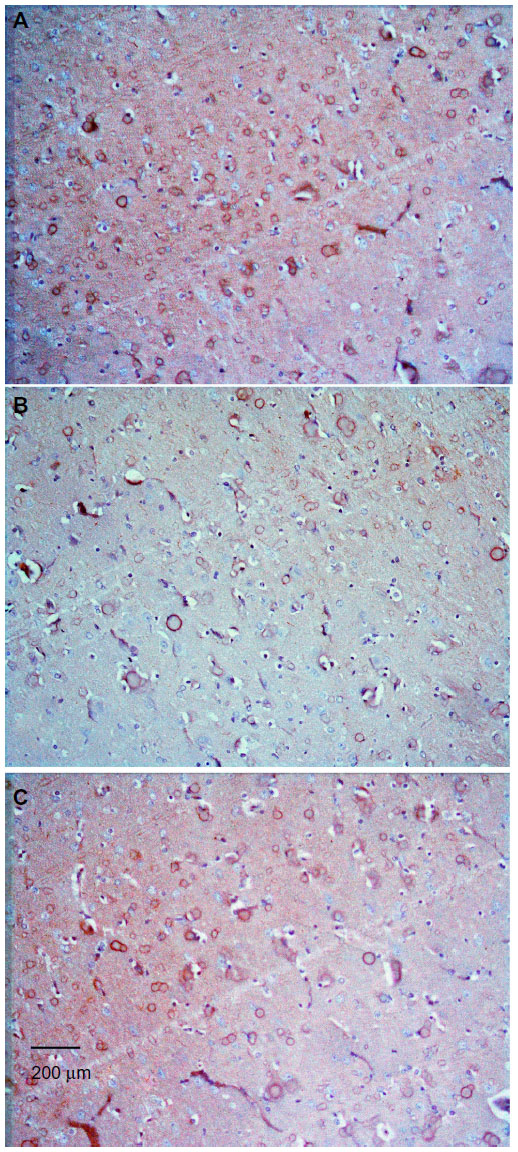
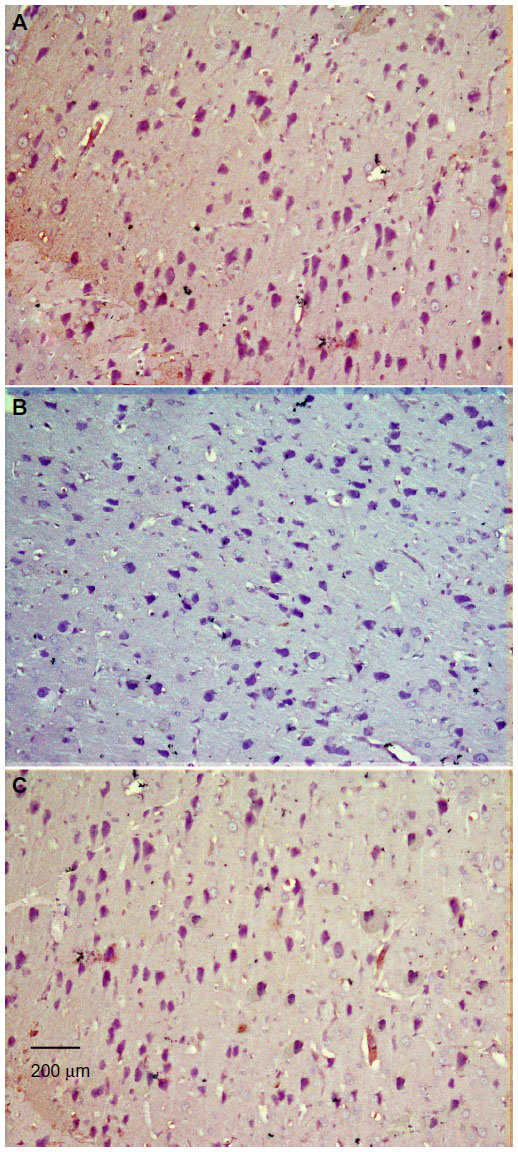 | Figure 9 Representative light microphotographs of TH expression in the striatum of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B), or rotenone and 20 mg/kg methylene blue (C). |
 | Figure 10 Representative light microphotographs of tyrosine hydroxylase expression in the striatum of rats treated with (A) vehicle (dimethyl sulfoxide), (B–C) rotenone, or (D) rotenone and 20 mg/kg methylene blue. |
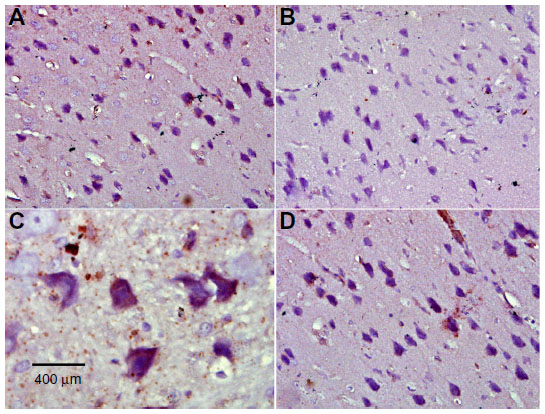
 | Figure 11 Representative light microphotographs of TH expression in the cortex of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B), or rotenone and 20 mg/kg methylene blue (C). |
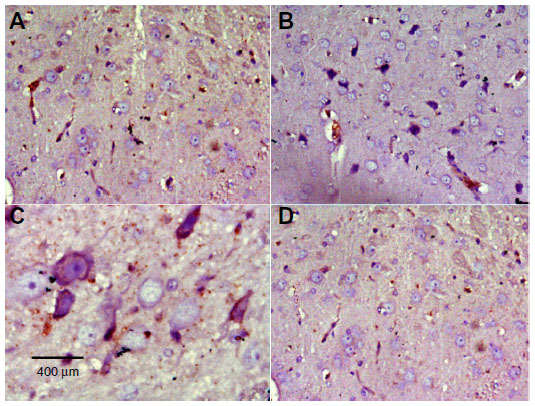 | Figure 12 Representative light microphotographs of tyrosine hydroxylase expression in the cortex of rats treated with (A) vehicle (dimethyl sulfoxide), (B–C) rotenone or (D) rotenone and 20 mg/kg methylene blue. |
Tumor necrosis factor-alpha
No TNF-α immunostaining was detected in the cortex or striatum in the control group (Figures 13A and 14A). Marked TNF-α expression was detected in the inflammatory cells for the group treated with rotenone (Figures 13B, 13C, 14B and 14C). Methylene blue 20 mg/kg attenuated the expression of TNF-α when compared with the rotenone only group (Figures 13D and 14D). Most TNF-α-positive cells had a shrunken and dark-stained morphology on hematoxylin and eosin staining. Table 5 shows the effect of methylene blue on the optical density measurements of TNF-α-positive cells in the striatum and cortex of rotenone-treated rats.
 | Table 5 Effect of methylene blue on the optical density measurements of TNF-α immunoreactivity in the striatum and cortex of rats treated with rotenone |
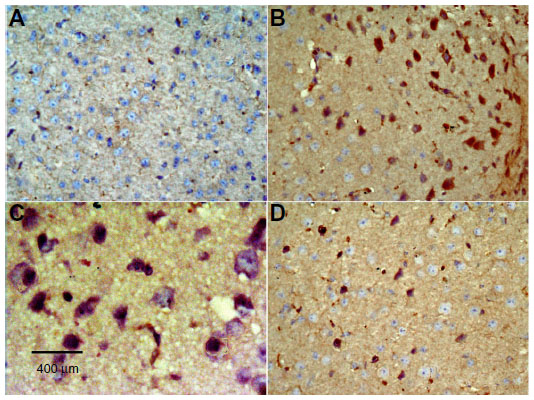 | Figure 13 Representative light microphotographs of TNF-α immunoreactivity in the cortex of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B–C), or rotenone and 20 mg/kg methylene blue (D). |
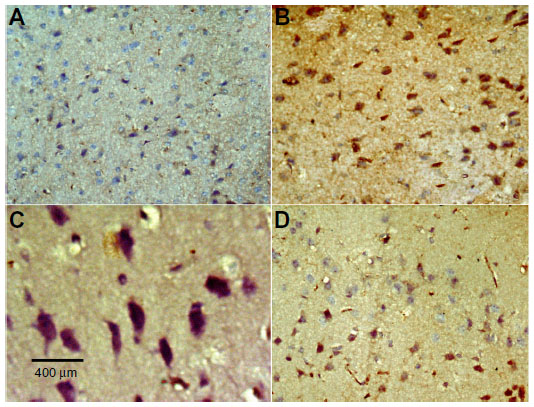 | Figure 14 Representative light microphotographs showing TNF-α immunoreactivity in the striatum of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B–C), or rotenone and 20 mg/kg methylene blue (D). |
Caspase-3
Caspase-3 expression was undetectable in the cortex and striatum of control brains (Figures 15A and 16A). In the rotenone only group, strong caspase-3 immunostaining was observed in both the cortex and striatum (Figure 16B and 16C). Sections from rats treated with rotenone and methylene blue 20 mg/kg showed very few caspase-3-positive cells (Figures 15D and 16D). Most caspase-3-positive cells had a shrunken and dark-stained morphology on hematoxylin and eosin staining. Table 6 shows the effect of methylene blue on optical density measurements of caspase-3-positive cells in the striatum and cortex of rotenone-treated rats.
 | Table 6 Effect of methylene blue on optical density measurements of caspase-3 immunoreactivity in the striatum and cortex of rats treated with rotenone |
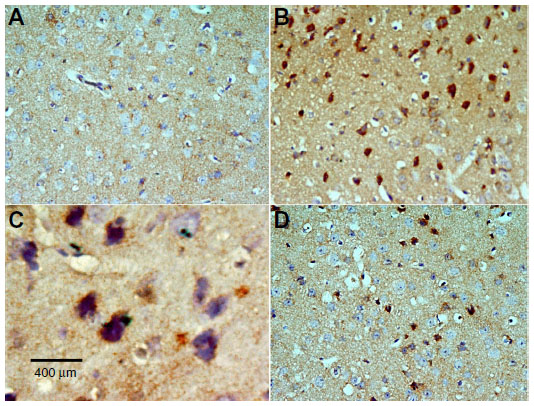 | Figure 15 Representative light microphotographs of caspase immunoreactivity in the cortex of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B–C), or rotenone and 20 mg/kg methylene blue (D). |
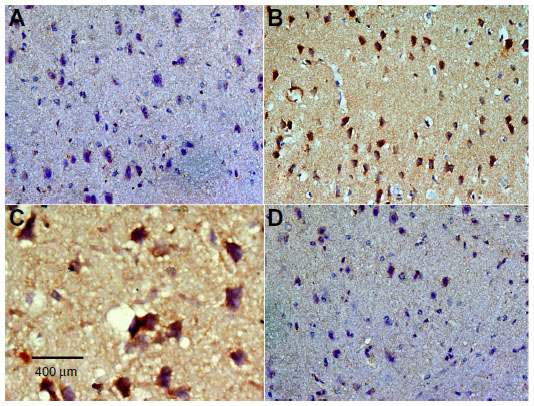 | Figure 16 Representative light microphotographs showing caspase immunoreactivity in the striatum of rats treated with vehicle (dimethyl sulfoxide) (A), rotenone (B–C), or rotenone and 20 mg/kg methylene blue (D). |
Discussion
The findings of the present study provide evidence that treatment with methylene blue can decrease neurodegeneration evoked by rotenone, a pesticide and complex I inhibitor, in rats. Systemic administration of this pesticide in rodents has been widely used as a model of human PD. In these studies, many of the pathological features could be replicated, namely dopaminergic neurodegeneration,45 α-synucleinopathy,46 and motor dysfunction.47 It has also been shown that the neurotoxicity induced by rotenone involves oxidative stress. In mice, subcutaneous rotenone increased generation of reactive oxygen species in the forebrain and midbrain regions.48 Immunoreactivity of acrolein, a product of lipid peroxides, increased in the brains of rotenone-treated mice.49 In rodent midbrain culture, rotenone increased protein carbonyl levels, indicative of oxidative damage to proteins.50 Rotenone also causes microglial activation.51–53 Once activated by rotenone, microglia show increased production of reactive oxygen species, particularly HOCl.53 Nicotinamide adenine dinucleotide phosphate oxidase-derived superoxide might be a factor mediating this microglia-enhanced rotenone neurotoxicity.52
In the present study, injection of rotenone resulted in increased oxidative stress in a number of brain areas including the cerebral cortex, striatum, hippocampus, and medulla, as shown by elevated levels of malondialdehyde, a marker of lipid peroxidation, which indicates increased free radical production with consequent attack on membrane lipids.54 The level of nitric oxide (measured as nitrite) was also increased, and most likely due to increased nitric oxide production due to induction of the inducible form of nitric oxide synthase (iNOS). We have previously found a marked increase in inducible nitric oxide synthase immunoreactivity in the mouse striatum after systemic administration of rotenone.55 Increased levels of nitric oxide are associated with development of neuronal damage via formation of reactive nitrogen oxide species and development of oxidative or nitrosative stress. The most important is the interaction between nitric oxide and the superoxide anion (O2–), resulting in formation of peroxynitrite (ONOO–), a potent oxidant and nitrating agent that is able to modify proteins, lipids, and nucleic acids.56 Nitric oxide fosters dopamine depletion and is associated with neurotoxic processes underlying PD.57 Glutathione is an intracellular tripeptide of glycine, glutamic acid, and cysteine, the reduced form of which is important in protecting cells from oxidative damage and maintaining a reducing intracellular environment.58 The increase in oxidative stress induced by rotenone was also demonstrated by decreased levels of reduced glutathione, suggesting consumption of this scavenger molecule by free radical excess.
The effects of rotenone on oxidative stress in the brain were substantially reduced by coadministration of methylene blue. Thus, methylene blue resulted in a dose-related decrease in brain malondialdehyde, restoration of reduced glutathione, as well as marked inhibition of nitrite concentrations in the different brain areas studied, suggesting decreased oxidative and nitrosative stress on administration of methylene blue. Several in vitro and in vivo studies indicate an antioxidant and neuroprotective effect for methylene blue.25,26,59 When infused into the rat striatum along with rotenone, methylene blue (8.8 μg) prevented a decrease in cytochrome oxidase activity and any perilesional increase in oxidative stress (dihydroethidium fluorescence).60 In rats treated subcutaneously with rotenone 5 mg/kg for 8 days, methylene blue given at 500 μg/kg improved locomotor deficits and prevented striatal dopamine depletion.25 In vitro, methylene blue at concentrations of 10 nM–10 μM increased the viability of murine hippocampal cells incubated with 5 μM rotenone.26
Several mechanisms of action have been proposed to account for the neuroprotective effects of methylene blue. When administered at low doses (1–4 mg/kg), this dye has been shown to increase oxygen consumption in the brain,27 increase oxidation of cytochrome c in the brain,28,61 and to restore the impaired mitochondrial electron transport caused by a cytochrome oxidase inhibitor in rats.62 However, it is unlikely that increasing oxygen utilization in the brain could account for the neuroprotective effects observed in the present study, since the doses were considerably higher. It has been suggested that cycling of methylene blue between oxidized and reduced forms might block production of oxidants by the mitochondria, thereby protecting the mitochondria against oxidative stress.24 It has also been suggested that methylene blue functions as an alternative electron carrier, which accepts electrons from nicotinamide adenine dinucleotide dehydrogenase and transfers them to cytochrome c and bypasses complex I–III blockade. In this way, methylene blue rescues the effects of rotenone on mitochondrial complex I–III inhibition and free radical overproduction.25 Methylene blue acts as a direct inhibitor of nitric oxide synthase,63 and thus might prevent the deleterious effects of elevated nitric oxide levels on neuronal integrity, which has been shown in inflammatory conditions or ischemic injury in the brain.64,65 The dye has also been shown to inhibit the stimulation of N-methyl-D-aspartate receptors induced by nitric oxide.66 Thus, methylene blue can have a neuroprotective action via decreasing excitotoxic neuronal damage.
Interestingly, methylene blue is also capable of inducing oxidative stress; when combined with visible light of the appropriate wave length, methylene blue, acting as a photosensitizer, generates toxic reactive oxygen species and induces cell damage via oxidative stress.67 Photodynamic therapy mediated by methylene blue is effective against several pathogens, including bacteria,68 fungi,69 and herpes virus,70 and has also been shown to aid in wound healing.71
Methylene blue is a reversible competitive inhibitor of both monoamine oxidase (MAO)-A and MAO-B.72 Moreover, the dye is metabolized to yield N-demethylated products of which azure B, the monodemethyl species, is the major metabolite. Azure B is a more potent MAO-A inhibitor than is methylene blue. Azure B also reversibly inhibits the MAO-B isozyme.73 This ability of methylene blue to inhibit MAO-A can precipitate serotonin syndrome in patients taking selective serotonin reuptake inhibitors.74 On the other hand, inhibition of MAO-B would be an additional advantage of using methylene blue in PD. In the present study, methylene blue was used at doses of 10 mg/kg and 20 mg/kg. The same or higher dosages were used for other studies in rats or mice.35–37 In humans, methylene blue has been administered intravenously in doses of 1.5–2 mg/kg to treat methemoglobinemia and hypotension after surgery, and doses of 36–72 mg/kg have been used in malaria and doses as high as 250 mg/day have also been used.75,76
The finding in the present study that systemic administration of rotenone decreases paraoxonase 1 activity in the brain is consistent with our previous report.55 The ability of methylene blue to increase paraoxonase 1 activity in several brain regions suggests that decreased paraoxonase 1 activity is one mechanism by which rotenone induces neuronal injury. Paraoxonases are a group of enzymes involved in the hydrolysis of organophosphates. Human paraoxonase 1 is a high-density lipoprotein-associated serum enzyme which possesses organophosphatase, arylesterase, and lactonase activity and hydrolyzes many substrates and xenobiotics.77 The enzyme functions as an antioxidant,42 and its activity in serum has been shown to be decreased in several neurological disease processes in which increased oxidative stress is evident.78–80 Paraoxonase 1 status has been shown to be important in the etiology of PD. Carriers of the “slower metabolizer” MM PON1-55 genotype exposed to organophosphates have been reported to be at increased risk of developing PD.81 Higher paraoxonase 1 levels might reduce this risk.82
In the present study, a significant decrease in cortical AChE activity was documented in rotenone-treated rats. The main neurotransmitter in the cholinergic system is acetylcholine, which is important in motor control and in the autonomic, enteric, and central nervous systems. The central cholinergic system is considered to be the most important neurotransmitter involved in the regulation of cognitive function.83 In cholinergic neurotransmission, the transmitted signal is terminated by cleavage of the transmitter, acetylcholine, yielding acetate and choline. This cleavage is mediated by AChE, an enzyme of the α/β-fold family of proteins. The main motor symptoms of PD are largely due to the loss of dopaminergic tone and a consequent imbalance between dopaminergic and cholinergic modulation of striatal output.84 Anticholinergic drugs thus have a role in alleviating the symptoms of PD, particularly the associated tremor.85 Therefore, inhibition of AChE activity is likely to worsen the motor features of PD. In light of our findings, it could be suggested that restoration of cortical AChE activity induced by methylene blue could relieve many of the locomotor, cognitive, and memory impairments induced by rotenone.
Apoptosis, or programmed cell death, is likely to be a contributing pathophysiological mechanism in the neurodegeneration caused by rotenone. Caspase-3 immunoreactivity was markedly increased in the striatum of mice treated with systemic rotenone.54 In the present study, there was marked caspase-3 immunostaining in the cerebral cortex and striatum after treatment with rotenone. Caspase proteins are cysteine proteases that act downstream of the Bcl-2 family by initiating cellular breakdown during apoptosis.86 The Bcl-2 family of proteins includes proapoptotic and antiapoptotic factors acting at the mitochondrial and microsomal membranes. The function of Bcl-2 and related proteins is due to their ability to interfere with mitochondrial apoptosis pathways. Apoptosis involves loss of integrity of the outer mitochondrial membrane and release of mitochondrial cytochrome c and other proteins from the intermembrane space into the cytosol, which in turn activate death-driving proteolytic caspases that orchestrate the dismantling of the cell, and this process is controlled by Bcl-2 family proteins. By preventing redistribution of the proapoptotic protein, Bax, to the mitochondria, Bcl-2 prevents release of cytochrome c from the mitochondria and subsequent activation of caspase proteins.87,88 Bcl-2 has been implicated as having strong antioxidant properties and thus may provide a common survival function in apoptotic and oxidative stress patterns of cellular injury.89 Rotenone has been shown to decrease Bcl-2 expression in a human-derived dopaminergic neuroblastoma cell line90 and in human dopaminergic SH-SY5Y cells.91 In the present study, the level of the Bcl-2 antiapoptotic protein in striatal tissue was evaluated using an enzyme-linked immunosorbent assay, with the results demonstrating decreased Bcl-2 in the striatum following systemic administration of rotenone. Methylene blue resulted in increased Bcl-2 levels in rotenone-treated rats, although the effect was significant only at the dose of 20 mg/kg. Therefore, methylene blue might interfere with rotenone-induced apoptosis by increasing Bcl-2.
Neuronal damage and loss of tyrosine hydroxylase immunoreactivity neurons in the substantia nigra and striatum has been reported after systemic administration of rotenone in rodents.45,92,93 In the current study, systemic administration of rotenone resulted in nigrostriatal cell damage, together with loss of pigmented neurons and decreased tyrosine hydroxylase immunoreactivity in the substantia nigra and striatum. Evidence of neuronal damage was also observed in the cerebral cortex and hippocampus. Caspase-3 and TNF-α immunoreactivity were observed in the cytoplasm of cortical and striatal neurons. These findings are consistent with our previously reported observations that subcutaneous administration of rotenone in mice resulted in neuronal damage in the cerebral cortex and hippocampus in addition to the striatum and substantia nigra.55 Other researchers have also found that the effect of subcutaneously injected rotenone in rats is widespread.94 The damage inflicted by rotenone on the brain is thus not limited to the nigrostriatal pathway. Nevertheless, in this study, the rotenone-induced pathological alterations and the decline in tyrosine hydroxylase immunoreactivity were mitigated by cotreatment with methylene blue.
In conclusion, the present study indicates a potentially beneficial effect of methylene blue on nigrostriatal damage and dopaminergic cell loss evoked by rotenone in the rat. This neuroprotective effect of methylene blue is likely to involve reduction of oxidative stress and decreased levels of the proinflammatory cytokine TNF-α, as well as an antiapoptotic effect.
Disclosure
The authors report no conflicts of interest in this work.
References
Zhang ZX, Román GC. Worldwide occurrence of Parkinson’s disease: an updated review. Neuroepidemiology. 1993;12(4):195–208. | |
de Rijk MC, Tzourio C, Breteler MM, et al. Prevalence of parkinsonism and Parkinson’s disease in Europe: the EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1997;62(1):10–15. | |
Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55(3):181–184. | |
Berardelli A, Rothwell JC, Thompson PD, Hallett M. Pathophysiology of bradykinesia in Parkinson’s disease. Brain. 2001;124 Pt 11:2131–2146. | |
Pankratz N, Foroud T. Genetics of Parkinson disease. Genet Med. 2007;9(12):801–811. | |
Kumar KR, Djarmati-Westenberger A, Grünewald A. Genetics of Parkinson’s disease. Semin Neurol. 2011;31(5):433–440. | |
Drechsel DA, Patel M. Role of reactive oxygen species in the neurotoxicity of environmental agents implicated in Parkinson’s disease. Free Radic Biol Med. 2008;44(11):1873–1876. | |
Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26 Suppl 1:S1–S58. | |
Priyadarshi A, Khuder SA, Schaub EA, Priyadarshi SS. Environmental risk factors and Parkinson’s disease: a meta-analysis. Environ Res. 2001;86(2):122–127. | |
Vanacore N, Nappo A, Gentile M, et al. Evaluation of risk of Parkinson’s disease in a cohort of licensed pesticide users. Neurol Sci. 2002;23 Suppl 2:S119–S120. | |
Jenner P. Oxidative stress in Parkinson’s disease. Ann Neurol. 2003;53 Suppl 3:S26–S36. | |
Miller RL, James-Kracke M, Sun GY, Sun AY. Oxidative and inflammatory pathways in Parkinson’s disease. Neurochem Res. 2009;34(1):55–65. | |
Halliwell B, Gutteridge JM. Free Radicals in Biology and Medicine. 2nd ed. Oxford, UK: Clarendon Press; 1989. | |
Sies H. Oxidative stress: introduction. In: Sies H, editor. Oxidative Stress: Oxidants and Antioxidants. London, UK: Academic Press; 1991. | |
Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18(9):685–716. | |
Dexter DT, Holley AE, Flitter WD, et al. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9(1):92–97. | |
Alam ZI, Daniel SE, Lees AJ, Marsden DC, Jenner P, Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson’s but not incidental Lewy body disease. J Neurochem. 1997;69(3):1326–1329. | |
Zhang J, Perry G, Smith MA, et al. Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol. 1999;154(5):1423–1429. | |
Murray J, Taylor SW, Zhang B, Ghosh SS, Capaldi RA. Oxidative damage to mitochondrial complex I due to peroxynitrite: identification of reactive tyrosines by mass spectrometry. J Biol Chem. 2003;278(39):37223–37230. | |
Keeney PM, Xie J, Capaldi RA, Bennett JP Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26(19):5256–5264. | |
Sian J, Dexter DT, Lees AJ, et al. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann Neurol. 1994;36(3):348–355. | |
Clifton J 2nd, Leikin JB. Methylene blue. Am J Ther. 2003;10(4):289–291. | |
Betten DP, Vohra RB, Cook MD, Matteucci MJ, Clark RF. Antidote use in the critically ill poisoned patient. J Intensive Care Med. 2006;21(5):255–277. | |
Atamna H, Nguyen A, Schultz C, et al. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008;22(3):703–712. | |
Wen Y, Li W, Poteet EC, et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J Biol Chem. 2011;286(18):16504–16515. | |
Poteet E, Winters A, Yan LJ, et al. Neuroprotective actions of methylene blue and its derivatives. PLoS One. 2012;7(10):e48279. | |
Riha PD, Bruchey AK, Echevarria DJ, Gonzalez-Lima F. Memory facilitation by methylene blue: dose-dependent effect on behavior and brain oxygen consumption. Eur J Pharmacol. 2005;511(2–3):151–158. | |
Wrubel KM, Riha PD, Maldonado MA, McCollum D, Gonzalez-Lima F. The brain metabolic enhancer methylene blue improves discrimination learning in rats. Pharmacol Biochem Behav. 2007;86(4):712–717. | |
Peter C, Hongwan D, Kupfer A, Lauterburg BH. Pharmacokinetics and organ distribution of intravenous and oral methylene blue. Eur J Clin Pharmacol. 2000;56(3):247–250. | |
Sontag EM, Lotz GP, Agrawal N, et al. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington’s disease models. J Neurosci. 2012;32(32):11109–11119. | |
Necula M, Breydo L, Milton S, et al. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry. 2007;46(30):8850–8560. | |
Medina DX, Caccamo A, Oddo S. Methylene blue reduces aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011;21(2):140–149. | |
Zhang X, Rojas JC, Gonzalez-Lima F. Methylene blue prevents neurodegeneration caused by rotenone in the retina. Neurotox Res. 2006;9(1):47–57. | |
Bardakci H, Kaplan S, Karadeniz U, et al. Methylene blue decreases ischemia-reperfusion (I/R)-induced spinal cord injury: an in vivo study in an I/R rabbit model. Eur Surg Res. 2006;38(5):482–488. | |
Lougheed R, Turnbull J. Lack of effect of methylene blue in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. PLoS One. 2011;6(10):e23141. | |
Klamer D, Engel JA, Svensson L. Phencyclidine-induced behaviour in mice prevented by methylene blue. Basic Clin Pharmacol Toxicol. 2004;94(2):65–72. | |
Weinbroum AA, Goldin I, Kluger Y, Szold A. Methylene blue in preventing hemodynamic and metabolic derangement following superior mesenteric artery clamping/unclamping: an intratracheal versus intraperitoneal dose-response study. Shock. 2002;17(5):372–376. | |
Ruiz-Larrea MB, Leal AM, Liza M, Lacort M, de Groot H. Antioxidant effects of estradiol and 2-hydroxyestradiol on iron induced lipid peroxidation of rat liver microsomes. Steroids. 1994;59(6):383–388. | |
Ellman GL. Tissue sulfhydryl groups. Arch Biochem. 1959;82(1):70–77. | |
Moshage H, Kok B, Huizenga JR, Jansen PL. Nitrite and nitrate determination in plasma: a critical evaluation. Clin Chem. 1995;41(6 Pt 1):892–896. | |
Higashino K, Takahashi Y, Yamamura Y. Release of phenyl acetate esterase from liver microsomes by carbon tetrachloride. Clin Chim Acta. 1972;41:313–320. | |
Watson AD, Berliner JA, Hama SY, et al. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest. 1995;96(6):2882–2891. | |
Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. | |
Gorun V, Proinov I, Baltescu V, Balaban G, Barzu O. Modified Ellman procedure for assay of cholinesterases in crude enzymatic preparation. Anal Biochem. 1978;86(1):324–326. | |
Sherer TB, Kim JH, Betarbet R, Greenamyre JT. Subcutaneous rotenone exposure causes highly selective dopaminergic degeneration and alpha-synuclein aggregation. Exp Neurol. 2003;179(1):9–16. | |
Uversky VN. Neurotoxicant-induced animal models of Parkinson’s disease: understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissue Res. 2004;318(1):225–241. | |
Alam M, Schmidt WJ. Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res. 2002;136(1):317–324. | |
Parameshwaran K, Irwin MH, Steliou K, Pinkert CA. Protection by an antioxidant of rotenone-induced neuromotor decline, reactive oxygen species generation and cellular stress in mouse brain. Pharmacol Biochem Behav. 2012;101(3):487–492. | |
Kaneko K, Hineno A, Yoshida K, Ikeda S. Increased vulnerability to rotenone-induced neurotoxicity in ceruloplasmin-deficient mice. Neurosci Lett. 2008;446(1):56–58. | |
Testa CM, Sherer TB, Greenamyre JT. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res Mol Brain Res. 2005;134(1):109–118. | |
Sherer TB, Betarbet R, Testa CM, et al. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23(34):10756–10764. | |
Gao HM, Hong JS, Zhang W, Liu B. Synergistic dopaminergic neurotoxicity of the pesticide rotenone and inflammogen lipopolysaccharide: relevance to the etiology of Parkinson’s disease. J Neurosci. 2003;23(4):1228–1236. | |
Chang CY, Song MJ, Jeon SB, et al. Dual functionality of myeloperoxidase in rotenone-exposed brain-resident immune cells. Am J Pathol. 2011;179(2):964–979. | |
Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin Chem. 1995;41(12 Pt 2):1819–1828. | |
Abdel-Salam OM, Omara EA, El-Shamarka MS, Hussein JS. Nigrostriatal damage after systemic rotenone and/or lipopolysaccharide and the effect of cannabis. Comp Clin Pathol. DOI:10.1007/s00580-013-1788-3. | |
Korhonen R, Lahti A, Kankaanranta H, Moilanen E. Nitric oxide production and signaling in inflammation. Curr Drug Targets Inflamm Allergy. 2005;4(4):471–479. | |
Ebadi M, Sharma SK. Peroxynitrite and mitochondrial dysfunction in the pathogenesis of Parkinson’s disease. Antioxid Redox Signal. 2003;5(3):319–335. | |
Wang W, Ballatori N. Endogenous glutathione conjugates: occurrence and biological functions. Pharmacol Rev. 1998;50(3):335–356. | |
Rojas JC, John JM, Lee J, Gonzalez-Lima F. Methylene blue provides behavioral and metabolic neuroprotection against optic neuropathy. Neurotox Res. 2009;15(3):260–273. | |
Rojas JC, Simola N, Kermath BA, Kane JR, Schallert T, Gonzalez-Lima F. Striatal neuroprotection with methylene blue. Neuroscience. 2009;163(3):877–889. | |
Callaway NL, Riha PD, Bruchey AK, Munshi Z, Gonzalez-Lima F. Methylene blue improves brain oxidative metabolism and memory retention in rats. Pharmacol Biochem Behav. 2004;77(1):175–181. | |
Callaway NL, Riha PD, Wrubel KM, McCollum D, Gonzalez-Lima F. Methylene blue restores spatial memory retention impaired by an inhibitor of cytochrome oxidase in rats. Neurosci Lett. 2002;332(2):83–86. | |
Mayer B, Brunner F, Schmidt K. Inhibition of nitric oxide synthesis by methylene blue. Biochem Pharmacol. 1993;45(2):367–374. | |
Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21(17):6480–6491. | |
Mander P, Borutaite V, Moncada S, Brown GC. Nitric oxide from inflammatory-activated glia synergizes with hypoxia to induce neuronal death. J Neurosci Res. 2005;79(1–2):208–215. | |
Martins-Pinge MC, Araújo GC, Lopes OU. Nitric oxide-dependent guanylyl cyclase participates in the glutamatergic neurotransmission within the rostral ventrolateral medulla of awake rats. Hypertension. 1999;34(4 Pt 2):748–751. | |
Tardivo JP, Giglio AD, Oliveira CS, et al. Methylene blue in photodynamic therapy: from basic mechanisms to clinical applications. Photodiagnosis Photodyn Ther. 2005;2:175–191. | |
Tubby S, Wilson M, Wright JA, Zhang P, Nair SP. Staphylococcus aureus small colony variants are susceptible to light activated antimicrobial agents. BMC Microbiol. 2013;13:201. | |
Scwingel AR, Barcessat AR, Núñez SC, Ribeiro MS. Antimicrobial photodynamic therapy in the treatment of oral candidiasis in HIV-infected patients. Photomed Laser Surg. 2012;30(8):429–432. | |
Marotti J, Sperandio FF, Fregnani ER, Aranha AC, de Freitas PM, Eduardo Cde P. High-intensity laser and photodynamic therapy as a treatment for recurrent herpes labialis. Photomed Laser Surg. 2010;28(3):439–444. | |
Sperandio FF, Simões A, Aranha AC, Corrêa L, Orsini Machado de Sousa SC. Photodynamic therapy mediated by methylene blue dye in wound healing. Photomed Laser Surg. 2010;28(5):581–587. | |
Oxenkrug GF, Sablin SO, Requintina PJ. Effect of methylene blue and related redox dyes on monoamine oxidase activity; rat pineal content of N-acetylserotonin, melatonin, and related indoles; and righting reflex in melatonin-primed frogs. Ann N Y Acad Sci. 2007;1122:245–252. | |
Petzer A, Harvey BH, Wegener G, Petzer JP. Azure B, a metabolite of methylene blue, is a high-potency, reversible inhibitor of monoamine oxidase. Toxicol Appl Pharmacol. 2012;258(3):403–409. | |
Ramsay RR, Dunford C, Gillman PK. Methylene blue and serotonin toxicity: inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. Br J Pharmacol. 2007;152(6):946–951. | |
Schirmera RH, Adlera H, Pickhard M, Mandelkow E. “Lest we forget you – methylene blue …”. Neurobiol Aging. 2011;32(12):2325.e7–e16. | |
Ginimuge PR, Jyothi SD. Methylene blue: revisited. J Anaesthesiol Clin Pharmacol. 2010;26(4):517–520. | |
La Du BN. Human serum paraoxonase: arylesterase. In: Kalow W, editor. Pharmacogenetics of Drug Metabolism. Elmford, NY, USA: Pergamon; 1992. | |
Jamroz-Wisniewska A, Beltowski J, Stelmasiak Z, Bartosik-Psujek H. Paraoxonase 1 activity in different types of multiple sclerosis. Mult Scler. 2009;15(3):399–402. | |
Ferretti G, Bacchetti T, Masciangelo S, Grugni G, Bicchiega V. Altered inflammation, paraoxonase-1 activity and HDL physicochemical properties in obese humans with and without Prader-Willi syndrome. Dis Model Mech. 2012;5(5):698–705. | |
Abdel-Salam OME, Khadrawy YA, Mohammed NA. Neuroprotective effect of nitric oxide donor isosorbide-dinitrate against oxidative stress induced by ethidium bromide in rat brain. EXCLI J. 2012;11:125–141. | |
Manthripragada AD, Costello S, Cockburn MG, Bronstein JM, Ritz B. Paraoxonase 1, agricultural organophosphate exposure, and Parkinson disease. Epidemiology. 2010;21(1):87–94. | |
Belin AC, Ran C, Anvret A, et al. Association of a protective paraoxonase 1 (PON1) polymorphism in Parkinson’s disease. Neurosci Lett. 2012;522(1):30–35. | |
Everitt BJ, Robbins TW. Central cholinergic systems and cognition. Annu Rev Psychol. 1997;48:649–684. | |
Aosaki T, Miura M, Suzuki T, Nishimura K, Masuda M. Acetylcholine-dopamine balance hypothesis in the striatum: an update. Geriatr Gerontol Int. 2010;10 Suppl 1:S148–S157. | |
Fox SH. Non-dopaminergic treatments for motor control in Parkinson’s disease. Drugs. 2013;73(13):1405–1415. | |
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–1316. | |
Belka C, Budach W. Anti-apoptotic Bcl-2 proteins: structure, function and relevance for radiation biology. Int J Radiat Biol. 2002;78(8):643–658. | |
Zhai D, Jin C, Huang Z, Satterthwait AC, Reed JC. Differential regulation of Bax and Bak by anti-apoptotic Bcl-2 family proteins Bcl-B and Mcl-1. J Biol Chem. 2008;283(15):9580–9586. | |
Lee L, Irani K, Finkel T. Bcl-2 regulates nonapoptotic signal transduction: inhibition of c-Jun N-terminal kinase (JNK) activation by IL-1 beta and hydrogen peroxide. Mol Genet Metab. 1998;64(1):19–24. | |
Hu LF, Lu M, Wu ZY, Wong PT, Bian JS. Hydrogen sulfide inhibits rotenone-induced apoptosis via preservation of mitochondrial function. Mol Pharmacol. 2009;75(1):27–34. | |
Kim HJ, Park HJ, Park HK, Chung JH. Tranexamic acid protects against rotenone-induced apoptosis in human neuroblastoma SH-SY5Y cells. Toxicology. 2009;262(2):171–174. | |
Feng Y, Liang ZH, Wang T, Qiao X, Liu HJ, Sun SG. alpha-Synuclein redistributed and aggregated in rotenone-induced Parkinson’s disease rats. Neurosci Bull. 2006;22(5):288–293. | |
Inden M, Kitamura Y, Tamaki A, et al. Neuroprotective effect of the antiparkinsonian drug pramipexole against nigrostriatal dopaminergic degeneration in rotenone-treated mice. Neurochem Int. 2009;55(8):760–767. | |
Lapointe N, St-Hilaire M, Martinoli MG, et al. Rotenone induces non-specific central nervous system and systemic toxicity. FASEB J. 2004;18(6):717–719. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.