")
Back to Journals » OncoTargets and Therapy » Volume 12
ROS generation and autophagosome accumulation contribute to the DMAMCL-induced inhibition of glioma cell proliferation by regulating the ROS/MAPK signaling pathway and suppressing the Akt/mTOR signaling pathway
Authors Wang Y, Zhang J, Yang Y, Liu Q, Xu G, Zhang R, Pang Q
Received 20 November 2018
Accepted for publication 1 February 2019
Published 7 March 2019 Volume 2019:12 Pages 1867—1880
DOI https://doi.org/10.2147/OTT.S195329
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Yanjun Wang,1 Jiachen Zhang,1 Yihang Yang,1 Qian Liu,2 Guangming Xu,1 Rui Zhang,1 Qi Pang1
1Department of Neurosurgery, Shandong Provincial Hospital Affiliated to Shandong University, Jinan, 250021, Shandong, China; 2Department of Histology and Embryology, Shandong University Cheeloo College Medicine, Jinan, 250012, Shandong, China
Purpose: Chemotherapy after surgery can prolong the survival of patients with gliomas. Dimethylaminomicheliolide (DMAMCL), a novel chemotherapeutic agent, exhibited antitumor properties in acute myeloid leukemia stem cells and showed an increased drug concentration in the brain. This study aims to investigate the specific anticancer activities and mechanisms of DMAMCL in glioma cells.
Materials and methods: In this study, the effects of DMAMCL were evaluated and characterized in U87-MG and U251 glioma cells. Cell viability was assessed by Cell Counting Kit-8. Apoptosis, mitochondrial membrane potential, and intracellular reactive oxygen species (ROS) generation were assessed by fluorescence microscopy. Autophagosome formation was observed with transmission electron microscopy, and the autophagy flux was measured by transfecting cells with mRFP-GFP-LC3 adenoviral vectors. Immunofluorescence and Western blot analyses were used to determine the expression of proteins.
Results: In the present study, treatment with DMAMCL decreased cell viability and induced apoptosis in U87-MG and U251 glioma cells. Additionally, DMAMCL activated autophagy-mediated cell death as evidenced by the formation of autophagosomes, accumulation of LC3B-II, inhibition of autophagy flux, and increase in cell viability after cotreatment with an autophagy inhibitor. Subsequent experiments showed that the DMAMCL-induced apoptosis and autophagy were possibly mediated by ROS generation and Akt/mTOR signaling pathway inhibition. On the other hand, the ROS scavenger N-acetyl-l-cysteine and the Akt activator insulin-like growth factor-1 attenuated the DMAMCL-induced autophagy and cell death.
Conclusion: Our findings revealed that DMAMCL induced apoptosis and autophagic cell death by regulating the ROS/mitogen-activated protein kinase signaling pathway and suppressing the Akt/mTOR signaling pathway in human glioma cells. DMAMCL may be a novel effective anticancer agent, which can target gliomas.
Keywords: DMAMCL, apoptosis, autophagy, ROS, glioma
Introduction
Gliomas are one of the most malignant and lethal primary brain cancers, which can occur in any part of the central nervous system. The overall incidence rate is 6.03 per 100,000.1 Due to the high invasiveness of gliomas, it is difficult to completely resect them. People with these neoplasms generally have a poor prognosis and poor quality of life as the disease progresses.2,3 Even after considerable research, there is little progress in limiting the development of gliomas and prolonging the median overall survival of patients. The high recurrence rate and the development of drug resistance contribute to this situation.4 The current clinical treatments for malignant gliomas are surgery, adjuvant postoperative radiotherapy, and chemotherapy. Although these therapies can prolong overall survival, the poor curative effects due to the inherent apoptosis-resistant phenotype of the malignancy and the extremely negative systemic side effects result in unsatisfactory outcomes.5,6 Hence, more effective treatments are still urgently needed.
Recent studies have revealed that sesquiterpene lactone compounds have many antitumor and anti-inflammatory effects.7–9 Parthenolide (PTL), derived from Tanacetum parthenium, is one of these compounds. Many reports have revealed that PTL and its soluble analog dimethylamino parthenolide have cytotoxic effects on breast, prostate, pancreatic, glioma, lung, and bladder cancer cells, in addition to hematologic malignancies. As these compounds are unstable in both acidic and basic conditions, their therapeutic applications have been restricted.10–16 Micheliolide, which is much more stable than PTL, was isolated from the Michelia compressa and Michelia champaca plants and showed remarkable therapeutic efficacy in nonobese diabetic/severe combined immunodeficiency AML models.17 Dimethylaminomicheliolide (DMAMCL), as a novel chemotherapeutic agent, has been reported to suppress inflammation in cases of intestinal disease and sepsis.18 In addition, it was proven to prolong the lifespan of a mouse model of human acute myelogenous leukemia.19 The distribution analysis in the DMAMCL-treated rats showed that the drug concentration in the brain was higher than in the plasma, and it was innocuous to the main organs.20
Apoptosis, also called type I programmed cell death, plays an important role in the progression of chemotherapy. Apoptosis is caspase-dependent and is characterized by some conspicuous changes in the cell death process; for example, cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, DNA fragmentation, and apoptotic body formation.21 However, in many cases, chemotherapy can induce autophagic cell death by activating the lysosome-dependent proteolytic pathway.22 Autophagy, a conservative process, enables cells to isolate the damaged or surplus organelles into autophagosomes and deliver them to lysosomes to degrade. However, autophagy has conflicting roles in various cell types under different cellular states.23
Reactive oxygen species (ROS) play an important role in the development of cancers. However, superfluous ROS have cytotoxicity against diverse targets, such as proteins, DNA, and lipids. In many exogenous stress conditions, ROS are important signaling molecules that induce apoptosis and autophagy and activate cellular signaling kinases.24 Some chemical drugs targeting ROS-related signaling pathways were proven to be effective in the treatment of human cancers, including ROS/mitogen-activated protein kinase (MAPK) signaling pathways, which was a momentous discovery. However, the effects of DMAMCL-induced ROS damage and the regulation of related signaling pathways in human glioma cancer cells remain unclear.
In the present study, we aimed to determine the anticancer activities and potential mechanisms of DMAMCL in two different human glioma cell lines. We found that DMAMCL could induce not only apoptosis through ROS generation, mitochondrial dysfunction, and caspase activation but also autophagy through the inhibition of the Akt/mTOR signaling pathways in the U87-MG and U251 cell lines. These novel findings provide a new perspective for DMAMCL in glioma chemotherapeutic interventions.
Materials and methods
Cell lines and cell culture and DMAMCL preparation
The human glioma cell lines U87-MG and U251 were obtained from the Chinese Academy of Sciences Cell Bank. These cell lines were both cultured in Dulbecco’s Modified Eagle’s Medium/HIGH glucose culture medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 mg/mL streptomycin. Cells were kept in the exponential growth phase and cultured at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. DMAMCL was a gift provided by Accendatech Co., Ltd. (Tianjin, China). The formula is C17H27NO3·C4H4O4. For the following experiments, DMAMCL was dissolved in water at a concentration of 10 mM as a stock solution and diluted to the indicated concentration with medium before use.
Cell viability assay
Cell viability was determined by Cell Counting Kit-8 (CCK-8) assays. Briefly, cells in the exponential phase of growth were harvested and seeded into 96-well plates at a density of 5,000 cells per well. After 24 hours of incubation, the cells were treated with different concentrations of DMAMCL or medium for another 24, 48, and 72 hours. Then, 10 μL CCK-8 solution was added into each well. One hour later, the absorbance was determined using a microplate reader (EL340; BioTek Instruments, Waltham, MA, USA) at 450 nm.
Cell apoptosis analysis by flow cytometry
The Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) kit was used to determine the effect of DMAMCL on apoptosis. First, the cells were exposed to different concentrations of DMAMCL for 48 hours, and then the cells were collected. Next, the cells were resuspended in binding buffer and incubated with 5 μL Annexin V-FITC and 5 μL PI for 15 minutes in the dark. The results were analyzed using a FACS Calibur or an EPICS XL flow cytometer (BD Biosciences).
Live/dead assay
U87-MG and U251 cells were treated with various concentrations of DMAMCL for 48 hours and then the LIVE/DEAD™ Cell Imaging Kit was used according to the instructions. After another 15 minutes of culture in the dark, the results were observed using fluorescence microscopy (Olympus).
Transmission electron microscopy
U87-MG and U251 cells were treated with DMAMCL (40 μM) for 48 hours. Afterward, the cells were fixed with 3% glutaraldehyde and 2% paraformaldehyde in a 0.1 M phosphate-buffered saline buffer for 30 minutes and postfixed with 1% osmium tetroxide for 1.5 hours. Then, the cells were washed and stained in 3% aqueous uranyl acetate for 1 hour. Subsequently, the cells were dehydrated in a graded series of ethanol and acetone and then embedded in Epon-Araldite resin. Ultrathin sections were cut by a Reichert ultramicrotome, double-stained with 0.3% lead citrate, and examined using a JEOL 1200EX electron microscope (Japan).
Confocal microscopy
U87-MG and U251 cells were seeded into 96-well plates, and when they reached 70% confluence, they were transfected with mRFP-GFP-LC3 adenoviral vectors (purchased from HanBio Technology, Shanghai, China) following the manufacturer’s instructions. Then, the cells were treated with DMAMCL for 48 hours. Finally, the LC3 puncta were examined with a confocal microscope (USA).
Measurement of intracellular ROS generation
Intracellular ROS were detected by the Total ROS Assay Kit. Briefly, 5,000 cells per well were cultured for 24 hours in a 96-well plate, and then the cells were incubated with 2′,7′-dichlorofluorescein-diacetate (DCFH-DA) for 1 hour. Following the treatment, the cells were washed with serum-free medium three times and exposed to the indicated treatments. The level of ROS was analyzed and imaged using a fluorescence microscope (Olympus).
Measurement of mitochondrial membrane potential
The mitochondrial membrane potential (MMP) was measured with the JC-1 Assay Kit. A total of 5 × 105 cells per well were cultured for 24 hours in a six-well plate. Then, the cells were treated with different concentrations of DMAMCL, ranging from 10 to 40 μM, for another 48 hours. The cells were washed three times, collected, and then incubated for 20 minutes with the JC-1 probe at 37°C. The MMPs were measured using a flow cytometer (BD Biosciences).
Western blot analysis
Cells were lysed in cold RIPA lysis buffer containing 1% phosphatase inhibitor and 1% protease inhibitor for 30 minutes. Protein samples were collected and centrifuged at 12,000 rpm for another 30 minutes at 4°C. The supernatants were collected for continued analysis. The Bradford protein method and the bicinchoninic acid protein assay kit were used to determine the protein concentration according to the manufacturer’s directions. A total of 40 μg protein was separated, electrophoresed using a bis-Tris polyacrylamide gel and transferred onto a polyvinylidenedifluoride membrane. Membranes were incubated with primary antibodies overnight at 4°C after being blocked in 5% nonfat dry milk. Then, the antigen–antibody binding reaction was performed using horseradish peroxidase-conjugated secondary antibodies for 1 hour at room temperature. Immunoblots were visualized using enhanced chemiluminescence (LAS-4000).
Immunofluorescence
Cell slides were prepared and exposed to various treatments. Cells were then fixed with 4% paraformaldehyde for 20 minutes and permeabilized with 0.3% Triton for 15 minutes at room temperature. Next, the smears were blocked with goat serum solution for 1 hour, followed by primary antibody incubation at 4°C overnight in a wet box. Cells were thoroughly rinsed and stained with a fluorescent secondary antibody for 1 hour. Finally, nuclei were stained with DAPI for 10 minutes and imaged using a fluorescence microscope (Olympus).
Statistical analysis
The experiments were performed independently at least three times. The data were statistically analyzed using t-test, chi-squared test, or Fisher’s exact tests using SPSS version 19.0 software (IBM SPSS, NY, USA). P-values <0.05 were considered statistically significant.
Results
DMAMCL inhibited the viability of glioma cells and induced cell death
To investigate the inhibitory effects of DMAMCL on human glioma cells, U87-MG and U251 cells were treated with different concentrations of DMAMCL. Cell viability was determined by the CCK-8 assay (Figure 1A). The results showed that the viability of these cells was reduced in a dose-dependent manner at 24, 48, and 72 hours. The ratios between the live and dead cells were further determined with the LIVE/DEAD Cell Imaging Kit. As shown in Figure 1B and C, the number of live cells (green) was significantly reduced, and the density of dead cells (red) was increased compared with the control group. Next, we further examined the effects of different treatments on cell viabilities. As shown in Figure 1D, co-treated 3-MA or IGF-1 with DMAMCL and pretreated with NAC could increase the cell viability compared with DMAMCL alone.
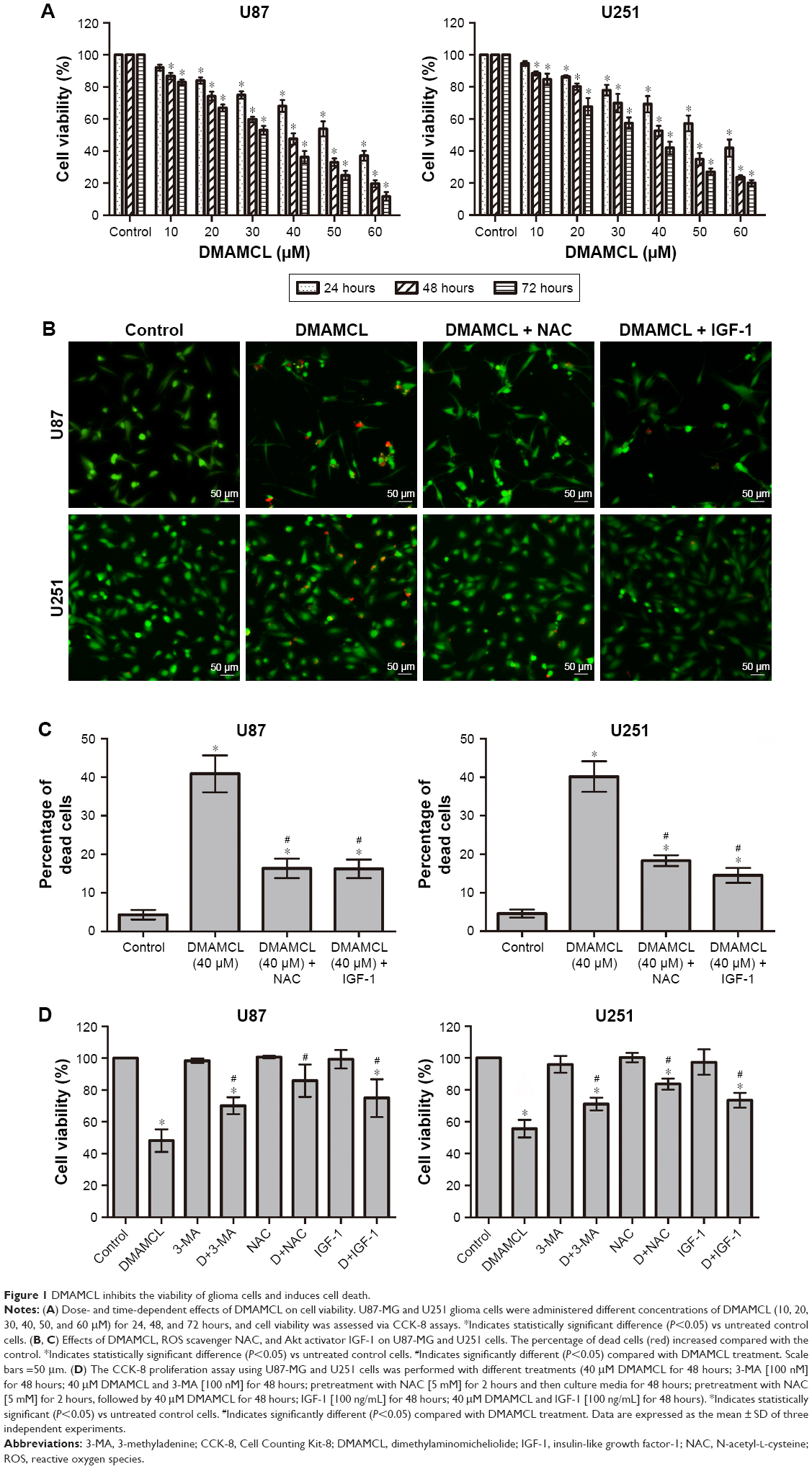 | Figure 1 DMAMCL inhibits the viability of glioma cells and induces cell death. |
DMAMCL induced apoptosis in glioma cells
To determine whether DMAMCL induced cell death via apoptosis, we measured the MMPs of the U87-MG and U251 cells with different concentrations of DMAMCL through a flow cytometric analysis using JC-1 as a mitochondrial fluorescent probe. As shown in Figure 2A and B, DMAMCL reduced the MMP in the U87-MG and U251 cells in a dose-dependent manner, as indicated by a decrease in the red–green fluorescence intensity ratio. To quantify the level of apoptosis, we used Annexin V-FITC/PI double staining. The results demonstrated that DMAMCL increased the numbers of both early and late apoptotic cells in a dose-dependent manner (Figure 3A and B). We also determined the expression of the main mitochondrion-dependent apoptotic proteins, including caspase-3, Bcl-2, and Bax, by Western blotting analysis. As shown in Figure 3C, the levels of procaspase-3 and Bcl-2 were decreased, while the levels of cleaved caspase-3 and Bax were significantly increased in the DMAMCL-treated U87-MG and U251 cells. Overall, these results indeed suggested that DMAMCL contributed to apoptosis in human glioma cells.
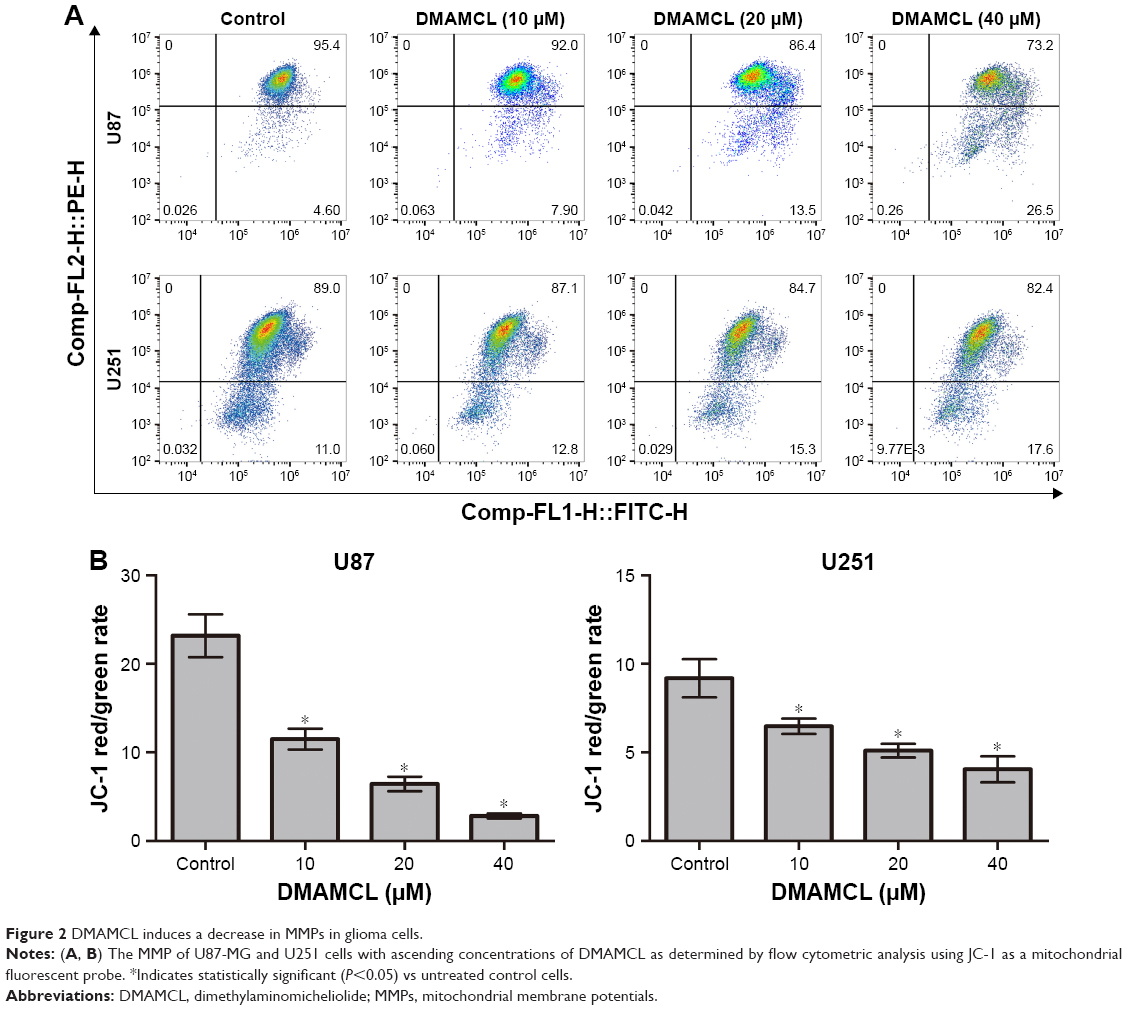 | Figure 2 DMAMCL induces a decrease in MMPs in glioma cells. |
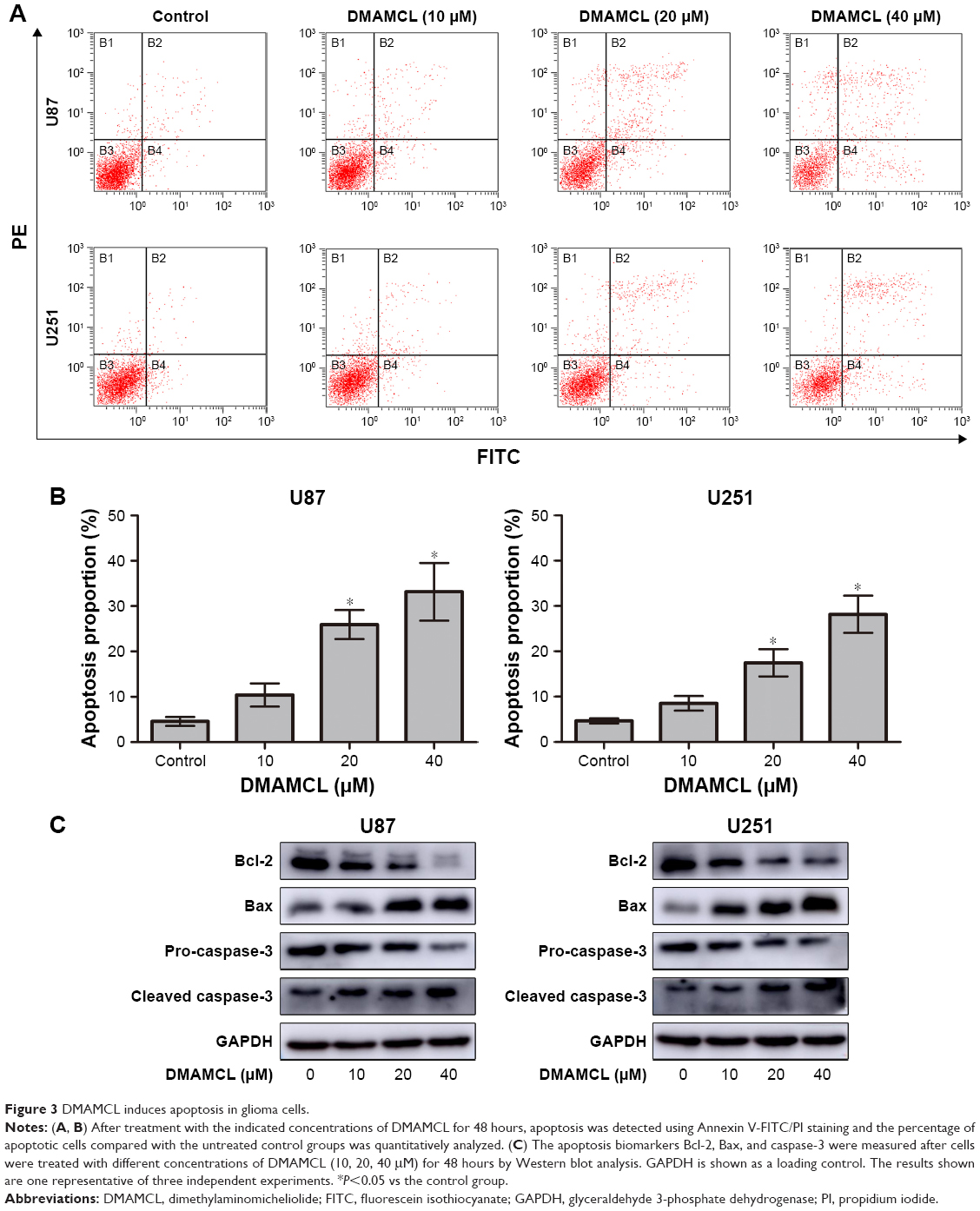 | Figure 3 DMAMCL induces apoptosis in glioma cells. |
DMAMCL induced autophagic activity in glioma cells, which subsequently promoted cell death
Autophagy is also an important type of programmed cell death; therefore, we next examined whether DMAMCL-induced cell death was due to autophagy. Transmission electron microscopy (TEM) was used to directly detect the formation of the autophagosome after DMAMCL treatment (Figure 4A). We found that the level of double membrane autophagosome formation was increased in the DMAMCL-treated group compared with the untreated group. Next, the expression of autophagy-related proteins was investigated by Western blotting after treatment with different concentrations of DMAMCL. As shown in Figure 4B, DMAMCL increased the levels of LC3B-II and beclin-1 in the U87-MG and U251 cells compared with the control cells, while p62 expression was upregulated. To determine whether the change in LC3B resulted from an increase in autophagosome formation or the inhibition of autophagosome–lysosome fusion, we used mRFP-GFP-LC3 adenovirus transfection to determine the autophagic flux with or without DMAMCL treatment. The results showed that the levels of autophagosomes (yellow point in merged images) were significantly increased, while autophagosome–lysosomes (red point in merged images) were not significantly changed after DMAMCL treatment in the U87-MG and U251 cells (Figure 4C–F). This suggested that the increased expression of LC3B resulted from the formation and accumulation of autophagosomes. Additionally, DMAMCL treatment might inhibit late-stage autophagy.
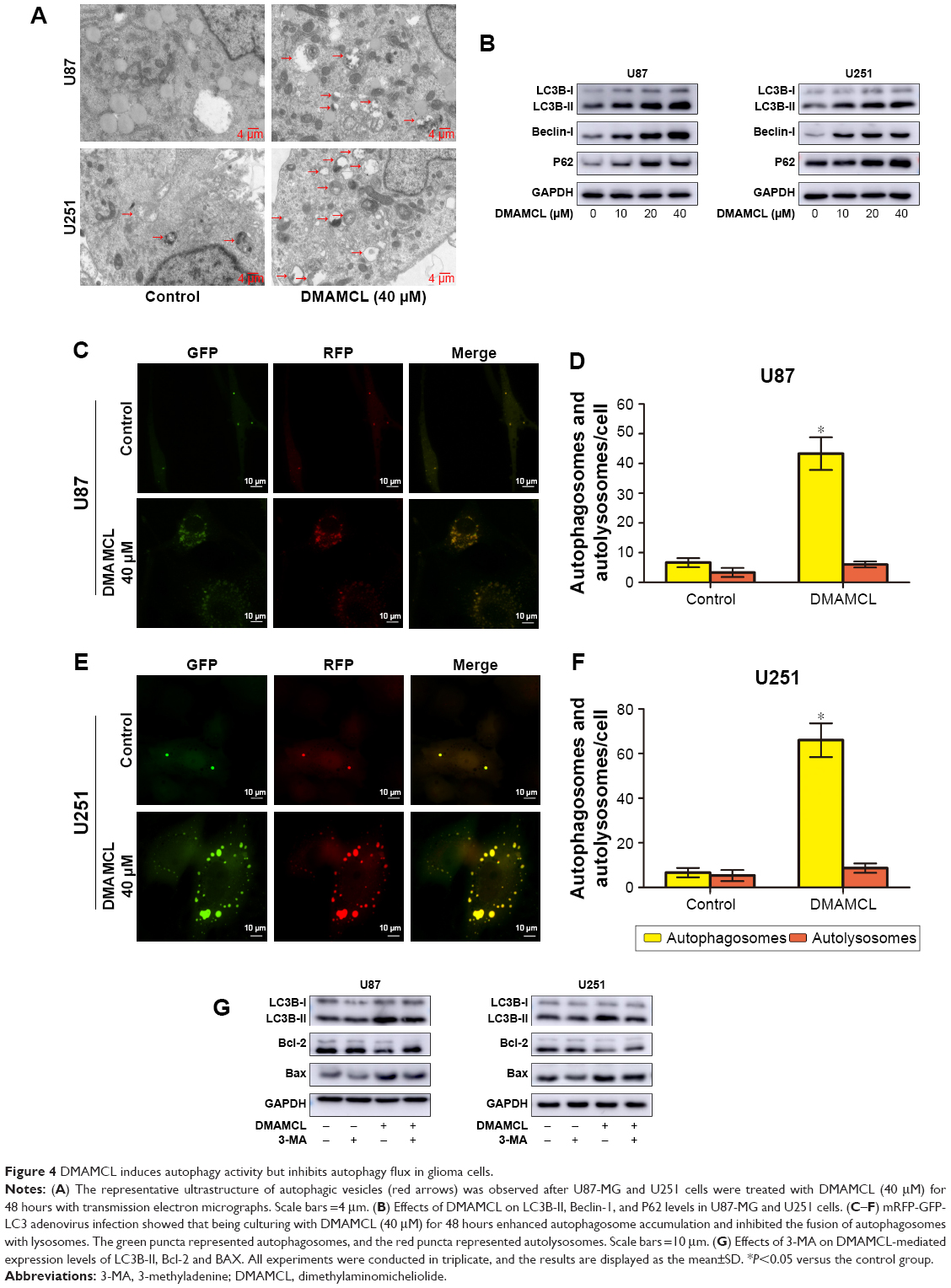 | Figure 4 DMAMCL induces autophagy activity but inhibits autophagy flux in glioma cells. |
Autophagy is a double-edged sword in the process of cell death;25 thus, the autophagy inhibitor 3-methyladenine (3-MA) was also used in the CCK-8 assays to elucidate the exact role of DMAMCL in the autophagic activity of glioma cells. The cell viabilities, after combined treatment with DMAMCL and 3-MA increased 18.21%±3.34% and 16.08%±1.75% than DMAMCL treatment in U87-MG and U251 cells, respectively; cotreatment with 3-MA indeed alleviated the effects on the cell viability induced by DMAMCL treatment. Also, treatment with 3-MA reduced the apoptotic effects of DMAMCL (Figure 4G). All the above results indicated that DMAMCL could induce autophagy-related cell death.
DMAMCL induced ROS generation
The balance of intracellular ROS is of vital importance in determining the fate of cancer cells, and this is done by regulating different important signaling pathways. Since our previous report clarified that MCL, the active ingredient of DMAMCL, exerted cytotoxic effects by generating ROS, we determined the levels of ROS in U87-MG and U251 cells after treatment with DCFH-DA probes. The results showed that DMAMCL dramatically increased ROS accumulation compared with the control group (Figure 5A). Subsequent analysis indicated that the levels of Nrf-2, heme oxygenase 1 (HO-1), and TXNRD expressions were significantly elevated, further suggesting the activation of oxidative stress response-related signaling pathways (Figure 5B and C). Additionally, when the cells were pretreated with N-acetyl-L-cysteine (NAC) for 2 hours, the DMAMCL-induced inhibition of cell viability was significantly alleviated. The same results were also obtained with the live/dead assay (Figure 1B–D). Overall, these experiments confirmed the results that the DMAMCL-induced cell death was ROS-dependent in U87-MG and U251 cells.
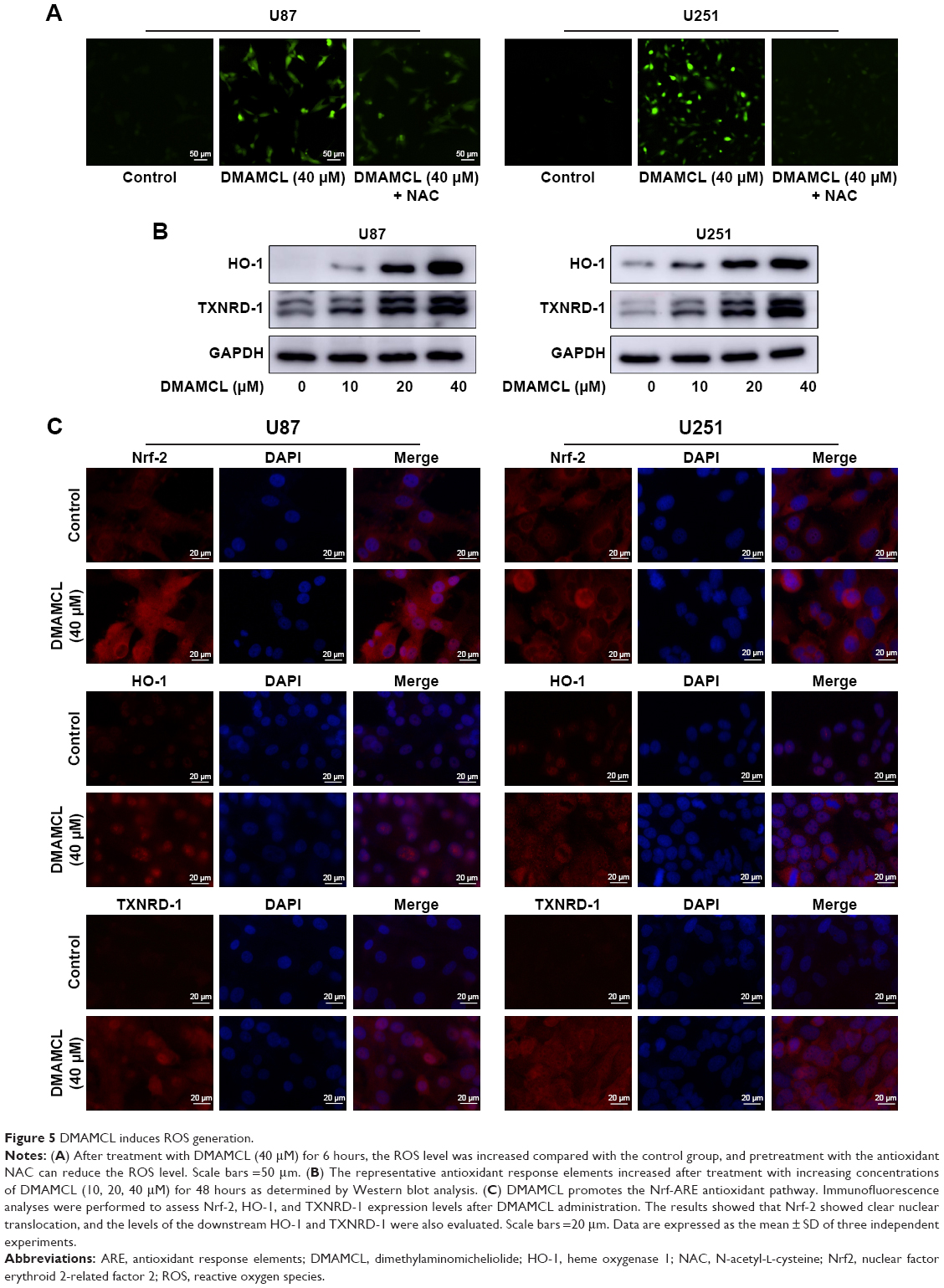 | Figure 5 DMAMCL induces ROS generation. |
DMAMCL regulated the ROS/MAPK signaling pathways and blocked the Akt/mTOR signaling pathway
As shown in Figure 6, Western blotting results showed that DMAMCL dose-dependently downregulated the phosphorylation levels of Akt and mTOR. Additionally, the MAPK pathway was also affected, that is, the phosphorylation of extracellular signal-regulated kinase (ERK) and p38 was increased, but the phosphorylation of c-Jun N-terminal kinase (JNK) was suppressed. To investigate whether the ROS/MAPK and Akt/mTOR signaling pathways were involved in DMAMCL-induced cell death, NAC (an ROS scavenger) and insulin-like growth factor-1 (IGF-1; an Akt activator) were introduced for further exploration. As shown in Figure 1D, after pretreatment with NAC for 2 hours and cotreatment with IGF-1 for 48 hours, the cell viability was significantly elevated. The same results could also be obtained with the live/dead assay (Figure 1B and C). Simultaneously, the level of LC3B was downregulated compared with the DMAMCL-only treatment group (Figure 7A and B). All the results shown above proved that DMAMCL-induced autophagic cell death might be associated with the ROS/MAPK and Akt/mTOR signaling pathways.
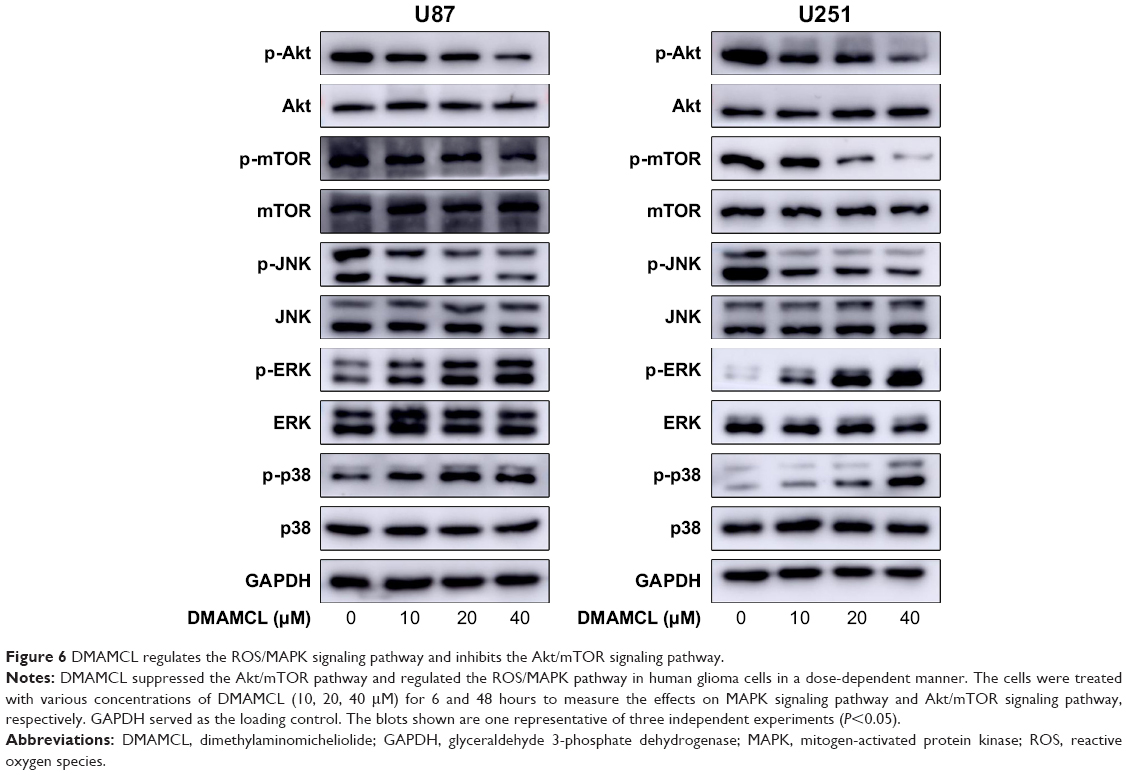 | Figure 6 DMAMCL regulates the ROS/MAPK signaling pathway and inhibits the Akt/mTOR signaling pathway. |
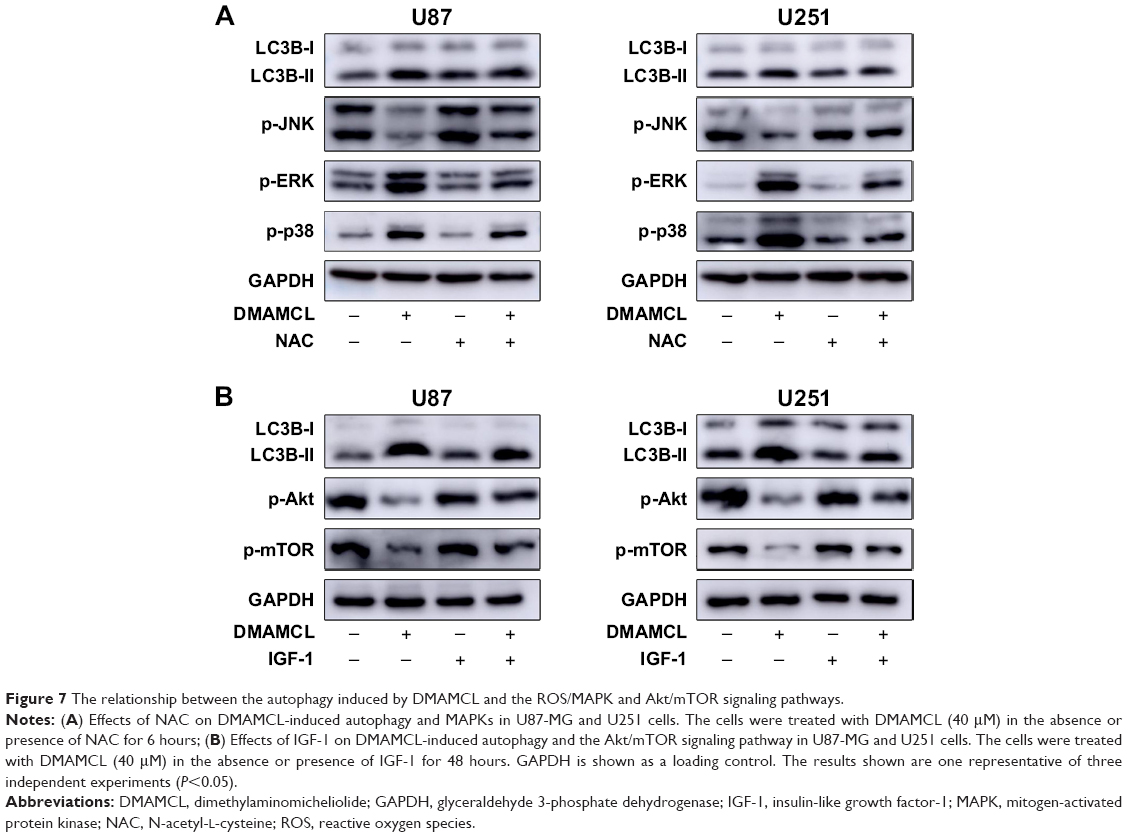 | Figure 7 The relationship between the autophagy induced by DMAMCL and the ROS/MAPK and Akt/mTOR signaling pathways. |
Discussion
In the present study, we examined whether DMAMCL, as a novel anticancer chemotherapeutic reagent, could exhibit cytotoxic activities in U87-MG and U251 cells and investigated the possible mechanisms. Briefly, we studied cell growth, apoptosis, autophagy, and the possible pathways impacted by different doses of DMAMCL in two glioma cell lines. Our results clearly indicated that DMAMCL induced cytotoxicity by interrupting autophagy flux, mediating autophagosome accumulation, inducing mitochondrial dysfunction, and stimulating excessive ROS generation. Collectively, we confirmed that DMAMCL inhibited the growth of U87-MG and U251 cells by promoting apoptosis and autophagosome accumulation through the ROS/MAPK and Akt/mTOR pathways.
Many anticancer compounds can induce not only apoptosis but also autophagic cell death. Both are well-known mechanisms of cell death. In many previous studies, PTL, the natural compound of DMAMCL, induced cancer cell death through apoptosis, autophagy, or both.26–28 However, the possible anticancer mechanisms of DMAMCL in glioma cells have rarely been reported. Apoptosis is the main process of antitumor, drug-induced cell death. Recent studies have shown that apoptosis can be induced by multiple signaling pathways, but the mitochondria-mediated pathways are also of vital importance.29 In our study, we found that the MMP was strikingly decreased following DMAMCL treatment for 48 hours, illustrating that mitochondrial membrane depolarization occurred. It is well known that mitochondrial membrane depolarization can induce cytochrome c release and activate the cytosolic caspases.30 Our subsequent experiments confirmed these results by showing that the ratio of apoptotic cells was increased after DMAMCL administration, while the pro-apoptotic proteins Bax and cleaved caspase-3 were upregulated and the anti-apoptotic proteins Bcl-2 and pro-caspase-3 were downregulated.
Autophagy, on the other hand, is an evolutionarily conserved pathway involved in cellular homoeostasis that is activated during stress and includes two main processes: autophagosome formation to sequester misfolded proteins and damaged organelles and autophagosome-to-lysosome fusion and degradation.31 Therefore, we next investigated, using TEM, whether autophagy was involved in DMAMCL-induced U87-MG and U251 cell death. In our study, we first detected an increased level of double membrane autophagosome formation after treatment with 40 μM DMAMCL for 48 hours. Subsequently, we found by Western blotting analyses that the expression levels of the quantitative autophagic biomarkers LC3B-II and Beclin-1 proteins were also upregulated in a dose-dependent manner. The p62/SQSTM1 protein, as an autophagic cargo protein, was degraded when the autophagosomes fused with the lysosomes to form autolysosomes. Surprisingly, p62/SQSTM1 was upregulated after DMAMCL administration, indicating that the final step of autophagy was inhibited and autophagosome degradation was reduced. According to Bernadette’s research, p62 not only acted as a cargo carrier but also stimulated the progress of autophagosome formation.32 For this reason, levels of autophagosomes could be further increased. This hypothesis was further confirmed by mRFP-GFP-LC3 adenovirus transfection. Consistent with the previous results,33–35 late-stage autophagy flux inhibition and autophagosome accumulation led to glioma cell death. To further illustrate this point, 3-MA, the most commonly used autophagy inhibition reagent, was used. 3-MA can interfere with the PI3KC3 signaling pathway during the formation of autophagosomes. Notably, our research data showed that the combined treatment of DMAMCL and 3-MA could alleviate U87-MG and U251 cell death. Consequently, we concluded that DMAMCL indeed played a key role in inducing both apoptosis and autophagy-related cell death in U87-MG and U251 cells.
Many anticancer drugs can induce oxidative stress, which is an important mechanism during autophagy and apoptosis.36 A surplus of ROS, including hydroxyl radical (•OH), superoxide (•O2−), and hydrogen peroxide (H2O2), can be generated to induce the oxidative damage of various macromolecules, including proteins, lipids, and DNA, which in turn promotes cell death.37 Increased ROS generation in cancer cells could therefore be a strategy for cancer therapy. However, the role of ROS in the induction of DMAMCL-treated cell death remains to be illustrated. Thorpe et al38 demonstrated that ROS can upregulate autophagy genes. Nuclear factor erythroid 2-related factor 2 (Nrf2), a redox-sensitive transcription factor, has an important function under oxidative stress by translocating into the nucleus and interacting with antioxidant response elements (ARE) to induce the subsequent expression of downstream genes, such as HO-1, thioredoxin reductase (TXNRD-1), NADPH dehydrogenase, and thioredoxin. P62, a selective autophagy adaptor, could activate the Nrf-ARE antioxidant pathway by degrading Kelch-like-ECH-associated protein (keap1).39 Consistent with the previous results, we found that DMAMCL administration in the U87-MG and U251 cells could induce ROS generation, as detected by the DCFH-DA probe. Furthermore, the nuclear translocation of Nrf2 and the upregulation of the downstream genes HO-1 and TXNRD-1 enhanced ROS generation in response to DMAMCL treatment. To determine whether ROS generation was related to autophagy and apoptosis, NAC, an ROS scavenger, was used before DMAMCL treatment. As the results show, NAC alleviated the DMAMCL-induced autophagy and cell death, suggesting that ROS mediated DMAMCL-induced autophagic cell death and apoptosis.
Among the many signaling pathways, the Akt-mTOR pathway is usually suppressed and autophagy is stimulated by anticancer agents.40,41 Targeting the Akt-mTOR signaling pathway has therefore been a novel therapeutic approach to curing gliomas.42 In our study, after DMAMCL treatment for 48 hours, phosphorylated Akt and phosphorylated mTOR were markedly suppressed along with the upregulation of autophagy markers and apoptosis. To confirm the biological effects of the Akt-mTOR pathway within these processes, IGF-1, an Akt activator, was coadministered with DMAMCL in U87-MG and U251 cells. We found that IGF-1 could not only reverse the levels of phosphorylated Akt and mTOR but also inhibit the induction of autophagy by DMAMCL. These findings indicated that the suppression of the Akt-mTOR pathway was associated with DMAMCL-mediated autophagy in inducing cell death.
The MAPKs include three main groups of proteins: ERK1/2, the JNK/stress-activated protein kinases (SAPK), and the p38 kinases. MAPKs are generally involved in response to stress stimuli.43 Chemopreventive compounds may inhibit cell proliferation by modulating these pathways. Different MAPK pathways are diversely regulated in different cells and compounds. For example, in gliomas, CIL-102 inhibited human astrocytoma cell growth by ERK1/2 activity.44 Evodiamine inhibited cell growth by activating the JNK and p38 pathways.45 On the other hand, nobiletin suppressed glioma cell proliferation by inhibiting p38 and JNK phosphorylation.46 We examined the activity of the MAPK pathway after DMAMCL treatment for 6 hours and found that the phosphorylation levels of p38 and ERK1/2 were increased, but the phosphorylation of JNK was significantly decreased. When the cells were pretreated with NAC, the changes in MAPKs could be reversed. We, therefore, concluded that the ROS/MAPK pathway played a role in DMAMCL-induced growth inhibition. Further studies are required to understand the detailed mechanism.
Conclusion
Our findings indicate that DMAMCL can induce autophagy, autophagosome accumulation, and apoptosis by regulating the Akt/mTOR and ROS/MAPK pathways, leading to inhibited proliferation of the U87-MG and U251 glioma cells (as shown in Figure 8). These novel findings provide a new perspective for DMAMCL in glioma chemotherapeutic interventions.
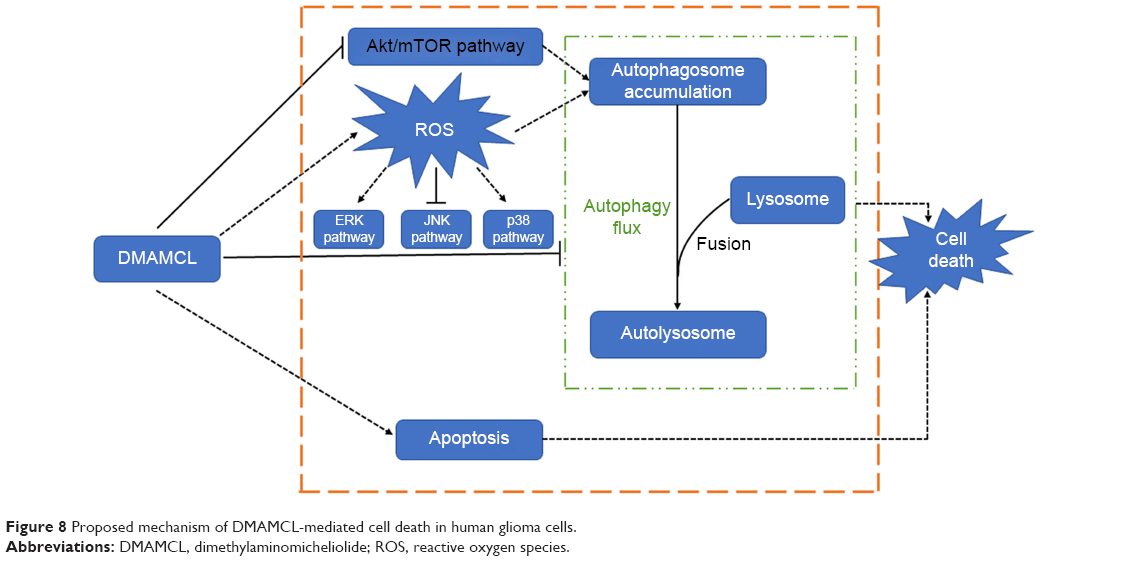 | Figure 8 Proposed mechanism of DMAMCL-mediated cell death in human glioma cells. |
Acknowledgments
This work was supported by Natural Science Foundation of China (Grant No 81771270), Shandong Provincial Natural Science Foundation (Grant No ZR2015HM015), and Jinan Science and Technology Bureau (Grant No 201602170).
Disclosure
The authors report no conflicts of interest in this work.
References
Wang G, Liu M, Wang H, et al. Centrosomal protein of 55 regulates glucose metabolism, proliferation and apoptosis of glioma cells via the Akt/mTOR signaling pathway. J Cancer. 2016;7(11):1431–1440. | ||
Huse JT, Holland EC. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer. 2010;10(5):319–331. | ||
Alexander BM, Cloughesy TF. Adult glioblastoma. J Clin Oncol. 2017;35(21):2402–2409. | ||
Alifieris C, Trafalis DT. Glioblastoma multiforme: pathogenesis and treatment. Pharmacol Ther. 2015;152:63–82. | ||
Jiang Y, Jiao Y, Wang Z, et al. Sinomenine hydrochloride inhibits human glioblastoma cell growth through reactive oxygen species generation and autophagy-lysosome pathway activation: an in vitro and in vivo study. Int J Mol Sci. 2017;18(9):pii:E1945. | ||
Wang Y, Wang K, Zhao J, et al. Multifunctional mesoporous silica-coated graphene nanosheet used for chemo-photothermal synergistic targeted therapy of glioma. J Am Chem Soc. 2013;135(12):4799–4804. | ||
Zhao Y, Chen SJ, Wang JC, et al. Sesquiterpene lactones inhibit advanced oxidation protein product-induced MCP-1 expression in podocytes via an IKK/NF-κB-dependent mechanism. Oxid Med Cell Longev. 2015;2015:1–13. | ||
Ren Y, Yu J, Kinghorn AD. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr Med Chem. 2016;23(23):2397–2420. | ||
Viennois E, Xiao B, Ayyadurai S, et al. Micheliolide, a new sesquiterpene lactone that inhibits intestinal inflammation and colitis-associated cancer. Lab Invest. 2014;94(9):950–965. | ||
Mendonca MS, Turchan WT, Alpuche ME, et al. DMAPT inhibits NF-κB activity and increases sensitivity of prostate cancer cells to x-rays in vitro and in tumor xenografts in vivo. Free Radic Biol Med. 2017;112:318–326. | ||
Fuchs O. Transcription factor NF-κB inhibitors as single therapeutic agents or in combination with classical chemotherapeutic agents for the treatment of hematologic malignancies. Curr Mol Pharmacol. 2010;3(3):98–122. | ||
Shanmugam R, Kusumanchi P, Appaiah H, et al. A water soluble parthenolide analog suppresses in vivo tumor growth of two tobacco-associated cancers, lung and bladder cancer, by targeting NF-κB and generating reactive oxygen species. Int J Cancer. 2011;128(10):2481–2494. | ||
Carlisi D, Buttitta G, di Fiore R, et al. Parthenolide and DMAPT exert cytotoxic effects on breast cancer stem-like cells by inducing oxidative stress, mitochondrial dysfunction and necrosis. Cell Death Dis. 2016;7:e2194. | ||
Hexum JK, Becker CM, Kempema AM, Ohlfest JR, Largaespada DA, Harki DA. Parthenolide prodrug LC-1 slows growth of intracranial glioma. Bioorg Med Chem Lett. 2015;25(12):2493–2495. | ||
Shi C, Wang Y, Guo Y, Chen Y, Liu N. Cooperative down-regulation of ribosomal protein L10 and NF-κB signaling pathway is responsible for the anti-proliferative effects by DMAPT in pancreatic cancer cells. Oncotarget. 2017;8(21):35009–35018. | ||
Jin P, Madieh S, Augsburger LL. The solution and solid state stability and excipient compatibility of parthenolide in feverfew. AAPS PharmSciTech. 2007;8(4):E105. | ||
Zhang Q, Lu Y, Ding Y, et al. Guaianolide sesquiterpene lactones, a source to discover agents that selectively inhibit acute myelogenous leukemia stem and progenitor cells. J Med Chem. 2012;55(20):8757–8769. | ||
Qin X, Jiang X, Jiang X, et al. Micheliolide inhibits LPS-induced inflammatory response and protects mice from LPS challenge. Sci Rep. 2016;6(1):23240. | ||
Ji Q, Ding YH, Sun Y, et al. Antineoplastic effects and mechanisms of micheliolide in acute myelogenous leukemia stem cells. Oncotarget. 2016;7(40):65012–65023. | ||
An Y, Guo W, Li L, et al. Micheliolide derivative DMAMCL inhibits glioma cell growth in vitro and in vivo. PLoS One. 2015;10(2):e0116202. | ||
Burgess DJ. Apoptosis: refined and lethal. Nat Rev Cancer. 2013;13(2):79. | ||
Wang G, Zhang T, Sun W, et al. Arsenic sulfide induces apoptosis and autophagy through the activation of ROS/JNK and suppression of Akt/mTOR signaling pathways in osteosarcoma. Free Radic Biol Med. 2017;106:24–37. | ||
Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1(2):66–74. | ||
Zou J, Zhang Y, Sun J, et al. Deoxyelephantopin induces reactive oxygen species-mediated apoptosis and autophagy in human osteosarcoma cells. Cell Physiol Biochem. 2017;42(5):1812–1821. | ||
Lin L, Baehrecke EH. Autophagy, cell death, and cancer. Mol Cell Oncol. 2015;2(3):e985913. | ||
Carlisi D, Lauricella M, D’Anneo A, et al. The synergistic effect of SAHA and parthenolide in MDA-MB231 breast cancer cells. J Cell Physiol. 2015;230(6):1276–1289. | ||
D’Anneo A, Carlisi D, Lauricella M, et al. Parthenolide generates reactive oxygen species and autophagy in MDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis. 2013;4:e891. | ||
Liu W, Wang X, Sun J, Yang Y, Li W, Song J. Parthenolide suppresses pancreatic cell growth by autophagy-mediated apoptosis. Onco Targets Ther. 2017;10:453–461. | ||
Booth LA, Tavallai S, Hamed HA, Cruickshanks N, Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell Signal. 2014;26(3):549–555. | ||
Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5(11):876–885. | ||
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. | ||
Carroll B, Otten EG, Manni D, et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat Commun. 2018;9(1):256. | ||
Dolma S, Selvadurai HJ, Lan X, et al. Inhibition of dopamine receptor D4 impedes autophagic flux, proliferation, and survival of glioblastoma stem cells. Cancer Cell. 2016;29(6):859–873. | ||
Li C, Liu Y, Liu H, et al. Impact of autophagy inhibition at different stages on cytotoxic effect of autophagy inducer in glioblastoma cells. Cell Physiol Biochem. 2015;35(4):1303–1316. | ||
Wang X, Qiu Y, Yu Q, et al. Enhanced glioma therapy by synergistic inhibition of autophagy and tyrosine kinase activity. Int J Pharm. 2018;536(1):1–10. | ||
Chen Y, Mcmillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008;15(1):171–182. | ||
Helfinger V, Schröder K. Redox control in cancer development and progression. Mol Aspects Med. 2018;63:88–98. | ||
Thorpe GW, Fong CS, Alic N, Higgins VJ, Dawes IW. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proc Natl Acad Sci U S A. 2004;101(17):6564–6569. | ||
Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73(17):3221–3247. | ||
Wang J, Liu X, Hong Y, et al. Ibrutinib, a Bruton’s tyrosine kinase inhibitor, exhibits antitumoral activity and induces autophagy in glioblastoma. J Exp Clin Cancer Res. 2017;36(1):96. | ||
Poornima P, Weng CF, Padma VV. Neferine from Nelumbo nucifera induces autophagy through the inhibition of PI3K/Akt/mTOR pathway and ROS hyper generation in A549 cells. Food Chem. 2013;141(4):3598–3605. | ||
Sami A, Karsy M. Targeting the PI3K/Akt/mTOR signaling pathway in glioblastoma: novel therapeutic agents and advances in understanding. Tumour Biol. 2013;34(4):1991–2002. | ||
Genestra M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell Signal. 2007;19(9):1807–1819. | ||
Teng C-C, Kuo H-C, Cheng H-C, Wang T-C, Sze C-I. The inhibitory effect of CIL-102 on the growth of human astrocytoma cells is mediated by the generation of reactive oxygen species and induction of ERK1/2 MAPK. Toxicol Appl Pharmacol. 2012;263(1):73–80. | ||
Wang X, Zou S, Lan Y-L, Xing J-S, Lan X-Q, Zhang B. Solasonine inhibits glioma growth through anti-inflammatory pathways. Am J Transl Res. 2017;9(9):3977–3989. | ||
Lien L-M, Wang M-J, Chen R-J, et al. Nobiletin, a polymethoxylated flavone, inhibits glioma cell growth and migration via arresting cell cycle and suppressing MAPK and Akt pathways. Phytother Res. 2016;30(2):214–221. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.