")
Back to Journals » OncoTargets and Therapy » Volume 9
Role of tyrosine-kinase inhibitors in myeloproliferative neoplasms: comparative lessons learned
Authors Pinilla-Ibarz J, Sweet K, Corrales-Yepez G, Komrokji R, Linder B
Received 15 December 2015
Accepted for publication 8 April 2016
Published 10 August 2016 Volume 2016:9 Pages 4937—4957
DOI https://doi.org/10.2147/OTT.S102504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Javier Pinilla-Ibarz, Kendra L Sweet, Gabriela M Corrales-Yepez, Rami S Komrokji
Department of Malignant Hematology, H Lee Moffitt Cancer Center and Research Institute, Tampa, FL, USA
Abstract: An important pathogenetic distinction in the classification of myeloproliferative neoplasms (MPNs) is the presence or absence of the BCR–ABL fusion gene, which encodes a unique oncogenic tyrosine kinase. The BCR–ABL fusion, caused by the formation of the Philadelphia chromosome (Ph) through translocation, constitutes the disease-initiating event in chronic myeloid leukemia. The development of successive BCR–ABL-targeted tyrosine-kinase inhibitors has led to greatly improved outcomes in patients with chronic myeloid leukemia, including high rates of complete hematologic, cytogenetic, and molecular responses. Such levels of treatment success have long been elusive for patients with Ph-negative MPNs, because of the difficulties in identifying specific driver proteins suitable as drug targets. However, in recent years an improved understanding of the complex pathobiology of classic Ph-negative MPNs, characterized by variable, overlapping multimutation profiles, has prompted the development of better and more broadly targeted (to pathway rather than protein) treatment options, particularly JAK inhibitors. In classic Ph-negative MPNs, overactivation of JAK-dependent signaling pathways is a central pathogenic mechanism, and mutually exclusive mutations in JAK2, MPL, and CALR linked to aberrant JAK activation are now recognized as key drivers of disease progression in myelofibrosis (MF). In clinical trials, the JAK1/JAK2 inhibitor ruxolitinib – the first therapy approved for MF worldwide – improved disease-related splenomegaly and symptoms independent of JAK2V617F mutational status, and prolonged survival compared with placebo or standard therapy in patients with advanced MF. In separate trials, ruxolitinib also provided comprehensive hematologic control in patients with another Ph-negative MPN – polycythemia vera. However, complete cytogenetic or molecular responses with JAK inhibitors alone are normally not observed, underscoring the need for novel combination therapies of JAK inhibitors and complementary agents that better address the complexity of the pathobiology of classic Ph-negative MPNs. Here, we discuss the role of tyrosine-kinase inhibitors in the current MPN-treatment landscape.
Keywords: chronic myeloid leukemia, myelofibrosis, tyrosine-kinase inhibitors, ruxolitinib, JAK inhibitor
Introduction
Myeloproliferative neoplasms (MPNs) are a heterogeneous group of hematologic malignancies characterized by the increased clonal proliferation of one or more myeloid lineages.1,2 Classic MPNs include chronic myeloid leukemia (CML), primary myelofibrosis (PMF), polycythemia vera (PV), and essential thrombocythemia (ET), whereas nonclassic MPNs include less common malignancies, such as chronic neutrophilic leukemia (CNL), chronic eosinophilic leukemia not otherwise specified (CEL-NOS), mast-cell disease (mastocytosis), and MPNs unclassifiable.3 MPNs are further divided into two groups based on the presence or absence of the Philadelphia chromosome (Ph), formed by a translocation event and housing the constitutively activated fusion tyrosine kinase (TK) gene BCR–ABL.2 BCR–ABL positivity is exclusively associated with CML, whereas Ph-negative MPNs are a heterogeneous group that includes all other MPNs.2
The identification of the BCR–ABL fusion gene in the pathogenesis of CML and the subsequent development of TK inhibitors (TKIs) that target the BCR–ABL fusion protein were major breakthroughs in cancer therapy and recognized as one of the best examples of rationally developed targeted drugs in oncology. Since then, characterization of mutations in other MPNs has improved the understanding of the pathogenesis of these diseases; however, unlike CML, the disease may be driven by more than one mutation or mechanism. Consequently, clinical development of specific TKIs has focused on targeting the abnormal signaling (eg, JAK pathway) involved in the pathogenesis of Ph-negative MPNs. The objective of this review is to provide the basis to understand the role of TKIs in the treatment of MPNs and why the mechanisms and benefits of therapeutic action differ between Ph-negative and Ph-positive diseases.
Pathobiology of MPNs
Classic Ph-positive MPNs: CML
CML is a myeloid malignancy originating from the clonal proliferation of a pluripotent stem cell in the bone marrow4 and involving one or all myeloid lineages.5 CML is an exemplary paradigm of a neoplasm that is essentially defined by a singular, unique cytogenetic abnormality, the Ph chromosome, resulting from a reciprocal chromosomal translocation between the long arms of chromosomes 9 and 22.6 The molecular consequence of this t(9;22) translocation is the creation of BCR–ABL, a chimeric protein with constitutive oncogenic TK activity.7 Expression of BCR–ABL results in the upregulation of multiple downstream oncogenic pathways,8 and has been shown to be the initiating event for the leukemogenesis of CML.9 Additionally, several lines of evidence indicate that disease progression from chronic phase (CP) to blast phase is driven by amplification of BCR–ABL in cooperation with other pathways, such as β-catenin.10,11
CML has a triphasic natural history: the initial indolent CP precedes an accelerated phase and blast phase. Most patients are diagnosed in CP, with 20%–40% of newly diagnosed patients being asymptomatic at presentation.4 The estimated yearly incidence of CML is approximately 1.8 per 100,000 persons in the US, accounting for about 15% of all adults with leukemia.12,13 Prior to the introduction of TKIs for CML, patients had a dismal prognosis, with an 8-year survival rate of 42%–65% from 1983–2000 for patients in CP.14
Classic Ph-negative MPNs: PV, ET, and PMF
PV, ET, and PMF are Ph-negative neoplastic hematopoietic stem cell disorders characterized by the uncontrolled clonal proliferation of myeloid progenitor cells affecting one or more myeloid lineages.1,2 These classic MPNs are relatively rare, and have a prevalence that varies from four to six per 100,000 persons for myelofibrosis (MF) to 45–57 and 39–57 per 100,000 persons for PV and ET, respectively.15 Although classic Ph-negative MPNs are distinct diseases, they have a common pathobiology of hyperactivation of the JAK–STAT pathway, and may share specific molecular and clinical features, including somatic stem cell mutations, elevated blood cell counts resulting from aberrant myeloproliferation, and constitutional symptoms. However, patients with ET, PV, and PMF differ regarding life expectancy at diagnosis, risk of leukemic transformation,16 rate of disease progression, and clinical presentation. Patients with any of these Ph-negative MPNs have a risk of progression to acute myeloid leukemia, which ranges from 2%–7% for PV and ET to 8%–30% for PMF.16 Patients with PV and ET have an approximate 10%–20% probability of evolving to post-PV MF or post-ET MF.16 In recognition of the largely indistinguishable clinical manifestations, clinical course, and histopathological features of PMF, post-PV MF, and post-ET MF,17 these diseases are collectively referred to as MF in this review.
The clinical presentation of MF is heterogeneous, with patients experiencing various degrees of bone marrow fibrosis, extramedullary hematopoiesis, spleen enlargement, anemia, and MF-related symptoms, many of which appear to be linked to the excessive production of inflammatory cytokines. The often-debilitating symptom burden of MF has a profound negative effect on quality of life and may shorten survival.18–20 PV is characterized by an increase in red blood cell production, low serum erythropoietin, and megakaryocyte pleomorphism in the bone marrow, whereas ET is associated with increased platelet production and increased megakaryocytic mass.21,22
Unlike CML, classic Ph-negative MPNs are characterized by complex and variable mutation profiles and clonal evolution with unknown disease-initiating event(s) (Figure 1).23 In the past decade, the identification and characterization of specific mutations associated with PV, ET, and PMF (Table 1) have begun to elucidate the underlying molecular pathobiology of classic Ph-negative MPNs and have prompted the development of targeted agents and disease-specific diagnostic tools.24–30
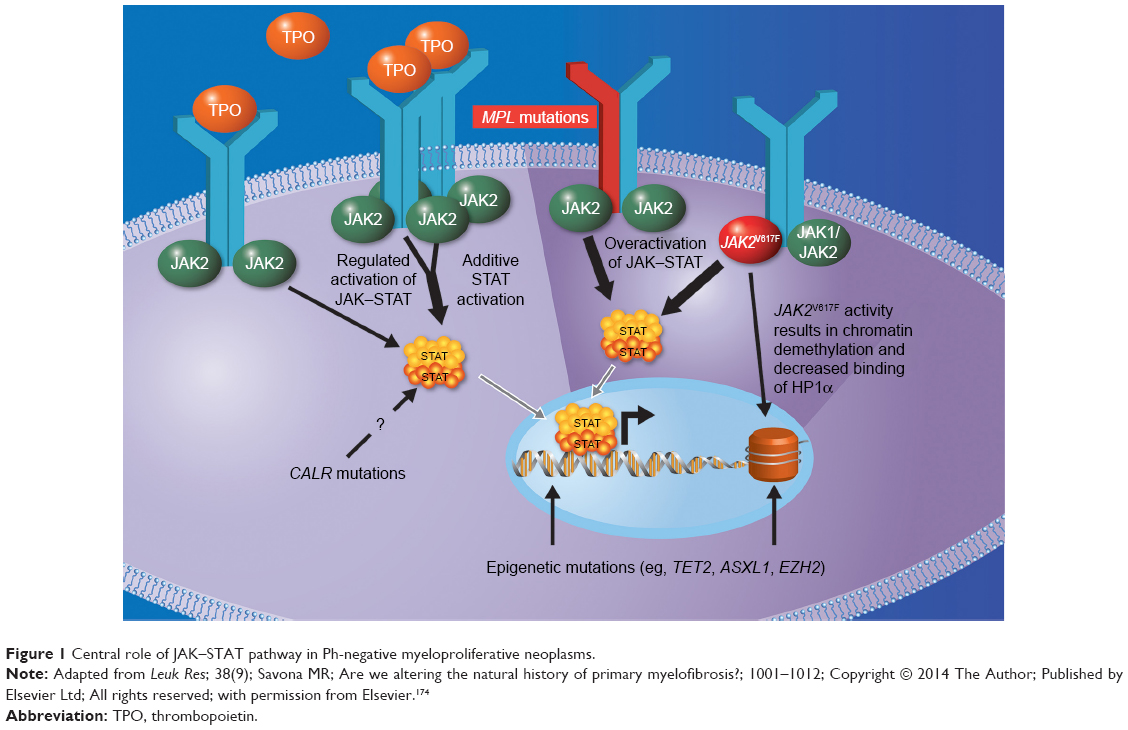 | Figure 1 Central role of JAK–STAT pathway in Ph-negative myeloproliferative neoplasms. |
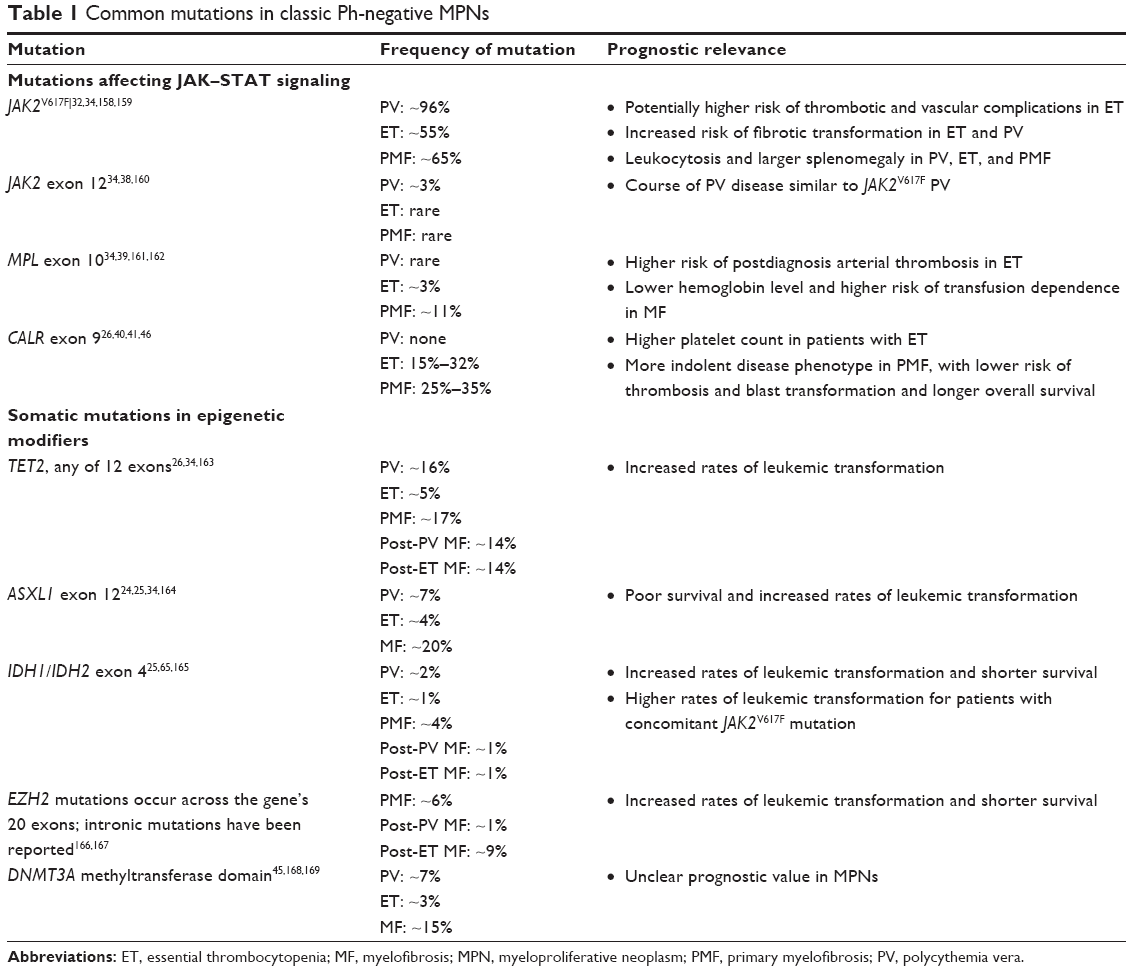 | Table 1 Common mutations in classic Ph-negative MPNs |
Mutations affecting the JAK–STAT pathway
JAK2-mediated transduction of signals by activated hematopoietic cytokine receptors is crucial for hematopoiesis. The identification of the JAK2V617F gain-of-function mutation in patients with classic Ph-negative MPNs31 provided the first clue to the pathogenic mechanism of these diseases. As a somatic mutation in hematopoietic stem cells that enables constitutive JAK2 activity in the absence of cytokines binding to their cognate receptors, this seemed to be an ideal candidate to explain the pathogenesis of MPNs, prompting numerous development efforts for JAK2-targeted therapies. However, patients without this mutation also develop Ph-negative MPNs and have evidence of increased JAK–STAT activity. The JAK2V617F mutation is present in >95% of patients with PV and >50% of patients with ET or PMF (Table 1).32–34 Moreover, even when present, the JAK2V617F mutation does not appear to be sufficient as a disease-initiating event.26
Intensive research in the last decade has not only revealed an unexpected genetic complexity of these diseases,31 but also suggests that aberrant JAK–STAT activation is a central defect, even in patients without the JAK2V617F mutation.28 Interestingly, disease-related hyperactivation of JAK–STAT signaling appears not to be limited to the mutant clonal stem cells, and is seen in “normal” nonclonal cells, particularly in MF, which is characterized by high circulating levels of inflammatory cytokines. The postulated role of JAK–STAT signaling in maintaining a state of excess inflammation and catabolism that promotes constitutional symptoms and cachexia appears to be validated by clinical evidence showing that JAK inhibition substantially reduces the levels of inflammatory cytokines, reverses signs of cachexia (eg, weight loss), and significantly reduces symptom burden compared with placebo or conventional therapy.35,36
In addition to JAK2V617F, a number of mutations have been identified that lead to hyperactivation of the JAK–STAT pathway in classic Ph-negative MPNs (Table 1).37 Approximately 3% of all patients with PV have mutations in JAK2 exon 12,34,38 and patients with PMF or ET may have mutations in the thrombopoietin-receptor gene MPL39 or in the CALR gene. With few exceptions, CALR, JAK2, and MPL mutations appear to be mutually exclusive within the same patient.24,26,40,41 Although the precise mechanism of action of the CALR mutation remains unclear, evidence from an integrated genomic analysis28 and preclinical studies26,41 suggests that CALR mutations facilitate cytokine-independent JAK–STAT pathway activation. This provides a strong rationale for the use of JAK-inhibitor therapy in MPN patients with CALR mutations, and may at least in part be responsible for the clinical responses to JAK-inhibitor therapy observed in patients with JAK2V617F-negative MF.29,35,36,42,43
Mutations in epigenetic modifiers
Genetic aberrations present in patients with classic Ph-negative MPNs often include somatic mutations in epigenetic modifiers, such as TET2, ASXL1, DNMT3A, EZH2, or IDH1 (Table 1), which may be acquired prior to or after JAK2V617F or CALR mutations. Recent evidence suggests that the order of acquiring mutations may determine the specific MPN phenotype that a patient develops.44 Although no particular epigenetic mutation has a reported frequency >20%, overall, mutations in genes encoding epigenetic regulators appear to be common in classic Ph-negative MPNs. The prognostic and clinical relevance of these mutations in different MPNs is still being elucidated;26,32 however, there is evidence that some mutations in epigenetic modifiers may increase the risk of leukemic transformation and/or shorten survival.26,45 Moreover, the number of mutations occurring in addition to those in JAK2, MPL, or CALR has been shown to affect outcomes. Patients harboring more than one mutation have a markedly increased probability of leukemic transformation and shortened survival.26 In patients with MF, “triple-negative” mutation status (ie, no mutation in JAK2, MPL, or CALR) is associated with the worst outcome, and the presence of ASXL1 mutation is an additional independent prognostic factor for poor survival.24,46
Nonclassic Ph-negative MPNs
Nonclassic MPNs are less common Ph-negative hematologic disorders that include CNL, CEL-NOS, mastocytosis, and MPNs unclassifiable.3,47 Recent discoveries of genetic abnormalities in nonclassic Ph-negative MPNs have paved the way for the development of targeted therapies that may impact the therapeutic approach to these rarer diseases that have historically had few treatment options.
Mutations in the CSF3R gene have recently been identified in 59% of patients with CNL or atypical CML (aCML).48 Functional genomic data indicate the existence of two types of CSF3R mutations with differential downstream effects: truncation mutations that activate the TNK2 and Src pathways, and membrane-proximal mutations that activate the JAK–STAT pathway.48 This has provided the rationale for the investigation of JAK or Src inhibitors to target membrane-proximal or truncation CSF3R mutations, respectively, in patients with CNL and aCML.48
Approximately 93% of adult patients with indolent and aggressive systemic mastocytosis carry the KITD816V mutation, which results in constitutive activation of the KIT TK and unrestricted cellular proliferation.49,50 Presence of this mutation is associated with higher bone marrow mast-cell burden and the presence of C-findings, including marrow dysfunction with cytopenias, hepatomegaly, splenomegaly, osteolysis, pathological fractures, weight loss, and leukemic transformation, which may be indicative of poor prognosis.51 Additional pathways also have been implicated in the pathobiology of the disease, supporting the rationale for a multitargeted therapeutic approach.50
Compared with the pathogenesis of CML, which is essentially driven by a de novo oncoprotein resulting from a unique chromosomal translocation, the pathobiology of classic Ph-negative MPNs is both variable and highly complex, often involving point mutations in multiple genes that are difficult to target without affecting nononcogenic pathways. The JAK2V617F mutation found in the majority of patients with these diseases is neither essential nor specific for any of these MPNs. Moreover, the JAK2V617F mutation is not a disease-initiating event, and many questions remain regarding the precise pathogenic mechanisms of other mutations. However, dysregulation of JAK–STAT signaling in and beyond hematopoietic stem cells is a dominant defect in classic Ph-negative MPNs, and current evidence validates the continued targeting of JAK–STAT signaling in MPNs, even in patients without mutations in JAK2.
Because of the overall lack of specificity of patients’ mutation profiles and overlapping clinical features at presentation, particularly at early disease stages, a combination of genetic, clinical, cytogenetic, and histomorphologic criteria is required for the definitive diagnosis of classic MPNs.
Diagnostic criteria
CML
Detectable presence of the Ph chromosome is required to confirm a CML diagnosis and to distinguish it from all other MPNs.52 The initial evaluation of patients with suspected CML should include physical examination, including spleen measurements, complete blood count with differential, chemistry profile, bone marrow morphology and cytogenetics, and real-time quantitative polymerase chain reaction (PCR) using the international scale for BCR–ABL1 (Table 2).4,12 Most patients are diagnosed in CP and have distinct laboratory features, such as leukocytosis with all stages of maturation, basophilia, thrombocytosis, and hypercellularity in the bone marrow.4
 | Table 2 Diagnostic criteria for classic MPNs |
Although the presence of BCR–ABL can be assessed by fluorescence in situ hybridization or quantitative reverse-transcriptase PCR in the peripheral blood, cytogenetic evaluation of bone marrow aspirate is recommended to detect cytogenetic abnormalities and perform accurate staging into the chronic phase, accelerated phase, or blast phase.12,53 Multiple staging criteria for CML have been developed; however, the different thresholds used to distinguish between the chronic phase, accelerated phase, and blast phase have not been validated in clinical trials. Most CML trials have used the staging system developed by the MD Anderson Cancer Center, which defines accelerated phase as ≥15% blasts, ≥30% blasts plus promyelocytes, ≥20% basophils in blood or bone marrow, platelets <100×109/L unrelated to therapy, or cytogenetic clonal evolution; and the blast phase as ≥30% blasts in blood or bone marrow in the presence of extramedullary disease with localized immature blasts.53,54
Ph-negative MPNs
Diagnosis of classic Ph-negative MPNs is based on the evaluation of clinical, histopathologic, morphologic, molecular, and cytogenetic variables (Table 2).3 Unlike the presence of BCR–ABL (which is sufficient for a CML diagnosis), the presence of JAK2 mutations is used to support the diagnosis of Ph-negative MPNs, but cannot differentiate between subtypes.3 Owing to the lack of strictly disease-specific mutations in these MPNs and the possibility of phenotypic resemblance at early disease stage, histopathologic and morphologic examination of the bone marrow is necessary for a definitive diagnosis of ET and PMF.3 In addition, although bone marrow histomorphology is a minor diagnostic criterion for PV, a bone marrow biopsy showing age-adjusted hypercellularity with trilineage growth (panmyelosis) is very specific for PV and necessary to exclude reactive polycythemias in patients with JAK2 mutation-negative PV3 and ET in patients with masked PV.55
Nonclassic Ph-negative MPNs
Diagnosis of nonclassic MPNs can be difficult, and generally requires absence of BCR–ABL, dyserythropoiesis, and granulocyte dysplasia.3,47 Additional molecular and histologic parameters are used to discern between nonclassic MPNs, with particular focus on morphology, which is a key criterion in the diagnosis of these rare malignancies. The major criteria for diagnosis are listed in Table 2. Notably, many major criteria mandate confirmation that the patient has no other Ph-negative MPN or CML.
Prognostic risk stratification
CML
Certain clinical characteristics at diagnosis may have prognostic value in patients with CML, and may guide optimal treatment selection and clinical management of the disease.56 For over 30 years, prognostic scoring systems for risk stratification of patients with CML have been widely used to predict outcome. Their development reflects the increased understanding of the disease and the evolution of therapeutic strategies available for patients through the years, from conventional therapy in the 1980s to the use of TKIs in the new millennium.56 Current prognostic scoring systems, including the Sokal, Hasford (also known as Euro), and EUTOS systems, classify patients into low-, intermediate-, and high-risk groups, although the EUTOS system does not include a definition for the intermediate-risk category (Table 3).57–59
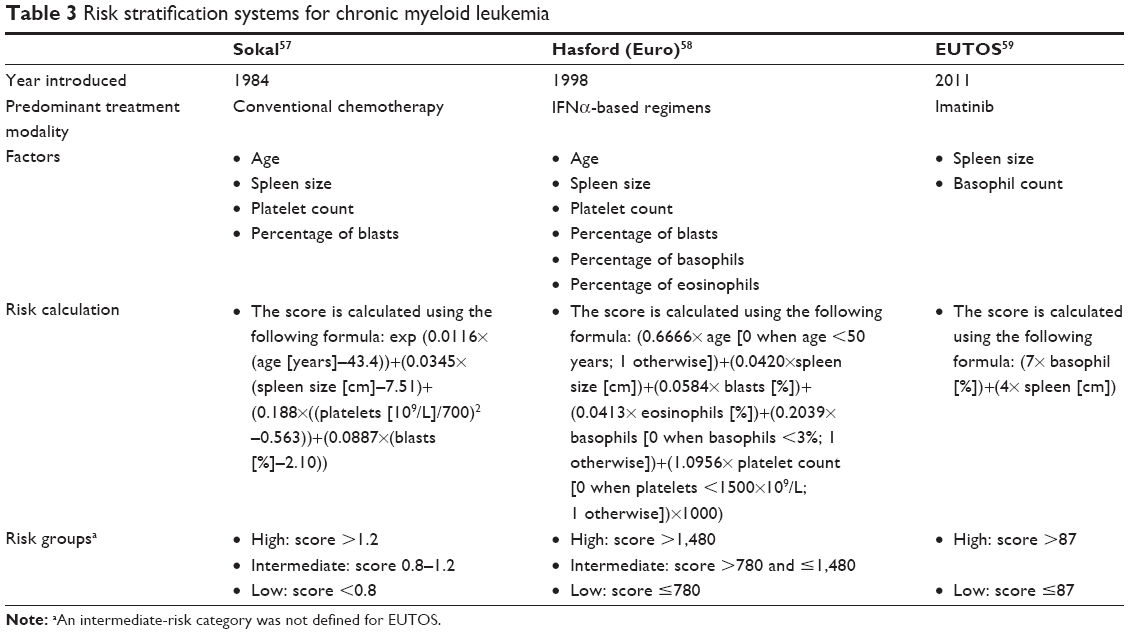 | Table 3 Risk stratification systems for chronic myeloid leukemia |
The Sokal and Hasford scoring systems were developed before the introduction of TKIs, and account for a patient’s age, spleen size, platelet count, and percentage of blasts at diagnosis.57,59 In addition to these categories, the Hasford system also includes percentage of basophils and eosinophils.57,59 In a study of 813 patients diagnosed between 1962 and 1981, the Sokal system estimated a median survival of 60 and 30 months for low- and high-risk patients, respectively.57 Similar results were obtained with the Hasford system in a meta-analysis of 1,303 patients treated between 1983 and 1994, which estimated median survival of 96 and 42 months for low- and high-risk patients, respectively.59 The EUTOS scoring system is the latest prognostic tool, and was developed after imatinib therapy became the standard of care. This system considers only spleen size and basophil count at the time of diagnosis.58
The prognostic validity of the Sokal, Hasford, and EUTOS systems needs further clarification, given the current use of TKIs as the standard of care in CML. In a study comparing the three scoring systems in 2,060 patients receiving first-line imatinib, the EUTOS scoring system proved to be superior to the Sokal and Hasford systems in predicting 5-year progression-free survival (PFS) and the probability of reaching complete cytogenetic response (CCyR) at 18 months.58 Similarly, in two additional studies of front-line imatinib in 1,288 and 143 patients with CML, the EUTOS score was superior to the Sokal and Hasford scoring systems in predicting CCyR, event-free survival, and overall survival.60,61 However, another study in patients with early CP CML receiving first-line imatinib or second-generation TKIs did not find a correlation between the EUTOS risk stratification and outcomes.62 Currently, only the Sokal and Hasford scoring systems are recommended by the National Comprehensive Cancer Network (NCCN) guidelines.12 These scoring systems may require additional updates, given that the prognostic parameters in the current scoring systems are limited and do not account for additional genetic or molecular biomarkers.56
Ph-negative MPNs
Risk stratification of patients with PMF is determined according to the International Prognostic Scoring System (IPSS) at the time of diagnosis, and according to the dynamic IPSS (DIPSS) at any time during the course of the disease (Table 4).20,63 Both the IPSS and DIPSS estimate survival of patients with PMF by accounting for five independent factors: age >65 years, hemoglobin <10 g/dL, leukocyte count >25×109/L, peripheral blood blasts ≥1%, and presence of disease-related constitutional symptoms. In addition, the DIPSS assigns higher prognostic weight to the acquisition of anemia over time, given its negative effect on survival compared with all other parameters.20,63 A third prognostic tool is the DIPSS Plus scoring system, which predicts overall survival in patients with PMF by combining the risk factors used in the DIPSS with platelet count <100×109/L, transfusion dependence, and unfavorable karyotype during disease course. Additionally, unfavorable karyotype and platelet count <100×109/L have been demonstrated to be independent predictors of leukemia-free survival and leukemic transformation.64 All three prognostic scoring systems divide patients into four distinct risk categories according to each individual score: low-, intermediate 1-, intermediate 2-, or high-risk.20,63 However, the dynamic risk assessment provided by the DIPSS or DIPSS Plus may be a more valuable tool than the IPSS in guiding clinical choices that accurately address the patient’s disease course.63 Despite disease similarities between de novo and transformed MF and the common use of prognostic models for PMF to drive treatment decisions in patients with post-ET MF or post-PV MF, recent research has suggested that the IPSS may not accurately discriminate different risk categories in patients with post-ET MF or post-PV MF.24,27,65 A separate scoring system may be warranted to aid physicians in treatment of these patients.66 In addition, recent studies suggest that the specific mutation profiles may have a significant effect on prognosis in patients with PMF.24,27,65
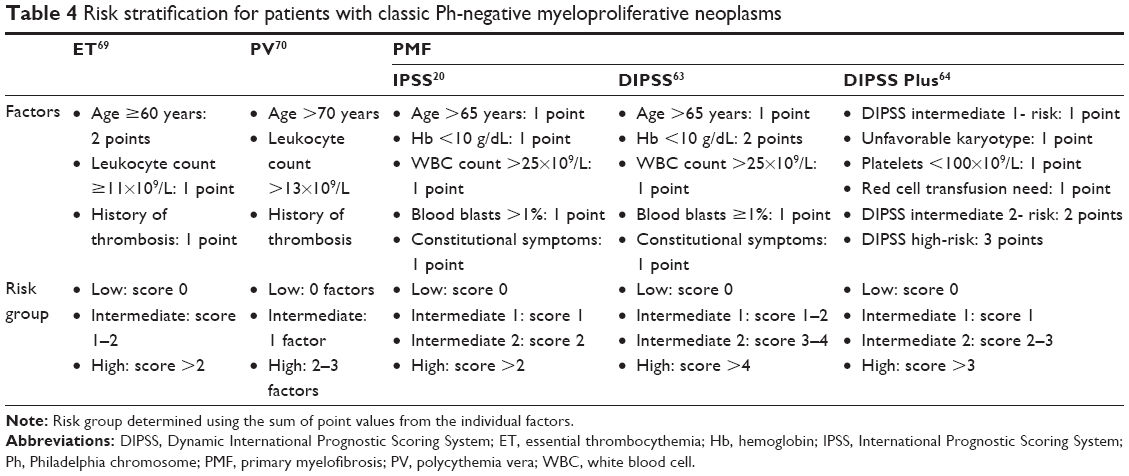 | Table 4 Risk stratification for patients with classic Ph-negative myeloproliferative neoplasms |
Owing to the increased risk of potentially fatal thrombosis and of microvascular vasomotor disturbances associated with ET and PV, prognostic stratification for these diseases aims to estimate the probability of thrombotic complications.67,68 The International Prognostic Score for ET predicts survival based on age, leukocyte count, and history of thrombosis (Table 4). According to the IPSS for ET, patients with ET are divided into three risk categories associated with substantial differences in survival: low-risk (median survival not reached), intermediate-risk (median survival 24.5 years), and high-risk (median survival 13.8 years).69 Patients with PV are categorized as high- or low-risk based on age, leukocyte count, and history of previous thrombosis.70 Abnormal karyotype has recently emerged as an additional prognostic factor correlated with shortened survival in these patients, but is not currently considered in prognostic models.71
TKI-based therapies for MPNs
CML
Owing to the involvement of BCR–ABL in disease initiation and progression, targeted therapeutic strategies have focused on the development of small molecules that inhibit the oncogenic constitutive activation of this TK. The role of BCR–ABL as the principal driver of CML has led to a high treatment success rate with these targeted inhibitors.
Imatinib
Imatinib was the first BCR–ABL inhibitor approved in 2001 for the treatment of patients with blast crisis, accelerated phase, or CP CML who failed IFNα therapy, and was subsequently approved in 2002 for newly diagnosed CP CML (Table 5).72 Imatinib was compared with standard IFNα plus low-dose cytarabine in the Phase III IRIS study of 1,106 patients with newly diagnosed CP CML.73 At the 18-month follow-up, imatinib was superior to IFNα plus low-dose cytarabine in terms of response (complete hematologic response [CHR], major CyR [MCyR], and CCyR), decreased likelihood of progression to accelerated phase or blast crisis, and tolerability (Table 6). Durable response rates, decreased progression to more advanced phases, and an overall survival benefit were maintained with long-term imatinib therapy compared with standard chemotherapy.74,75
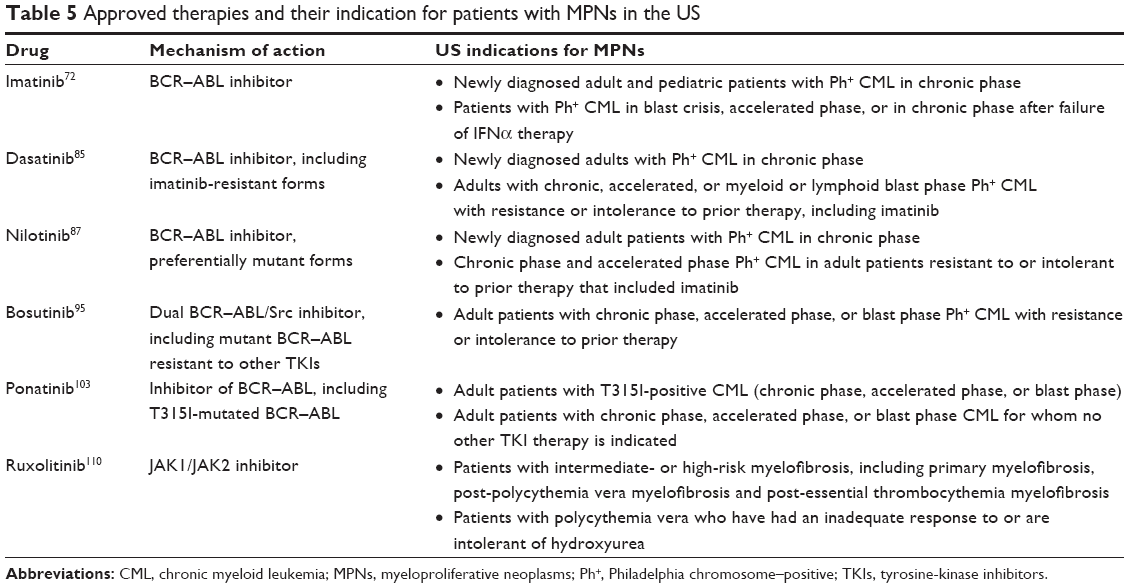 | Table 5 Approved therapies and their indication for patients with MPNs in the US |
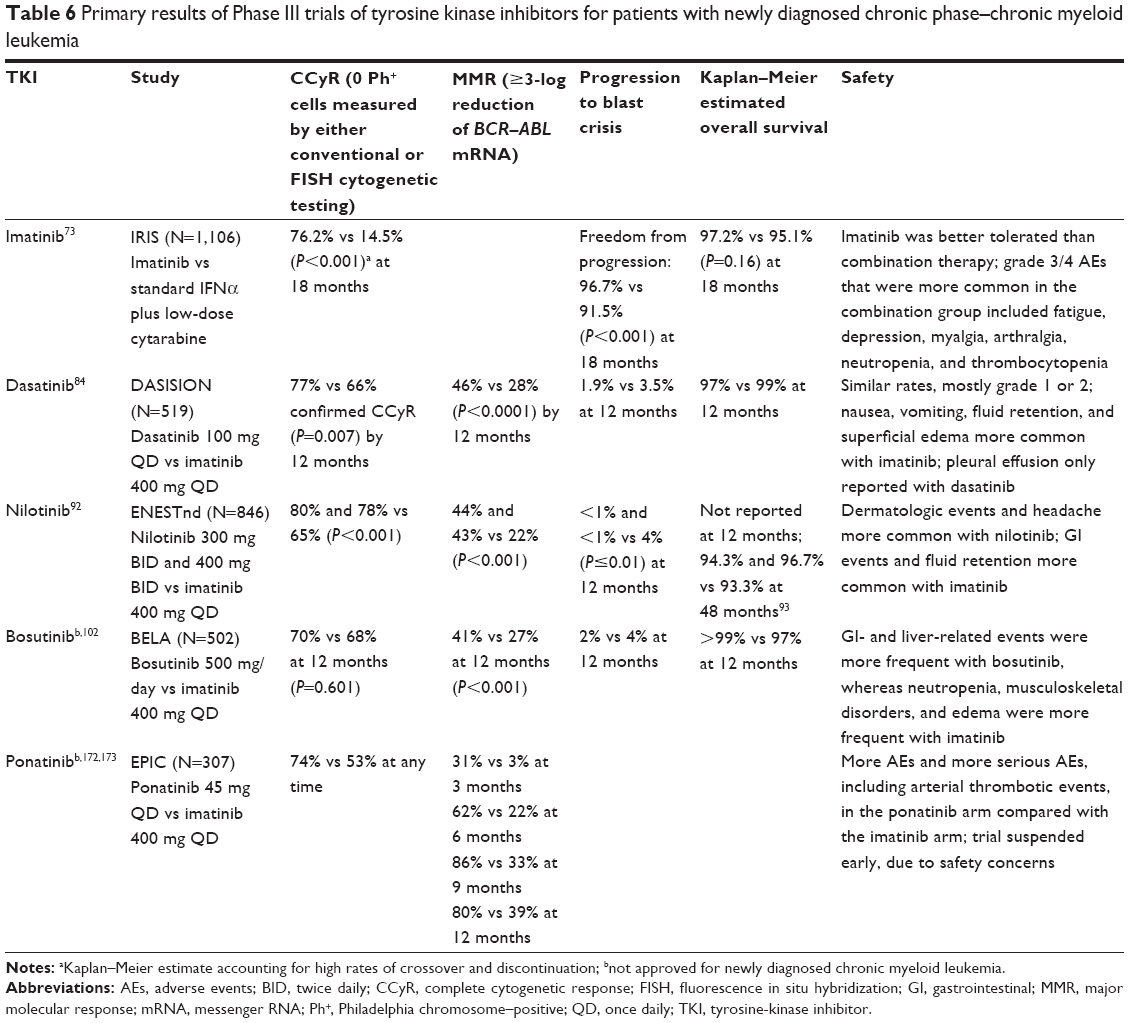 | Table 6 Primary results of Phase III trials of tyrosine kinase inhibitors for patients with newly diagnosed chronic phase–chronic myeloid leukemia |
Despite these promising results, an estimated 20%–30% of patients who achieve CHR and/or CCyR eventually develop secondary (acquired) resistance to imatinib.76,77 Approximately 40%–90% of imatinib-resistant patients develop secondary resistance as a result of mutations arising in the BCR–ABL kinase domain. These genetic aberrations include the T315I mutation, which changes the conformation of BCR–ABL, thus preventing imatinib from accessing the protein’s adenosine triphosphate-binding pocket.76–80 Furthermore, several additional BCR–ABL-independent mechanisms of imatinib resistance, such as intracellular drug uptake, Src overexpression, leukemic stem cell quiescence, and clonal evolution, are currently under investigation.76,77 Second- and third-generation TKIs, such as dasatinib, nilotinib, bosutinib, and ponatinib were designed to overcome imatinib resistance.
Dasatinib
Dasatinib is an oral BCR–ABL inhibitor that is more potent than imatinib and can inactivate several imatinib-resistant forms of BCR–ABL, with the exception of T315I/A, Q252H, E255K/V, F317L/I, and V299L.81,82 Dasatinib also inhibits Src family TKs, which have been shown to contribute to imatinib resistance through the activation of BCR–ABL-independent oncogenic pathways.79 In the Phase II START-C trial, dasatinib 70 mg twice daily (BID) resulted in durable hematologic and CyRs in patients (n=387) with CP CML resistant to or intolerant of imatinib. This included CCyR in 49% of patients (40% imatinib resistant and 75% imatinib intolerant) at a median follow-up of 15.2 months.83 Grade 3/4 adverse events (AEs) consisted mainly of myelosuppression, pleural effusion, and dyspnea. A Phase III follow-on study comparing four dosing regimens of dasatinib in 724 patients found that dasatinib 100 mg once daily (QD) resulted in comparable responses to the 70 mg BID dose and necessitated fewer dose reductions or interruptions.83 Furthermore, the 100 mg QD dose resulted in less toxicity, with significantly lower rates of any grade pleural effusion and grade 3/4 thrombocytopenia. Discontinuation due to toxicity occurred in 4% of patients receiving 100 mg QD versus 11% receiving 70 mg BID. PFS and overall survival rates at 24 months were 80% and 91%, respectively, for 100 mg QD, and 76% and 88%, respectively, for 70 mg BID.84 Based on the more favorable benefit–risk profile, the 100 mg QD dose of dasatinib was selected as the starting dose and approved for the treatment of patients with CP CML resistant or intolerant to imatinib.83,84
When dasatinib was compared directly with imatinib in 519 patients with newly diagnosed CP CML enrolled in the Phase III DASISION trial, responses occurred more rapidly and to a greater degree in the dasatinib arm. Significantly more patients treated with dasatinib achieved a CCyR or a major molecular response (MMR) compared with those treated with imatinib by 12 months (Table 6).84 Rates of disease progression were also numerically lower for dasatinib. Based on this trial, dasatinib was approved in 2010 as first-line treatment for patients with newly diagnosed CP CML (Table 5).85 At the 4-year follow-up, dasatinib continued to demonstrate higher and faster cytogenetic and molecular responses compared with imatinib, with no unexpected toxicity through year 4. However, no long-term differences in overall survival (93% vs 92%) or PFS (90% vs 90%) rates were reported.86
Nilotinib
Nilotinib is also an oral inhibitor of BCR–ABL indicated for first- and second-line treatment of patients with CP CML (Table 5).87 Nilotinib binds to BCR–ABL more tightly than imatinib, and inhibits most of the clinically relevant mutated forms of BCR–ABL, with the exception of T315I, Y253H, E255K/V, and F359V/C.88 In a Phase II study of 280 patients with CP CML after imatinib failure or intolerance, nilotinib 400 mg BID resulted in MCyR and CCyR at 6 months in 48% and 31% of patients, respectively.89 Nilotinib treatment was well tolerated, with a safety profile that included mainly low-grade dermatologic events and transient grade 3 or 4 myelosuppression. The 48-month estimated rate of overall survival was 78%, and was positively correlated with deeper levels of molecular responses at 3 and 6 months.90 Furthermore, long-term results indicated that patients who were in CHR at baseline had significantly higher PFS rates compared with those who did not have responses with initial therapy (71% vs 49%, P=0.001).90 Nilotinib was also effective in patients with imatinib-resistant or -intolerant accelerated phase CML.91 The MCyR rate was 29% in this population of patients who had limited therapeutic options, and overall survival was 79% after 12 months of follow-up. The most common grade ≥3 AEs were thrombocytopenia, neutropenia, and bilirubin and lipase elevations.
The Phase III ENESTnd trial in patients with newly diagnosed CP CML showed that nilotinib was associated with higher rates of CCyR and MMR at 12 months compared with imatinib (Table 6); these higher response rates were observed even among patients with Sokal high-risk CML.92 Furthermore, patients treated with nilotinib at either dose had significantly lower rates of disease progression compared with imatinib. At the 4-year follow-up, nilotinib treatment remained associated with significantly higher rates of MMR and freedom from progression to accelerated phase or blast phase compared with imatinib; however, overall survival and PFS rates with nilotinib and imatinib were similar.93 A recent analysis from this trial found that nilotinib was associated with a reduced incidence of treatment-emergent BCR–ABL mutations compared with imatinib.94
Despite the promising responses observed with long-term dasatinib and nilotinib, neither agent has been effective against the T315I mutation, and neither has demonstrated improved overall survival compared with imatinib. Furthermore, some patients develop resistance to or are intolerant of these first-line therapies. Additional therapeutic agents, described herein, have been approved for the treatment of patients with CML after failure of previous therapies and for the treatment of patients with the T315I mutation.
Bosutinib
Bosutinib is a dual BCR–ABL/Src TKI approved for second-line treatment of patients who did not respond to a previous TKI (Table 5).95 It is effective against many BCR–ABL mutations that are resistant to imatinib, dasatinib, and nilotinib, with the exception of T315I and V299L.96,97 In a Phase I/II study of 288 patients with CP CML who were resistant to or intolerant of imatinib therapy, bosutinib treatment was associated with CHR, MCyR, and CCyR rates of 86%, 53%, and 41%, respectively, after a median follow-up of 24.2 months.98 Bosutinib treatment was generally well tolerated, with mild-to-moderate transient myelosuppression, gastrointestinal toxicity, and rash. At 4 years, the rate of disease progression to accelerated phase or blast phase was 22% for patients with imatinib resistance and 10% for patients intolerant of imatinib therapy.99 In part 2 of this study, a separate cohort of 118 patients previously treated with imatinib followed by dasatinib and/or nilotinib were evaluated for response to bosutinib 500 mg/day.100 Third- or fourth-line treatment with bosutinib was associated with 73% CHR, 32% MCyR, and 24% CCyR. At a median follow-up of 28.5 months, five patients had progressed to accelerated phase or blast phase.100 Toxicities were similar to those seen in the patients treated with second-line bosutinib. Gastrointestinal toxicities, including diarrhea, nausea, and vomiting, occurred in 81%, 43%, and 32% of patients, respectively. Although most of these toxicities were grade 1 or 2, they may pose significant challenges for long-term therapy. Bosutinib was also effective in patients with accelerated phase or blast phase CML following imatinib intolerance or resistance,101 leading to its approval in patients with all three phases of CML who are resistant or intolerant to previous TKI therapy.
Bosutinib was compared with imatinib in the Phase III BELA trial enrolling patients with newly diagnosed CP CML.102 There was no significant difference between bosutinib and imatinib in terms of CCyR at 12 months (Table 6), and bosutinib was not approved for use in patients with newly diagnosed CP CML.
Ponatinib
Ponatinib is a BCR–ABL inhibitor indicated for use in patients with T315I-positive CML or in whom no other TKI is indicated (Table 5).103 The antileukemic property of ponatinib is based on its inhibitory activity of all mutated forms of BCR–ABL, including the T315I mutant, which is resistant to all other currently available BCR–ABL TKIs.104 The single-arm Phase II PACE trial assessed ponatinib in patients with chronic phase, accelerated phase, or blast phase CML who were resistant to or intolerant of dasatinib or nilotinib or who had the T315I mutation.105 For patients with CP CML, ponatinib treatment was associated with MCyR in 56%, CCyR in 46%, and MMR in 34% of patients at a median follow-up of 15 months. Responses were durable and occurred regardless of BCR–ABL mutation, with no mutations showing resistance to ponatinib. In addition, 57% of patients with accelerated phase and 32% with blast phase CML had major hematologic responses, and 34% and 18%, respectively, had MCyR, leading to ponatinib’s approval for treatment of all three phases of CML.105
Recently, the Phase III EPIC trial was suspended and marketing temporarily halted following an investigation of the drug’s safety profile.106 A report from the US Food and Drug Administration (FDA) found that approximately 24% of patients in the Phase II study and approximately 48% of patients in the Phase I study experienced some degree of cardiovascular AEs after median treatment duration of 1.3 years and 2.7 years, respectively.107 After implementation of additional recommended safety monitoring, ponatinib is recommended in the NCCN guidelines only for patients who have the T315I mutation or who have not responded to two or more TKIs.12 The US prescribing information contains a black-box warning regarding vascular occlusion, heart failure, and hepatotoxicity. Of note, rebastinib, a conformational control inhibitor of BCR–ABL1 that has demonstrated strong activity against the T315I mutation in preclinical studies, including inhibition of primary cells from patients with CML,108,109 is currently in early clinical development for T315I-positive CP CML (NCT00827138).
Beyond TKIs
Results from TKIs in CML have reinforced the key nature of constitutive BCR–ABL activation in driving the disease. Targeted inhibitors have led to high response rates and very good overall survival. However, point mutations in BCR–ABL can lead to acquired resistance in patients who have previously achieved a response. For patients with disease that fails multiple TKIs, other types of therapy are being developed, such as the novel, first-in-class protein-synthesis inhibitor omacetaxine. This approved agent has a unique mechanism of action that is independent of BCR–ABL and effective in patients with the T315I mutation.105
Ph-negative MPNs
Based on the discovery of the JAK2V617F mutation in patients with MPNs, TKIs have been developed to specifically target the JAK–STAT pathway. These have resulted in clinical responses and quality-of-life and survival improvements in many patients. However, because the responses do not appear to be dependent on the presence or absence of a single targetable mutation and because of intra- and interdisease variability of genetic aberrations among patients with Ph-negative MPNs, combination therapies of JAK inhibitors with other agents may be required to achieve the degree of success that was seen in CML with imatinib and its successors.
Ruxolitinib
Ruxolitinib is an orally administered JAK1/JAK2 inhibitor that modulates intracellular cytokine signaling by binding to the cytoplasmic JAK1 and JAK2 kinases, thus inhibiting their activity. Based on the results of two Phase III studies,35,36 ruxolitinib became the first-in-class drug to be approved for the treatment of patients with intermediate- or high-risk MF (Table 5).110 The COMFORT-I and COMFORT-II trials (NCT00952289 and NCT00934544, respectively) demonstrated that ruxolitinib therapy was associated with significant reduction of splenomegaly and improved MF-related symptoms compared with placebo or best-available therapy (BAT).35,36 Subsequent follow-up analyses have demonstrated that long-term ruxolitinib treatment provided sustained clinical benefit for up to 3 years.111–114 Importantly, this benefit was observed independent of JAK2V617F mutational status. In COMFORT-I, mean changes in spleen volume from baseline to week 24 were −34.5% for ruxolitinib-treated patients with the JAK2V617F mutation and −23.8% for those without the mutation. Patients in the placebo arm showed increases in spleen volume of 8.1% and 8.4% for those with and without the mutation, respectively.35 Among patients with JAK2V617F-positive MF in COMFORT-II, spleen volume reduction ≥35% by week 48 was observed in 33% of those receiving ruxolitinib versus 0% receiving BAT. The corresponding rates in patients who were JAK2V617F-negative were 14% and 0% for ruxolitinib and BAT, respectively.36 Furthermore, symptom improvement was not dependent on mutational status: the mean change in total symptom score was −52.6% (improvement) in the ruxolitinib arm of COMFORT-I and 42.8% (worsening) in the placebo arm among JAK2V617F-positive patients. Corresponding changes for JAK2V617F-negative patients were −28.1% and +37.2% in the ruxolitinib and placebo arms, respectively.
In the COMFORT studies, the significant clinical responses observed in patients with the JAK2V617F mutation were generally not accompanied by major allele burden reductions. In COMFORT-I, ruxolitinib treatment was associated with a mean decrease in JAK2V617F allele burden of 10.9% at week 24 and 21.5% at week 48, whereas patients in the placebo arm experienced a mean increase in JAK2V617F allele burden of 3.5% at week 24 and 6.3% at week 48.35 Despite the modest short-term effect on allele burden, ruxolitinib may have disease-modifying properties. Results of survival analyses from the COMFORT studies, including a pooled 3-year survival analysis from both trials,114 suggest that ruxolitinib was associated with a 30%–50% survival advantage compared with placebo or BAT that was consistently observed, despite continuing crossover from the control arms.35,36,111–113 In addition, six patients in the ruxolitinib arm of COMFORT-I achieved a complete molecular response after a median treatment duration of 27.5 months, and 20 patients in the study achieved a partial molecular response after a median treatment duration of 22.2 months.115
Anemia and thrombocytopenia were the most common AEs associated with ruxolitinib treatment in the COMFORT trials, typically observed in the first 8–12 weeks of treatment. These events were usually manageable with dose modification or brief treatment interruption, and rarely led to discontinuation.35,36,111–114
Based on the results of the randomized, open-label, multicenter Phase III RESPONSE trial,116 ruxolitinib was also recently approved by the FDA for the treatment of patients with PV who have had an inadequate response to or are intolerant of hydroxyurea (Table 5). RESPONSE enrolled 222 patients and compared the proportion of patients with both hematocrit control (one or more instance of phlebotomy eligibility during the first 8 weeks after randomization and no phlebotomy eligibility afterwards) and a ≥35% reduction in spleen volume from baseline to week 32 for those randomized to ruxolitinib versus those randomized to standard therapy. This composite primary end point was achieved by 20.9% and 0.9% of patients receiving ruxolitinib and standard therapy, respectively (P<0.001). Overall, 60%, 38%, and 49% in the ruxolitinib arm compared with 20%, 1%, and 5% in the standard therapy arm achieved hematocrit control, a ≥35% reduction in spleen volume, and a ≥50% reduction in total symptom score, respectively. Safety and tolerability profiles were similar for ruxolitinib and standard therapy. Nonhematologic AEs were mostly grade 1 or 2, and no grade 3 or 4 anemia was reported.116 Recent follow-up data from RESPONSE for a median exposure to ruxolitinib of 111 weeks showed that ruxolitinib provided durable and comprehensive hematologic control of hematocrit, white blood cell count, and platelet count.117
Preliminary results from an open-label, single-arm, multicenter Phase II study further suggest that combination therapy with IFNα2 and ruxolitinib may be effective in eliciting both hematologic responses and substantial reductions in JAK2V617F allele burden in patients with PV.118 Of the 20 patients with PV with available data, 15 (75%) achieved a complete response and five (25%) achieved a partial response during the first 3 months of therapy.118
Momelotinib
Momelotinib is a JAK1/JAK2 inhibitor currently in Phase III development for MF (SIMPLIFY 1, NCT01969838). A Phase I/II trial and subsequent long-term follow-up and extension study in patients with high- or intermediate-risk primary or post-PV/ET MF showed that the majority of evaluable patients treated with momelotinib had durable spleen response and symptom improvement, with a median treatment duration of 507 days.119 Momelotinib treatment was generally well tolerated, with thrombocytopenia, dizziness, and peripheral neuropathy being the main treatment-emergent AEs.119–121 Peripheral neuropathy generally manifested as grade 1 sensory symptoms in the feet and/or hands, and appeared to involve length-dependent sensorimotor large- and small-fiber neuropathy with axonal features.121 In contrast, Wernicke’s encephalopathy, a serious neurologic condition caused by thiamine deficiency, has been associated only with the JAK2 inhibitor fedratinib.122,123 Consequently, clinical development of fedratinib was discontinued, despite the drug’s demonstrated efficacy in advanced MF in a large Phase III clinical trial.122 In 100 consecutive patients with MF treated with momelotinib at the Mayo Clinic, presence of CALR mutations (hazard ratio 0.2, 95% confidence interval 0.04–0.6) and absence of ASXL1 mutations (hazard ratio 0.3, 95% confidence interval 0.1–0.8) were independently associated with better spleen response (as per International Working Group-Myeloproliferative Neoplasms Research and Treatment) in a multivariable analysis.124
Pacritinib
An in vitro kinome screening analysis of pacritinib (SB1518), a dual JAK2–FLT3 inhibitor,125 indicated that it inhibits both JAK2V617F and wild-type JAK2 at similar concentrations.126 Results of Phase II studies in patients with MF suggested that pacritinib treatment may provide spleen volume reduction and clinically significant improvement in symptom burden with minimal myelosuppression, including in patients with low baseline platelet counts.127,128 Recent results from PERSIST-1 (NCT01773187), a randomized controlled study of pacritinib versus BAT in patients with moderate or severe MF and any degree of anemia or thrombocytopenia (N=327), showed that the primary end point of ≥35% decrease in spleen volume at week 24 was achieved by 19.1% of patients randomized to pacritinib versus 4.7% of patients randomized to BAT (P=0.0003).129 The corresponding proportions among patients with a platelet count <50×109/L (15% of the total study population) were 22.9% versus 0%, demonstrating that pacritinib can provide clinically significant improvements in splenomegaly in patients with severe thrombocytopenia. Generally low-grade gastrointestinal disturbances were the most common AEs.129
Agents in early stages of clinical development
Additional TKIs are being investigated in Phase I and Phase II studies in patients with Ph-negative MPNs. These include gandotinib (LY2784544),130 a JAK2V617F-selective inhibitor; INCB39110,131 a JAK1-selective inhibitor; and the JAK2-selective inhibitor NS-018.132
Beyond TKIs
Overall, although the original rationale behind the development of TKIs for classic Ph-negative MPNs was based on the identification of the JAK2V617F mutation, clinical evidence suggests that JAK inhibitors can be effective regardless of JAK2V617F mutational status. Based on the complex genetic profile of classic Ph-negative MPNs, multitargeted therapies that inhibit multiple oncogenic pathways are likely to provide superior clinical benefit while reducing the possibility of the activation of mechanisms of resistance.133 For example, additional compounds in clinical development for the treatment of MF include the non-TKIs imetelstat,134 pomalidomide,135–137 panobinostat,138 and PRM-151,139 and some of these agents may prove to be useful in combination with JAK inhibitors.
Nonclassic Ph-negative MPNs
Because of the rarity of the nonclassic Ph-negative MPNs, clinical trial evidence is scarce for these diseases. Case reports indicate that treatment has drawn from knowledge of TKIs in other MPNs.
Imatinib
In a case study of a PDGFRA/B-negative CEL-NOS patient, imatinib treatment was associated with rapid eosinophil clearance, complete response, including normalization of bone marrow histomorphology, and improvement of disease-related symptoms.140 Other institutions have had limited success with imatinib in CEL-NOS, a rare and aggressive disease that does not respond well to conventional treatment.141
Imatinib has also been used to treat systemic mastocytosis, as have dasatinib, nilotinib, masitinib, and midostaurin, all of which can block activated KIT TK. However, the clinical efficacy of TKIs in patients with kinase-domain KITD816V mutations remains unclear.50,142,143 Further studies are needed to determine whether TKIs have a clinical advantage over cladribine or IFNα in mastocytosis.
Ruxolitinib
In preclinical studies, chronic myelomonocytic leukemia cells have demonstrated dependence on granulocyte macrophage colony-stimulating factor through activation of the JAK–STAT pathway.144 Based on these observations, the efficacy and safety of ruxolitinib are currently being investigated in a Phase I/II study in patients with chronic myelomonocytic leukemia.
In patients with CNL and aCML, the discovery of different classes of CSF3R mutations has provided the rationale for the investigation of JAK or Src inhibitors to target membrane proximal or truncation CSF3R mutations, respectively.48 Consistent with preclinical observations showing that ruxolitinib treatment inhibited colony formation of cells with CSF3R membrane-proximal mutations,48 ruxolitinib has been shown to provide hematologic responses in individual patients with CNL or aCML who had the JAK-activating membrane-proximal CSF3RT618I mutation.48,145–147 Dasatinib may be useful in patients with CNL who do not respond to ruxolitinib, based on preclinical data demonstrating that dasatinib inhibited colony formation of primary cells from patients with Src-activating CSF3R truncation mutations that were insensitive to ruxolitinib treatment.48
The use of TKIs in nonclassic Ph-negative MPNs may be a feasible approach for some patients harboring TK-activating mutations; however, the bulk of evidence for these rarer diseases has been obtained from small case studies and will need to be validated in larger clinical trials.
Treatment goals and expectations in CML and MF
Prior to the introduction of BCR–ABL-targeted TKI therapy, survival for patients with CML was poor. Based on the results of the imatinib trials, which reported unprecedented hematologic responses and CyRs, and mindful of the prognostic implications of these studies, CHR became the first treatment goal, as failure to achieve CHR by 3 months constituted failure as per European LeukemiaNet and NCCN criteria.12,148 As TKIs became more specific, more stringent CyR and molecular responses became the goals of therapy.149 CyR is the strongest predictor of outcome in patients with CML receiving TKIs, as achieving an early CCyR was associated with significantly better event-free and overall survival compared with patients who did not achieve CCyR.150 Once CCyR is achieved, more sensitive assays are used to quantify the presence of residual CML cells by PCR analysis of BCR–ABL messenger RNA expression levels. Achievement of MMR is associated with very low odds of progression to advanced disease and superior PFS compared with patients who do not achieve MMR,151 even among patients receiving second-line therapy after imatinib failure.152 Responses are assessed every 3 months, and if transcript levels of BCR–ABL increase, patients should be analyzed for mutations, which can help guide treatment decisions.4 Since the introduction of imatinib in 2001, survival has improved significantly in patients with CML, especially those with CP or accelerated phase disease.14 In addition, imatinib therapy minimized the impact of previous prognostic factors and Sokal risk on survival.
Although definitions of complete and partial cytogenetic or molecular remission were recently introduced for the evaluation of treatment efficacy in patients with MF, response in these patients remains essentially defined by clinical criteria, with complete and partial remission based on the degree of normalization of bone marrow findings and hematologic laboratory results, and resolution of splenomegaly and symptoms. In addition, to qualify as response, any clinical benefits have to last at least 12 weeks.153 Hematologic and clinical responses, such as spleen size reduction and symptom improvement, are not dependent on the presence of a specific mutation, and JAK-inhibitor therapy is able to provide rapid and durable clinical benefits in MPNs that have not been seen with conventional therapies. Most importantly, in addition to providing clinically significant reductions in splenomegaly and symptom burden in the majority of patients with MF, the JAK1/JAK2 inhibitor ruxolitinib is the first and only therapy in MF to be associated with survival advantages compared with no or standard therapy.111,113,114,154 Furthermore, long-term therapy with ruxolitinib was shown to result in gradual decreases in mutant allele burden, and in some cases complete molecular and/or histologic responses.115,155–157 Together, these observations are evidence of the ability of ruxolitinib to modify the natural history of MF.
Conclusion
There are profound differences between the mechanisms of disease in CML and MF. While the success of BCR–ABL-targeting TKIs in CML is contingent on the presence of the Ph chromosome and the BCR–ABL mutation, the clinical efficacy of JAK inhibitors in MF is independent of JAK2-mutational status, and has demonstrated positive results in patients with both wild-type and mutant JAK2. The oncogenic BCR–ABL TK is the driving event for CML initiation and progression, and is the molecular signature for diagnosis and response to treatment. TKIs, such as imatinib, dasatinib, nilotinib, bosutinib, and ponatinib, target this aberrant fusion protein kinase and some of its acquired mutated forms, and have demonstrated significant complete hematologic, cytogenetic, and molecular responses in patients with newly diagnosed or advanced CML.
In contrast, the molecular pathobiology of Ph-negative MPNs is complex, involving multiple mutations and multiple pathways. Unlike CML, classic Ph-negative MPNs have no singular disease signature mutation, but instead have a preponderance of mutations that produce disease initiation and progression. The identification of mutations affecting JAK2 leading to aberrant JAK–STAT signaling in patients with MPNs has propelled the development of JAK inhibitors, such as ruxolitinib, which in controlled clinical trials have shown successful control of disease-related symptoms, improvement in quality-of-life measures, and overall survival benefit in patients with MF. However, contrary to initial expectations, the clinical efficacy of JAK inhibitors in patients with Ph-negative MPNs is not dependent on the presence of any one specific mutation as a driver of the disease or on the reduction of JAK2V617F allele burden in patients who carry this mutation. Current evidence suggests that overactive JAK–STAT signaling is a central defect in Ph-negative MPNs that extends beyond the neoplastic clones carrying the somatic mutation, and is independent of the specific mutation background. By providing rapid and durable clinical benefit with concomitant quality-of-life improvement for patients with MF, JAK inhibitors constitute significant therapeutic progress over historically limited treatment options with at best transient benefits. In the near future, it is probable that novel combinations of targeted therapies that complement JAK inhibition will further improve the treatment landscape for classic Ph-negative MPNs by addressing additional components of the complex pathobiology of these diseases.
Acknowledgments
Editorial support during the development of this manuscript was provided by Alfredo Toschi, PhD, of Evidence Scientific Solutions (Philadelphia, PA, USA) and was funded by Incyte Corporation, the manufacturer of ruxolitinib.
Author contributions
All authors contributed to the conception of this review, critically reviewed and revised each draft for intellectual content, and approved the final version of the manuscript for publication.
Disclosure
JPI is an investigator for Novartis and Ariad and served as a consultant/advisor for Novartis, Bristol-Myers Squibb, and Ariad. KLS has received research funding from Incyte Corporation, is an adviser for Novartis and Ariad, and serves on the speaker bureau for Novartis, Ariad, and Pfizer. GMCY reports no conflict of interest in this work. RSK serves as a consultant/advisor and member of the speaker bureau for Incyte Corporation and Novartis, is a consultant/adviser for CTI Biopharma Corp and Celgene, and has received research funding from Celgene and GlaxoSmithKline. The authors report no other conflicts of interest in this work.
References
Fialkow PJ. Cell lineages in hematopoietic neoplasia studied with glucose-6-phosphate dehydrogenase cell markers. J Cell Physiol Suppl. 1982;1:37–43. | ||
Tefferi A, Thiele J, Vardiman JW. The 2008 World Health Organization classification system for myeloproliferative neoplasms: order out of chaos. Cancer. 2009;115(17):3842–3847. | ||
Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22(1):14–22. | ||
Vardiman JW. Chronic myelogenous leukemia, BCR-ABL1+. Am J Clin Pathol. 2009;132(2):250–260. | ||
Fialkow PJ, Jacobson RJ, Papayannopoulou T. Chronic myelocytic leukemia: clonal origin in a stem cell common to the granulocyte, erythrocyte, platelet and monocyte/macrophage. Am J Med. 1977;63(1):125–130. | ||
Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–293. | ||
Groffen J, Stephenson JR, Heisterkamp N, de Klein A, Bartram CR, Grosveld G. Philadelphia chromosomal breakpoints are clustered within a limited region, Bcr, on chromosome 22. Cell. 1984;36(1):93–99. | ||
Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5(3):172–183. | ||
Wetzler M, Talpaz M, Van Etten RA, Hirsh-Ginsberg C, Beran M, Kurzrock R. Subcellular localization of Bcr, Abl, and Bcr-Abl proteins in normal and leukemic cells and correlation of expression with myeloid differentiation. J Clin Invest. 1993;92(4):1925–1939. | ||
Quintás-Cardama A, Cortes J. Molecular biology of Bcr-Abl1-positive chronic myeloid leukemia. Blood. 2009;113(8):1619–1630. | ||
Zhao C, Blum J, Chen A, et al. Loss of β-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12(6):528–541. | ||
National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Chronic Myelogenous Leukemia. Version 3.2014. Fort Washington (PA): NCCN; 2014. Available from: http://williams.medicine.wisc.edu/cml.pdf. Accessed November, 6, 2015. | ||
Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review: 1975–2012. Bethesda (MD): National Cancer Institute; 2015. Available from: http://seer.cancer.gov/csr/1975_2012. Accessed November 6, 2015. | ||
Kantarjian H, O’Brien S, Jabbour E, et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: a single-institution historical experience. Blood. 2012;119(9):1981–1987. | ||
Mehta J, Wang H, Iqbal SU, Mesa R. Epidemiology of myeloproliferative neoplasms in the United States. Leuk Lymphoma. 2013;55(3):595–600. | ||
Passamonti F, Maffioli M, Caramazza D, Cazzola M. Myeloproliferative neoplasms: from JAK2 mutations discovery to JAK2 inhibitor therapies. Oncotarget. 2011;2(6):485–490. | ||
Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22(2):437–438. | ||
Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007;109(1):68–76. | ||
Mesa RA, Shields A, Hare T, et al. Progressive burden of myelofibrosis in untreated patients: assessment of patient-reported outcomes in patients randomized to placebo in the COMFORT-I study. Leuk Res. 2013;37(8):911–916. | ||
Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. | ||
Spivak JL. Polycythemia vera: myths, mechanisms, and management. Blood. 2002;100(13):4272–4290. | ||
Beer PA, Green AR. Pathogenesis and management of essential thrombocythemia. Hematology Am Soc Hematol Educ Program. 2009;2009(1):621–628. | ||
Jäger R, Kralovics R. Molecular basis and clonal evolution of myeloproliferative neoplasms. Haematologica. 2010;95(4):526–529. | ||
Tefferi A, Guglielmelli P, Lasho TL, et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: an international study of 570 patients. Leukemia. 2014;28(7):1494–1500. | ||
Vannucchi AM, Lasho TL, Guglielmelli P, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. 2013;27(9):1861–1869. | ||
Lundberg P, Karow A, Nienhold R, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood. 2014;123(14):2220–2228. | ||
Guglielmelli P, Lasho TL, Rotunno G, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804–1810. | ||
Rampal R, Al-Shahrour F, Abdel-Wahab O, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123(22):e123–e133. | ||
Passamonti F, Caramazza D, Maffioli M. JAK inhibitor in CALR-mutant myelofibrosis. N Engl J Med. 2014;370(12):1168–1169. | ||
Santos FP, Verstovsek S. JAK2 inhibitors for myelofibrosis: why are they effective in patients with and without JAK2V617F mutation? Anticancer Agents Med Chem. 2012;12(9):1098–1109. | ||
Viny AD, Levine RL. Genetics of myeloproliferative neoplasms. Cancer J. 2014;20(1):61–65. | ||
Cross NC. Genetic and epigenetic complexity in myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2011;2011(1):208–214. | ||
Vainchenker W, Delhommeau F, Constantinescu SN, Bernard OA. New mutations and pathogenesis of myeloproliferative neoplasms. Blood. 2011;118(7):1723–1735. | ||
Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia. 2010;24(6):1128–1138. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. | ||
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. | ||
Kilpivaara O, Levine RL. JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia. 2008;22(10):1813–1817. | ||
Tefferi A, Vainchenker W. Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J Clin Oncol. 2011;29(5):573–582. | ||
Vannucchi AM, Guglielmelli P, Tefferi A. Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J Clin. 2009;59(3):171–191. | ||
Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–2405. | ||
Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–2390. | ||
Patel KP, Newberry KJ, Luthra R, et al. Correlation of mutation profile and response in patients with myelofibrosis treated with ruxolitinib. Blood. 2015;126(6):790–797. | ||
Guglielmelli P, Rotunno G, Bogani C, et al. A retrospective analysis of safety and efficacy of ruxolitinib in CALR-positive patients with myelofibrosis. Blood. 2014;124(21):1853. | ||
Ortmann CA, Kent DG, Nangalia J, et al. Effect of mutation order on myeloproliferative neoplasms. N Engl J Med. 2015;372(7):601–612. | ||
Kim E, Abdel-Wahab O. Focus on the epigenome in the myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. 2013;2013(1):538–544. | ||
Tefferi A, Guglielmelli P, Larson DR, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. 2014;124(16):2507–2513; quiz 2615. | ||
Vardiman J, Hyjek E. World Health Organization classification, evaluation, and genetics of the myeloproliferative neoplasm variants. Hematology Am Soc Hematol Educ Program. 2011;2011(1): 250–256. | ||
Maxson JE, Gotlib J, Pollyea DA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. 2013;368(19):1781–1790. | ||
Garcia-Montero AC, Jara-Acevedo M, Teodosio C, et al. KIT mutation in mast cells and other bone marrow hematopoietic cell lineages in systemic mast cell disorders: a prospective study of the Spanish Network on Mastocytosis (REMA) in a series of 113 patients. Blood. 2006;108(7):2366–2372. | ||
Verstovsek S. Advanced systemic mastocytosis: the impact of KIT mutations in diagnosis, treatment, and progression. Eur J Haematol. 2013;90(2):89–98. | ||
Lim KH, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009;113(23):5727–5736. | ||
Tefferi A, Skoda R, Vardiman JW. Myeloproliferative neoplasms: contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol. 2009;6(11):627–637. | ||
Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood. 2012;120(7):1390–1397. | ||
Kantarjian HM, Keating MJ, Smith TL, Talpaz M, McCredie KB. Proposal for a simple synthesis prognostic staging system in chronic myelogenous leukemia. Am J Med. 1990;88(1):1–8. | ||
Barbui T, Thiele J, Carobbio A, et al. Discriminating between essential thrombocythemia and masked polycythemia vera in JAK2 mutated patients. Am J Hematol. 2014;89(6):588–590. | ||
Sweet K, Zhang L, Pinilla-Ibarz J. Biomarkers for determining the prognosis in chronic myelogenous leukemia. J Hematol Oncol. 2013;6(1):54. | ||
Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimination in “good-risk” chronic granulocytic leukemia. Blood. 1984;63(4):789–799. | ||
Hasford J, Baccarani M, Hoffmann V, et al. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011;118(3):686–692. | ||
Hasford J, Pfirrmann M, Hehlmann R, et al. A new prognostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. J Natl Cancer Inst. 1998;90(11):850–858. | ||
Uz B, Buyukasik Y, Atay H, et al. EUTOS CML prognostic scoring system predicts ELN-based ‘event-free survival’ better than Euro/Hasford and Sokal systems in CML patients receiving front-line imatinib mesylate. Hematology. 2013;18(5):247–252. | ||
Hoffmann VS, Baccarani M, Lindoerfer D, et al. The EUTOS prognostic score: review and validation in 1288 patients with CML treated frontline with imatinib. Leukemia. 2013;27(10):2016–2022. | ||
Jabbour E, Cortes J, Nazha A, et al. EUTOS score is not predictive for survival and outcome in patients with early chronic phase chronic myeloid leukemia treated with tyrosine kinase inhibitors: a single institution experience. Blood. 2012;119(19):4524–4526. | ||
Passamonti F, Cervantes F, Vannucchi AM, et al. Dynamic International Prognostic Scoring System (DIPSS) predicts progression to acute myeloid leukemia in primary myelofibrosis. Blood. 2010;116(15):2857–2858. | ||
Gangat N, Caramazza D, Vaidya R, et al. DIPSS Plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. | ||
Tefferi A, Jimma T, Sulai NH, et al. IDH mutations in primary myelofibrosis predict leukemic transformation and shortened survival: clinical evidence for leukemogenic collaboration with JAK2V617F. Leukemia. 2012;26(3):475–480. | ||
Hernández-Boluda JC, Pereira A, Gómez M, et al. The International Prognostic Scoring System does not accurately discriminate different risk categories in patients with post-essential thrombocythemia and post-polycythemia vera myelofibrosis. Haematologica. 2014;99(4):e55–e57. | ||
Barbui T, Barosi G, Birgegard G, et al. Philadelphia-negative classical myeloproliferative neoplasms: critical concepts and management recommendations from European LeukemiaNet. J Clin Oncol. 2011;29(6):761–770. | ||
Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013;88(6):507–516. | ||
Verstovsek S, Kantarjian HM, Estrov Z, et al. Long-term outcomes of 107 patients with myelofibrosis receiving JAK1/JAK2 inhibitor ruxolitinib: survival advantage in comparison to matched historical controls. Blood. 2012;120(6):1202–1209. | ||
Bonicelli G, Abdulkarim K, Mounier M, et al. Leucocytosis and thrombosis at diagnosis are associated with poor survival in polycythaemia vera: a population-based study of 327 patients. Br J Haematol. 2013;160(2):251–254. | ||
Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874–1881. | ||
Novartis. GLEEVEC® (imatinib mesylate) [prescribing information]. 2015. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/gleevec_tabs.pdf. Accessed March 18, 2016. | ||
O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. | ||
Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. | ||
Deininger M, O’Brien SG, Guilhot J, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood. 2009;114(22):1126. | ||
Milojkovic D, Apperley J. Mechanisms of resistance to imatinib and second-generation tyrosine inhibitors in chronic myeloid leukemia. Clin Cancer Res. 2009;15(24):7519–7527. | ||
Quintás-Cardama A, Kantarjian HM, Cortes JE. Mechanisms of primary and secondary resistance to imatinib in chronic myeloid leukemia. Cancer Control. 2009;16(2):122–131. | ||
O’Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood. 2007;110(7):2242–2249. | ||
Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7(5):345–356. | ||
Pricl S, Fermeglia M, Ferrone M, Tamborini E. T315I-mutated Bcr-Abl in chronic myeloid leukemia and imatinib: insights from a computational study. Mol Cancer Ther. 2005;4(8):1167–1174. | ||
Jabbour E, Hochhaus A, Cortes J, La Rosee P, Kantarjian HM. Choosing the best treatment strategy for chronic myeloid leukemia patients resistant to imatinib: weighing the efficacy and safety of individual drugs with BCR-ABL mutations and patient history. Leukemia. 2010;24(1):6–12. | ||
O’Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65(11):4500–4505. | ||
Hochhaus A, Baccarani M, Deininger M, et al. Dasatinib induces durable cytogenetic responses in patients with chronic myelogenous leukemia in chronic phase with resistance or intolerance to imatinib. Leukemia. 2008;22(6):1200–1206. | ||
Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362(24):2260–2270. | ||
Bristol-Myers Squibb. SPRYCEL® (dasatinib) [prescribing information]. 2015. Available from: http://packageinserts.bms.com/pi/pi_sprycel.pdf. Accessed March 18, 2016. | ||
Cortes JE, Hochhaus A, Kim DW, et al. Four-year (Yr) follow-up of patients (Pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) receiving dasatinib or imatinib: efficacy based on early response. Blood. 2013;122(21):653. | ||
Novartis. TASIGNA® (nilotinib) [prescribing information]. 2015. Available from: http://www.pharma.us.novartis.com/product/pi/pdf/tasigna.pdf. Accessed March 18, 2016. | ||
Weisberg E, Manley P, Mestan J, Cowan-Jacob S, Ray A, Griffin JD. AMN107 (nilotinib): a novel and selective inhibitor of BCR-ABL. Br J Cancer. 2006;94(12):1765–1769. | ||
Kantarjian HM, Giles F, Gattermann N, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood. 2007;110(10):3540–3546. | ||
Giles FJ, le Coutre PD, Pinilla-Ibarz J, et al. Nilotinib in imatinib-resistant or imatinib-intolerant patients with chronic myeloid leukemia in chronic phase: 48-month follow-up results of a phase II study. Leukemia. 2013;27(1):107–112. | ||
le Coutre P, Ottmann OG, Giles F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111(4):1834–1839. | ||
Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362(24):2251–2259. | ||
Saglio G, Hochhaus A, Hughes TP, et al. ENESTnd update: nilotinib (NIL) vs imatinib (IM) in patients (pts) with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) and the impact of early molecular response (EMR) and Sokal risk at diagnosis on long-term outcomes. Blood. 2013;122(21):92. | ||
Hochhaus A, Saglio G, Larson RA, et al. Nilotinib is associated with a reduced incidence of BCR-ABL mutations vs imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Blood. 2013;121(18):3703–3708. | ||
Pfizer. BOSULIF (bosutinib monohydrate) [prescribing information]. 2015. Available from: http://labeling.pfizer.com/ShowLabeling.aspx?id=884. Accessed March 18, 2016. | ||
Golas JM, Arndt K, Etienne C, et al. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003;63(2):375–381. | ||
Puttini M, Coluccia AM, Boschelli F, et al. In vitro and in vivo activity of SKI-606, a novel Src-Abl inhibitor, against imatinib-resistant Bcr-Abl+ neoplastic cells. Cancer Res. 2006;66(23):11314–11322. | ||
Cortes JE, Kantarjian HM, Brümmendorf TH, et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011;118(17):4567–4576. | ||
Brümmendorf T, Cortes J, Khoury H, et al. Bosutinib as therapy for chronic phase chronic myeloid leukemia following resistance or intolerance to imatinib: 48-month update. Blood. 2013;122(21):2723. | ||
Khoury HJ, Cortes JE, Kantarjian HM, et al. Bosutinib is active in chronic phase chronic myeloid leukemia after imatinib and dasatinib and/or nilotinib therapy failure. Blood. 2012;119(15):3403–3412. | ||
Gambacorti-Passerini C, Cortes JE, Khoury HJ, et al. Safety and efficacy of bosutinib in patients with AP and BP CML and ph+ ALL following resistance/intolerance to imatinib and other TKIs: update from study SKI-200. J Clin Oncol. 2010;28(15 Suppl):6509. | ||
Cortes JE, Kim DW, Kantarjian HM, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol. 2012;30(28):3486–3492. | ||
Ariad Pharmaceuticals. ICLUSIG® (ponatinib) [prescribing information]. 2015. Available from: http://iclusig.com/wp-content/uploads/2015/12/Iclusig_PackageInsert_DEC2015.pdf. Accessed March 18, 2016. | ||
Huang WS, Metcalf CA, Sundaramoorthi R, et al. Discovery of 3-[2-(imidazo[1,2-b]pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-y l)methyl]-3-(trifluoromethyl)phenyl}benzamide (AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region-Abelson (BCR-ABL) kinase including the T315I gatekeeper mutant. J Med Chem. 2010;53(12):4701–4719. | ||
Cortes JE, Kim DW, Pinilla-Ibarz J, et al. A phase 2 trial of ponatinib in Philadelphia chromosome-positive leukemias. N Engl J Med. 2013;369(19):1783–1796. | ||
US Food and Drug Administration. FDA Drug Safety Podcast: FDA asks manufacturer of the leukemia drug Iclusig (ponatinib) to suspend marketing and sales [podcast]. 2013. Available from: http://www.fda.gov/Drugs/DrugSafety/DrugSafetyPodcasts/ucm373240.htm. Accessed February 21, 2014. | ||
US Food and Drug Administration. FDA Drug Safety Communication: FDA requires multiple new safety measures for leukemia drug Iclusig; company expected to resume marketing. 2014. Available from: http://www.fda.gov/Drugs/DrugSafety/ucm379554.htm. Accessed February 21, 2014. | ||
Chan WW, Wise SC, Kaufman MD, et al. Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036. Cancer Cell. 2011;19(4):556–568. | ||
Eide CA, Adrian LT, Tyner JW, et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res. 2011;71(9):3189–3195. | ||
Incyte Corporation. Jakafi® (ruxolitinib) [prescribing information]. 2016. Available from: http://www.jakafi.com/pdf/prescribing-information.pdf. Accessed March 18, 2016. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: results of a median 2-year follow-up of COMFORT-I. Haematologica. 2013;98(12):1865–1871. | ||
Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3-year follow-up of COMFORT-I. Haematologica. 2015;100(4):479–488. | ||
Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047–4053. | ||
Vannucchi AM, Kantarjian HM, Kiladjian JJ, et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. 2015;100(9):1139–1145. | ||
Deininger M, Radich J, Burn TC, Huber R, Paranagama D, Verstovsek S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood. 2015;126(13):1551–1554. | ||
Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. | ||
Harrison CN, Masszi T, Zachee P, et al. Complete hematologic control with ruxolitinib in patients with polycythemia vera (PV) resistant to or intolerant of hydroxyurea. Poster presented at: 20th Congress of the European Hematology Association; June 11–14, 2015; Vienna, Austria. | ||
Mikkelsen SU, Kjær L, Skov V, et al. Safety and efficacy of combination therapy of interferon-alpha2 + JAK1-2 inhibitor in the Philadelphia-negative chronic myeloproliferative neoplasms: preliminary results from the Danish Combi-Trial – an open label, single arm, non-randomized multicenter phase II study. Blood. 2015;126(23):824. | ||
Pardanani A, Gotlib J, Gupta V, et al. Update on the long-term efficacy and safety of momelotinib, a JAK1 and JAK2 inhibitor, for the treatment of myelofibrosis. Blood. 2013;122(21):108. | ||
Pardanani A, Laborde RR, Lasho TL, et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia. 2013;27(6):1322–1327. | ||
Abdelrahman RA, Begna KH, Al-Kali A, et al. Momelotinib treatment-emergent neuropathy: prevalence, risk factors and outcome in 100 patients with myelofibrosis. Br J Haematol. 2015;169(1):77–80. | ||
Pardanani A, Harrison C, Cortes JE, et al. Safety and efficacy of fedratinib in patients with primary or secondary myelofibrosis: a randomized clinical trial. JAMA Oncol. 2015;1(5):643–651. | ||
Zhang Q, Zhang Y, Diamond S, et al. The Janus kinase 2 inhibitor fedratinib inhibits thiamine uptake: a putative mechanism for the onset of Wernicke’s encephalopathy. Drug Metab Dispos. 2014;42(10):1656–1662. | ||
Pardanani A, Abdelrahman RA, Finke C, et al. Genetic determinants of response and survival in momelotinib-treated patients with myelofibrosis. Leukemia. 2015;29(3):741–744. | ||
Hart S, Goh KC, Novotny-Diermayr V, et al. SB1518, a novel macrocyclic pyrimidine-based JAK2 inhibitor for the treatment of myeloid and lymphoid malignancies. Leukemia. 2011;25(11):1751–1759. | ||
Singer J, Al-Fayoumi S, Ma H, Komrokji RS, Mesa R, Verstovsek S. Comprehensive kinase profile of pacritinib, a non-myelosuppressive JAK2 kinase inhibitor in phase 3 development in primary and post ET/PV myelofibrosis. Blood. 2014;124(21):1874. | ||
Verstovsek S, Dean JP, Cernohous P, et al. Pacritinib, a dual JAK2/FLT3 inhibitor: an integrated efficacy and safety analysis of phase II trial data in patients with primary and secondary myelofibrosis (MF) and platelet counts ≤100,000/μL. Blood. 2013;122(21):395. | ||
Komrokji RS, Seymour JF, Roberts AW, et al. Results of a phase 2 study of pacritinib (SB1518), a JAK2/JAK2(V617F) inhibitor, in patients with myelofibrosis. Blood. 2015;125(17):2649–2655. | ||
Mesa RA, Egyed M, Szoke A, et al. Results of the PERSIST-1 phase III study of pacritinib (PAC) versus best available therapy (BAT) in primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF), or post-essential thrombocythemia-myelofibrosis (PET-MF). J Clin Oncol. 2015;33 Suppl:LBA7006. | ||
Verstovsek S, Mesa RA, Salama ME, et al. Phase I study of LY2784544, a JAK2 selective inhibitor, in patients with myelofibrosis (MF), polycythemia vera (PV), and essential thrombocythemia (ET). Blood. 2013;122(21):665. | ||
Mascarenhas JO, Talpaz M, Gupta V. Primary analysis results from an open-label phase II study of INCB039110, a selective JAK1 inhibitor, in patients with myelofibrosis. Poster presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San Francisco, CA. | ||
Verstovsek S, Talpaz M, Ritchie EK, et al. A phase 1/2, open-label, dose-escalation, multi-center study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of orally administered NS-018 in patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (postPV MF), or post-essential thrombocythemia myelofibrosis (postET MF) [abstract 1839]. Poster presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San Francisco, CA. | ||
Odenike O. Beyond JAK inhibitor therapy in myelofibrosis. Hematology Am Soc Hematol Educ Program. 2013;2013(1):545–552. | ||
Tefferi A, Begna K, Laborde RR, et al. Imetelstat, a telomerase inhibitor, induces morphologic and molecular remissions in myelofibrosis and reversal of bone marrow fibrosis. Blood. 2013;122(21):662. | ||
Tefferi A, Verstovsek S, Barosi G, et al. Pomalidomide is active in the treatment of anemia associated with myelofibrosis. J Clin Oncol. 2009;27(27):4563–4569. | ||
Daver N, Shastri A, Kadia T, et al. Phase II study of pomalidomide in combination with prednisone in patients with myelofibrosis and significant anemia. Leuk Res. 2014;38(9):1126–1129. | ||
Begna KH, Pardanani A, Mesa R, et al. Long-term outcome of pomalidomide therapy in myelofibrosis. Am J Hematol. 2012;87(1):66–68. | ||
DeAngelo DJ, Mesa RA, Fiskus W, et al. Phase II trial of panobinostat, an oral pan-deacetylase inhibitor in patients with primary myelofibrosis, post-essential thrombocythaemia, and post-polycythaemia vera myelofibrosis. Br J Haematol. 2013;162(3):326–335. | ||
Promedior. A phase 2 study of PRM-151 in subjects with myelofibrosis. Available from: http://clinicaltrials.gov/show/NCT01981850. NLM identifier: NCT01981850. Accessed November 11, 2014. | ||
Iurlo A, Fracchiolla NS, Ferla V, et al. Successful treatment with imatinib in a patient with chronic eosinophilic leukemia not otherwise specified. J Clin Oncol. 2014;32(10):e37–e39. | ||
Helbig G, Soja A, Bartkowska-Chrobok A, Kyrcz-Krzemień S. Chronic eosinophilic leukemia-not otherwise specified has a poor prognosis with unresponsiveness to conventional treatment and high risk of acute transformation. Am J Hematol. 2012;87(6):643–645. | ||
Jin B, Ding K, Pan J. Ponatinib induces apoptosis in imatinib-resistant human mast cells by dephosphorylating mutant D816V KIT and silencing β-catenin signaling. Mol Cancer Ther. 2014;13(5):1217–1230. | ||
Magliacane D, Parente R, Triggiani M. Current concepts on diagnosis and treatment of mastocytosis. Transl Med UniSa. 2014;8:65–74. | ||
Padron E, Painter JS, Kunigal S, et al. GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood. 2013;121(25):5068–5077. | ||
Gotlib J, Maxson JE, George TI, Tyner JW. The new genetics of chronic neutrophilic leukemia and atypical CML: implications for diagnosis and treatment. Blood. 2013;122(10):1707–1711. | ||
Dao KH, Solti MB, Maxson JE, et al. Significant clinical response to JAK1/2 inhibition in a patient with CSF3R-T618I-positive atypical chronic myeloid leukemia. Leuk Res Rep. 2014;3(2):67–69. | ||
Freedman JL, Desai AV, Bailey LC, et al. Atypical chronic myeloid leukemia in two pediatric patients. Pediatr Blood Cancer. 2016;63(1):156–159. | ||
Baccarani M, Castagnetti F, Gugliotta G, Palandri F, Soverini S. Response definitions and European LeukemiaNet management recommendations. Best Pract Res Clin Haematol. 2009;22(3):331–341. | ||
Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884. | ||
Jabbour E, Kantarjian H, O’Brien S, et al. The achievement of an early complete cytogenetic response is a major determinant for outcome in patients with early chronic phase chronic myeloid leukemia treated with tyrosine kinase inhibitors. Blood. 2011;118(17):4541–4546; quiz 4759. | ||
Breccia M, Alimena G. The significance of early, major and stable molecular responses in chronic myeloid leukemia in the imatinib era. Crit Rev Oncol Hematol. 2011;79(2):135–143. | ||
Boquimpani C, Schaffel R, Biasoli I, Bendit I, Spector N. Molecular responses at 3 and 6 months after switching to a second-generation tyrosine kinase inhibitor are complementary and predictive of long-term outcomes in patients with chronic myeloid leukemia who fail imatinib. Leuk Lymphoma. 2015;56(6):1787–1792. | ||
Tefferi A, Cervantes F, Mesa R, et al. Revised response criteria for myelofibrosis: International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) and European LeukemiaNet (ELN) consensus report. Blood. 2013;122(8):1395–1398. | ||
Passamonti F, Maffioli M, Cervantes F, et al. Impact of ruxolitinib on the natural history of primary myelofibrosis: a comparison of the DIPSS and the COMFORT-2 cohorts. Blood. 2014;123(12):1833–1835. | ||
Koehler A, Hubert K, Lange T, et al. JAK2V617F molecular remission in a primary myelofibrosis patient treated with ruxolitinib. Ann Hematol. 2015;94(11):1929–1930. | ||
Molica M, Serrao A, Saracino R, et al. Disappearance of fibrosis in secondary myelofibrosis after ruxolitinib treatment: new endpoint to achieve? Ann Hematol. 2014;93(11):1951–1952. | ||
Wilkins BS, Radia D, Woodley C, Farhi SE, Keohane C, Harrison CN. Resolution of bone marrow fibrosis in a patient receiving JAK1/JAK2 inhibitor treatment with ruxolitinib. Haematologica. 2013;98(12):1872–1876. | ||
Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26(47):6724–6737. | ||
Vannucchi AM, Pieri L, Guglielmelli P. JAK2 allele burden in the myeloproliferative neoplasms: effects on phenotype, prognosis and change with treatment. Ther Adv Hematol. 2011;2(1):21–32. | ||
Scott LM. The JAK2 exon 12 mutations: a comprehensive review. Am J Hematol. 2011;86(8):668–676. | ||
Kaushansky K. The molecular mechanisms that control thrombopoiesis. J Clin Invest. 2005;115(12):3339–3347. | ||
Guglielmelli P, Pancrazzi A, Bergamaschi G, et al. Anaemia characterises patients with myelofibrosis harbouring MplW515L/K mutation. Br J Haematol. 2007;137(3):244–247. | ||
Tefferi A, Pardanani A, Lim KH, et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia. 2009;23(5):905–911. | ||
Brecqueville M, Rey J, Bertucci F, et al. Mutation analysis of ASXL1, CBL, DNMT3A, IDH1, IDH2, JAK2, MPL, NF1, SF3B1, SUZ12, and TET2 in myeloproliferative neoplasms. Genes Chromosomes Cancer. 2012;51(8):743–755. | ||
Tefferi A, Lasho TL, Abdel-Wahab O, et al. IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia. 2010;24(7):1302–1309. | ||
Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7(3):299–313. | ||
Guglielmelli P, Biamonte F, Score J, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood. 2011;118(19):5227–5234. | ||
Stegelmann F, Bullinger L, Schlenk RF, et al. DNMT3A mutations in myeloproliferative neoplasms. Leukemia. 2011;25(7):1217–1219. | ||
Abdel-Wahab O, Pardanani A, Rampal R, Lasho TL, Levine RL, Tefferi A. DNMT3A mutational analysis in primary myelofibrosis, chronic myelomonocytic leukemia and advanced phases of myeloproliferative neoplasms. Leukemia. 2011;25(7):1219–1220. | ||
Vardiman JW, Melo JV, Baccarani M, et al. Chronic myelogenous leukaemia, BCR-ABL1 positive. In: Swerdlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. Vol 2. 4th ed. Lyon: IARC Press; 2008:32–37. | ||
Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. | ||
Ariad Pharmaceuticals. Ariad announces preliminary safety and efficacy data of ponatinib in patients with newly diagnosed CML from discontinued phase 3 EPIC trial [press release]. Cambridge (MA): Ariad Pharmaceuticals; 2014 [May 31]. Available from: http://investor.ariad.com/phoenix.zhtml?c=118422&p=irol-newsArticle&ID=1936051. Accessed November 14, 2014. | ||
Lipton JH, Chuah C, Guerci-Bresler A, et al. A phase 3 trial of ponatinib compared with imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CP-CML). Poster presented at: 56th ASH Annual Meeting and Exposition; December 6–9, 2014; San Francisco, CA. | ||
Savona MR. Are we altering the natural history of primary myelofibrosis? Leuk Res. 2014;38(9):1001–1012. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.