")
Back to Journals » Journal of Asthma and Allergy » Volume 15
Role of Thymus and Activation-Regulated Chemokine in Allergic Asthma
Authors Luu Quoc Q , Moon JY, Lee DH, Ban GY , Kim SH, Park HS
Received 30 November 2021
Accepted for publication 22 January 2022
Published 5 February 2022 Volume 2022:15 Pages 157—167
DOI https://doi.org/10.2147/JAA.S351720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
Quang Luu Quoc,1 Ji-Young Moon,1 Dong-Hyun Lee,1 Ga-Young Ban,2 Seung-Hyun Kim,3 Hae-Sim Park1
1Department of Allergy and Clinical Immunology, Ajou University School of Medicine, Suwon, South Korea; 2Department of Pulmonary, Allergy, and Critical Care Medicine, Kangdong Sacred Heart Hospital, Hallym University College of Medicine Institute for Life Sciences, Chuncheon, South Korea; 3Translational Research Laboratory for Inflammatory Disease, Clinical Trial Center, Ajou University Medical Center, Suwon, South Korea
Correspondence: Seung-Hyun Kim
Translational Research Laboratory for Inflammatory Disease, Clinical Trial Center, Ajou University Medical Center, Suwon, South Korea
, Tel +82 31-219-4264
, Fax +82 31-219-4265
, Email [email protected]
Hae-Sim Park
Department of Allergy and Clinical Immunology, Ajou University School of Medicine, 206 Worldcup-ro, Yeongtong-gu, Suwon, 16499, South Korea
, Tel +82 31-219-5000
, Fax +82 31-219-6380
, Email [email protected]
Background: Asthma is a chronic inflammatory airway disease characterized by the predominant infiltration of inflammatory cells in the airways. Thymus and activation-regulated chemokine/C–C motif chemokine 17 (TARC/CCL17) is a chemokine responsible for trafficking T helper 2 cells into sites of allergic inflammation.
Objective: To validate the role of TARC in association with clinical and inflammatory parameters in adult asthmatics.
Methods: We enrolled 128 asthmatic patients and 70 healthy controls (HCs). Asthma-related clinical and laboratory parameters, including lung function and eosinophil counts, were measured. Serum levels of TARC, free immunoglobulin E (IgE), and eosinophil-derived neurotoxin (EDN) were determined by using enzyme-linked immunosorbent assay; serum total IgE level was measured using ImmunoCAP. The levels of inflammatory lipid mediators, such as leukotriene E4 (LTE4), 15-hydroxyeicosatetraenoic acid (15-HETE), thromboxane B2 (TXB2), and prostaglandin F2α (PGF2α), were measured by liquid chromatography-tandem mass spectrometry.
Results: Serum TARC levels are significantly higher in asthmatics than in HCs and in allergic asthmatics than in HCs (P < 0.010 for all), with significantly negative correlations between serum TARC levels and FEV1%/MMEF% values (r = − 0.314, r = − 0.268, P < 0.050 for both). The patients with higher serum TARC levels had higher levels of serum total and free IgE levels (P < 0.050 for both) with positive correlations to serum levels of EDN, TXB2, and 15-HETE (r = 0.233, r = 0.264, and r = 0.223, respectively, P < 0.050 for all).
Conclusion: We suggest the role of TARC in allergic asthma via contributing to mast cell and eosinophilic inflammation.
Keywords: asthma, allergic asthma, thymus and activation-regulated chemokine, IgE, T cells, mast cells, eosinophils
Introduction
Asthma is a chronic heterogeneous airway disease that affects more than 300 million people worldwide.1 The prevalence of asthma in the general population was reported to range from 1.6% to 4.5% in various countries including South Korea.2,3 Efforts to classify asthma phenotype and endotype facilitate effective diagnosis, treatment, and management. Asthma phenotypes and endotypes could be divided into 1) non-allergic vs allergic asthma and 2) type 2-high/eosinophilic asthma vs type 2-low/non-eosinophilic asthma.4 Allergic asthma (also known as extrinsic/atopic asthma) is classified if asthmatics have identifiable allergens and their symptoms were developed in early life,5 while eosinophilic asthma is defined if asthmatics have higher blood/sputum eosinophilia (whether they have allergic or not) and their symptoms developed in adults.5 T helper (Th)2-mediated eosinophilic activation (initiated by causative allergens) is the major inflammatory mechanism in asthmatics with allergic backgrounds, while type 2 innate lymphoid cells (ILC2)-mediated eosinophil activation plays a role in those with the nonallergic background.
Eosinophils are considered key effector cells damaging airway epithelial cells, perpetuating type 2 airway inflammation in asthma.6 Two major pathways for eosinophil activations are 1) allergen-exposed epithelial cells could release alarmins (thymic stromal lymphopoietin, interleukin [IL]-25, and IL-33 initiating Th2 signaling pathways), 2) ILCs and T cells are differentiated into ILC2 and Th2 cells to release their cytokines, such as IL-4, IL-5, and IL-13, leading to eosinophil activation/recruitment. Mast cells are predominantly activated by the immunoglobulin E (IgE)-dependent pathway in allergic asthma.7 The crosstalk between mast cells and eosinophils is recognized. Mast cell degranulation-derived cytokines could recruit eosinophils from bone marrow and peripheral sources into the lungs.7 Furthermore, eosinophils could activate mast cells via releasing eosinophil cationic protein (ECP) and major basic protein (MBP) which further binds to MAS-related G protein-coupled receptor member X2 receptor expressed on the surface of mast cells.8 Although there have been a few type 2 biomarkers including eosinophilia, higher total IgE, and fractional exhaled nitric oxide (FeNO) levels, each represents different pathogenic mechanisms and they are not closely correlated.9 Therefore, further investigations to clarify various underlying mechanisms of allergic or eosinophilic asthma are required in order to support selecting suitable biomarkers for type 2 asthma, which helps avoid unnecessary use of high-dose corticosteroid and allow the most appropriate and cost-effective use of biologics or disease modifiers.
Thymus and activation-regulated chemokine/C–C motif chemokine 17 (TARC/CCL17) is mainly produced by immune cells (macrophages and dendritic cells), endothelial cells, keratinocytes, and fibroblasts.10 TARC had a high affinity for the C-C chemokine receptors CCR4 and CCR8 on CD4+ Th2 cell surface in order to work as a selective chemoattractant for T cells.11 TARC could be detected in the sera of patients with atopic dermatitis, and sputa (but not serum) of asthmatic patients.12,13 Additionally, suppression of the TARC/CCR4 axis by using anti-TARC antibody could reduce airway hyperresponsiveness (AHR) and eosinophilia in an ovalbumin-induced murine model of asthma.14 These findings suggest that TARC may be potential biomarkers for predicting the endotype of type 2 high/eosinophilic asthma. The present study aimed to elucidate the role of TARC in airway inflammatory mechanisms of asthma.
Materials and Methods
Study Subjects
One hundred twenty-eight asthmatic patients and 70 healthy controls (HCs) were enrolled from the Department of Allergy and Clinical Immunology at Ajou University Hospital, Suwon, South Korea. This study was approved by the Institutional Review Board of Ajou University Hospital (AJIRB-BMR-SUR-15-498), and all the subjects provided written informed consent. Blood total eosinophil counts (TEC), sputum cell (eosinophil and neutrophil) counts, pulmonary function tests (forced expiratory volume in 1 second [FEV1], percent and maximal mid-expiratory flow [MMEF]), and FeNO levels were measured. TEC in peripheral blood was counted using a hematology analyzer. For spontaneous sputum evaluation, the total number of cells was counted after exclusion of dead cells by using Trypan blue stain. The differential cell counts were determined by the Wright-Giemsa stain. Sputum eosinophil/neutrophil counts were calculated as % of eosinophil or neutrophil number per 100 leukocytes counted.15 FeNO was measured with a NIOX™ analyzer (Aerocrine AB) in accordance with American Thoracic Society/European Respiratory Society recommendations.16 FEV1/FVC/MMEF values were evaluated by spirometry, and their predictive values were expressed on the basis of the Morris method.17 The diagnosis of asthma was evaluated according to the recent Global Initiative for Asthma guideline.1 Briefly, asthma was defined if patients had suffered from recurrent respiratory symptoms (wheezing, dyspnea, and cough), airway reversibility, and AHR to methacholine (a decrease in FEV1% of 20% on methacholine challenge test). Type 2 high asthma was defined if patients have at least one among three criteria of type 2 markers including higher TEC (>300 cells/µL), higher FeNO (>20 ppb), and higher serum-free IgE levels (>85.7 ng/mL).18 All of the enrolled patients had maintained anti-asthmatic medications, including inhaled corticosteroid plus long-acting beta2 agonist (ICS+LABA) with/without leukotriene modifiers as most of them were in moderate-to-severe asthma. Severe asthma was defined if they had suffered from more than 2 times asthma exacerbation per year even with maintaining anti-inflammatory medications including medium-to-high doses of ICS+LABA according to the International European Respiratory Society/American Thoracic Society guidelines.19,20 Those with autoimmune disease or who had used type 2 biologics within 180 days were excluded. This study was conducted in accordance with the Declaration of Helsinki.
The phenotype of allergic asthma was determined if asthmatics had positive results to skin prick test and/or serum-specific IgE levels to at least more than 1 common inhalant allergen including Dermatophagoides pteronyssinus, Dermatophagoides farinae, cat, dog, cockroach, tree and grass pollen mixtures, mugwort, ragweed, Japanese hops, Aspergillus, and Alternaria (Bencard. Brentford, UK). The levels of serum total and specific IgE antibodies were measured using the ImmunoCAP system (ThermoFisher Scientific, Waltham, CA, USA). Serum-free IgE was measured by enzyme-linked immunosorbent assay (ELISA) as previously reported.18 Briefly, serum (diluted 1:10 in phosphate-buffered saline containing 1% bovine serum albumin) was loaded in the optimal conditions of IgETRAP protein (YH, Seoul, South Korea)-coated 96-well plates. Antihuman IgE antibody (ThermoFisher Scientific) which served as the detection antibody was added to each well and incubated for 1 hour. Then, samples were incubated with horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody (Novusbio, Littleton, CO, USA) for 1 hour. 3,3′,5,5′-Tetramethylbenzidine substrate (BD Biosciences, Franklin Lakes, NJ, USA) was added to develop the color. The reaction was stopped by the addition of H2SO4. Finally, absorbance values were read by using an ELISA reader (BioTek, Winooski, VT, USA) at 450 nm. Blood were collected in a BD Vacutainer® SST tube. Serum was separated by centrifugation and stored at −70 °C for further analysis.
Measurement of Inflammatory Lipid Metabolites in Sera
Inflammatory lipid metabolites, such as leukotriene E4 (LTE4), 15-hydroxyeicosatetraenoic acid (15-HETE), thromboxane B2 (TXB2), and prostaglandin F2α (PGF2α) were measured by the liquid chromatography-tandem mass spectrometry method. Particularly, sera collected from asthmatic patients and HCs were separated using a BD Vacutainer® SST tube (BD Biosciences). LTE4-d5 for LTE4, 15(S)-HETE-d8 for 15-HETE, 11-dehydro TXB2-d4 for 11-dehydro TXB2, and 8-iso PGF2α-d4 for PGF2α (Cayman Chemical Company, Ann Arbor, MI, USA) were used as the internal standards. A Waters Acquity UPLC system (Waters) with a Hypersil GOLD column (ThermoFisher Scientific) was used to separate chromatography. Data were obtained using an API5500 triple quadrupole mass spectrometer (AB Sciex, Framingham, MA, USA) equipped with an ESI source. Data analysis was performed using Analyst 1.5.2 software (AB Sciex).
Measurement of Serum TARC and Eosinophil-Derived Neurotoxin (EDN) Levels
The levels of TARC were measured in sera of the study subjects using ELISA according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA). Serum EDN levels were measured using the ELISA kit (SKIMS-BIO Co., Seoul, Korea) according to the manufacturer’s instructions.
Statistical Analysis
The test for normal distribution was performed by using D’Agostino-Pearson omnibus test. Differences between the two groups were analyzed by Mann–Whitney U-test for non-normally distributed variables and Pearson chi-square test for categorical variables. Comparisons of data from multiple groups were made by using Kruskal–Wallis test. Correlations among continuous clinical variables were calculated by Pearson’s or Spearman’s rank correlation coefficient. To evaluate the diagnostic value of the serum TARC level in discriminating between asthmatics and HCs, receiver operating characteristic (ROC) curve analysis was performed. Statistical analyses were performed using SPSS software version 22.0 (IBM Corp., Armonk, NY, USA). GraphPad Prism 6.0 software (GraphPad Inc., San Diego, CA, USA) was used to produce graphs.
Results
Clinical Characteristics of the Study Subjects
Table 1 shows the demographic characteristics of the subjects. Subjects were classified into the allergic and non-allergic asthma groups according to the results of the skin prick test and serum-specific IgE levels. The median age was older in the non-allergic asthma group (56.0 ranged from 48.0 to 63.0 years) than in the allergic asthma group (48.0 ranged from 31.0 to 60.0 years) or HCs (47.5 ranged from 39.0 to 56.5 years) (P = 0.008, P = 0.002, respectively). No significant differences were observed in inflammatory parameters (TEC, sputum eosinophil/neutrophil counts), lung function parameters, or FeNO levels between the allergic and non-allergic groups. Significantly higher levels of serum total and free IgE antibodies were noted in the allergic asthma group than in the non-allergic asthma group or HCs (P < 0.001 for both).
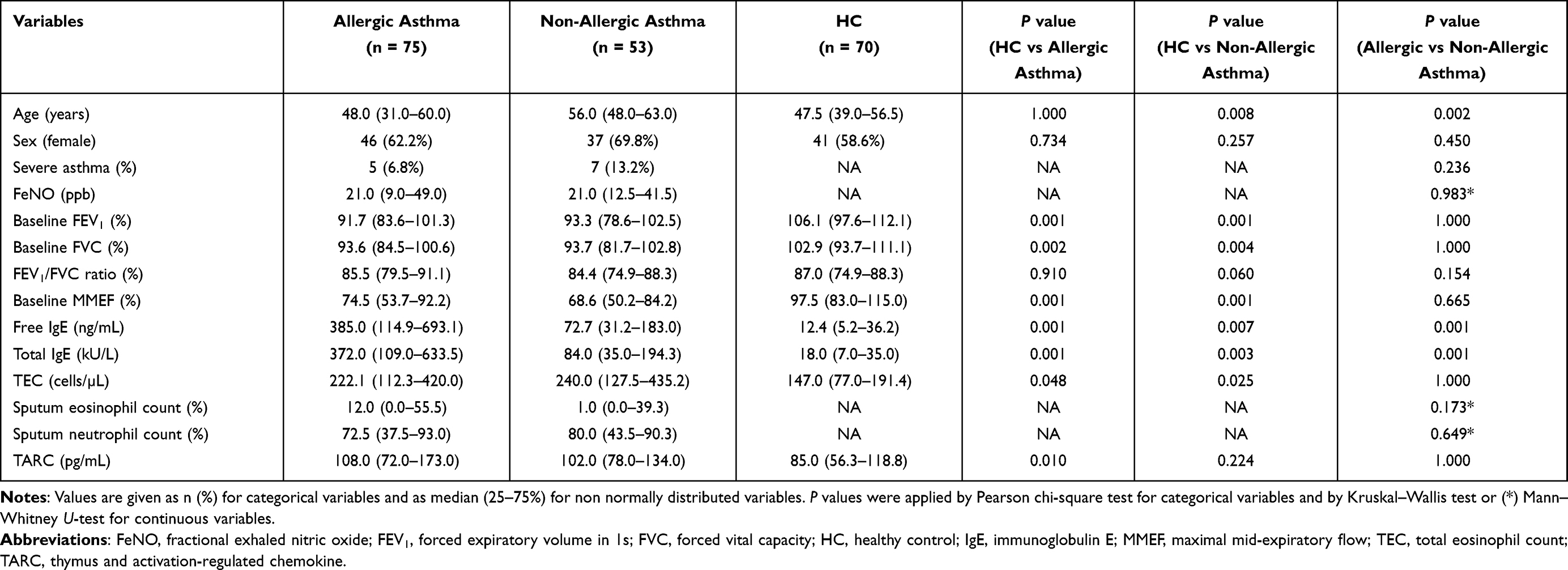 |
Table 1 Demographic Data of the Study Subjects |
Comparison of Serum TARC Levels in Asthmatics
Higher serum TARC levels were noted in asthmatics than in HCs 107.0 (75.5–155.5) pg/mL vs 85.0 (56.3–118.8) pg/mL (P = 0.004; Figure 1A), among which significantly higher TARC levels were noted in patients with allergic asthma than in HCs, 108.0 (72.0–173.0) pg/mL vs 85.0 (56.3–118.8) pg/mL (P = 0.010; Table 1 and Figure 1B), while no significant differences were noted between non-allergic asthmatics and HCs or between allergic asthmatics and non-allergic asthmatics (P > 0.050 for both, Table 1 and Figure 1B). The serum TARC level was able to differentiate the asthma group from HCs at the optimal cut-off value of 92.5 pg/mL with 60.0% sensitivity and 61.7% specificity (AUC = 0.622; 95% CI = 0.543–0.701, P = 0.005) as well as the allergic asthma group from HCs at the optimal cut-off value of 93.0 pg/mL with 60.0% sensitivity and 66.2% specificity (AUC = 0.639; 95% CI = 0.549–0.729, P = 0.004, data not shown). Negative correlations were noted between serum TARC levels and FEV1%/MMEF% values (r = −0.314; r = −0.268, respectively, P < 0.050 for both) (Figure 1C and D).
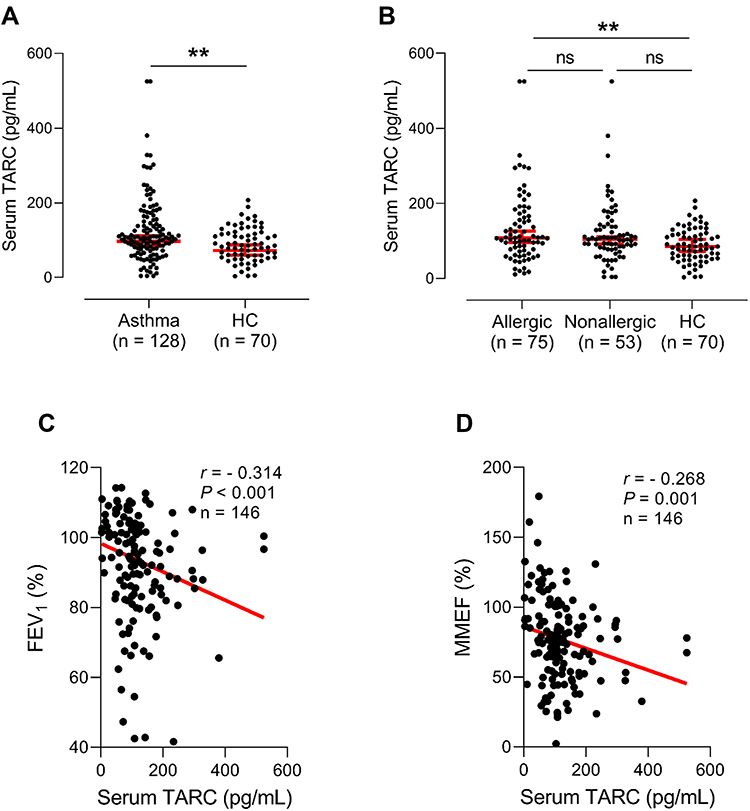 |
Figure 1 Increased serum thymus and activation-regulated chemokine (TARC) levels in asthmatic patients. Comparison of serum TARC levels (A) between asthmatics and healthy controls (HCs) as well as (B) among allergic asthmatics, nonallergic asthmatics, and HCs. Data are presented as Geometric mean with 95% CI. **P < 0.010 by Mann–Whitney U-test or Kruskal–Wallis test. ns, not significant. Significant negative correlations between serum TARC and (C) FEV1%/(D) MMEF%. Data are represented as Spearman correlation coefficient r (P value). FEV1, forced expiratory volume in 1 second; MMEF, maximal mid-expiratory flow. |
When asthmatics are classified into the type 2-high and type 2-low asthma groups according to TEC, FeNO, and free IgE profiles, the serum TARC level can be used to differentiate the type 2-high asthma group from HCs at the optimal cut-off value of 92.5 pg/mL with 60.0% sensitivity and 63.0% specificity (AUC = 0.622; 95% CI = 0.540–0.704, P = 0.006; Figure S1).
Association Between Serum TARC Levels and Inflammatory Mediators
When asthmatic patients were classified into the serum TARC-high and TARC-low groups according to the cutoff value derived from the mean+2 SD values of HCs, 178.1 pg/mL), the TARC-high group was younger (P = 0.013; Table 2), and had significantly higher serum-free IgE (P = 0.031), total IgE (P = 0.030), and 15-HETE levels (P = 0.046) (Table 2 and Figure 2A and B). No significant differences were observed in blood/sputum eosinophil counts or sputum neutrophils between the two groups. No differences were found in serum EDN/TXB2/PGF2α between the high- and low-TARC groups (P > 0.050 for all, Table 2).
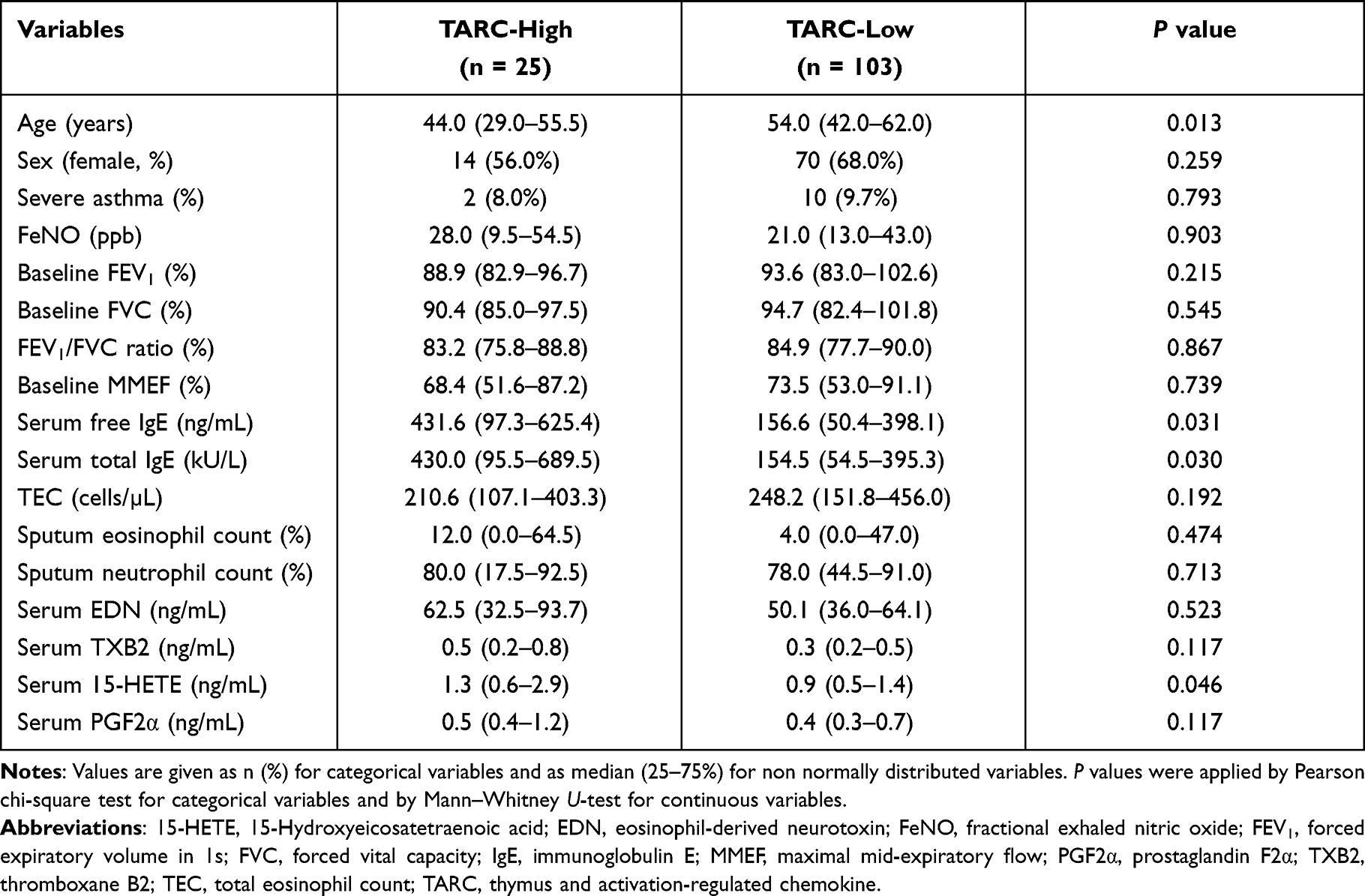 |
Table 2 Comparison of Demographic Characteristics Between the TARC-High and TARC-Low Groups |
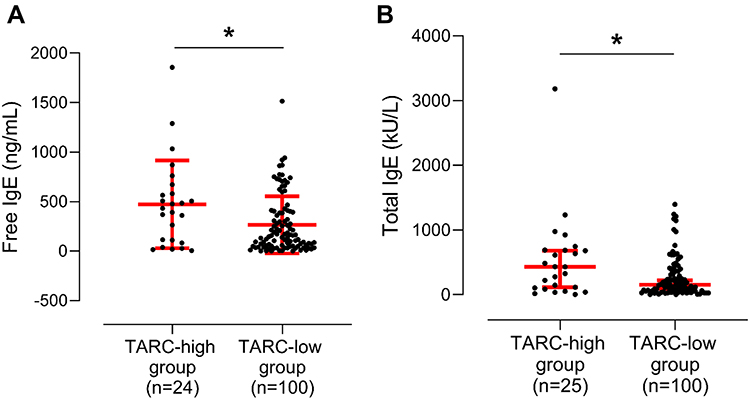 |
Figure 2 Comparison of serum free and total IgE levels between the activation-regulated chemokine (TARC)-high and TARC-low groups. Concentrations of serum levels of (A) free and (B) total IgE between the TARC-high and TARC-low groups. Data are presented as Geometric mean with 95% CI. *P < 0.050 by Mann–Whitney U-test. IgE, immunoglobulin E. |
Subsequently, we further analyzed correlations between serum TARC levels and inflammatory metabolites. There were positive correlations between serum TARC and EDN levels (an activation marker of eosinophils) as well as inflammatory lipid metabolites (15-HETE, TXB2) (r = 0.233, P = 0.024; r = 0.223, P = 0.007; r = 0.264, P = 0.036; respectively) (Figure 3A-C), while no significant difference was noted in the LTE4 level. In addition, significant positive correlations were noted between serum 15-HETE and serum-free IgE/total IgE levels (r = 0.261, P = 0.002; r = 0.232, P = 0.005; respectively) (Figure 3D-E).
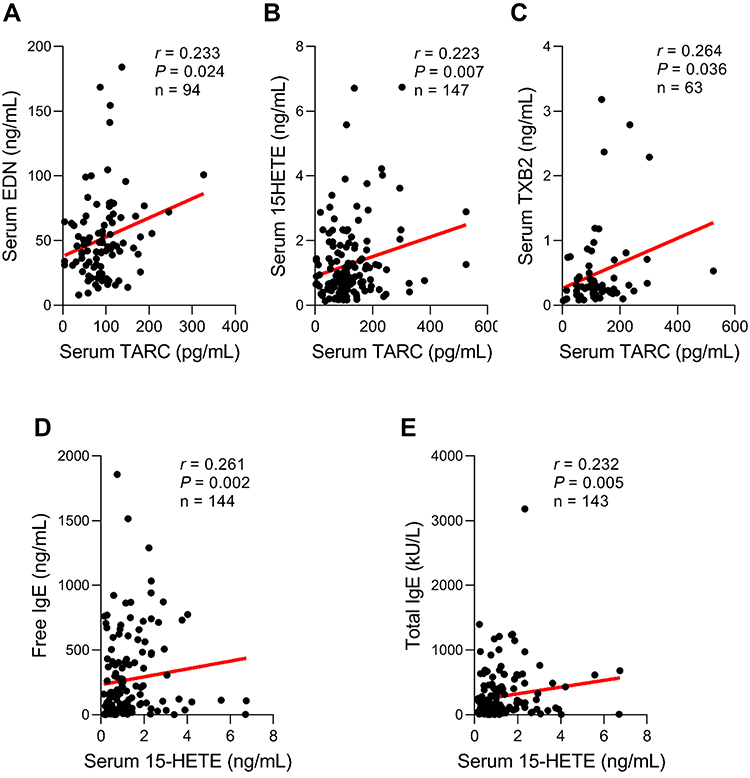 |
Figure 3 Correlations between serum thymus and activation-regulated chemokine (TARC) levels and EDN/lipid mediators. Positive correlations between serum TARC and (A) EDN/ (B) 15-HETE/ (C) TXB2 levels. Data are represented as Pearson correlation coefficient r (P value). Positive correlations between serum 15-HETE and (D) free IgE/ (E) total IgE levels. Data are represented as Spearman correlation coefficient r (P value). 15-HETE, 15-hydroxyeicosatetraenoic acid; EDN, eosinophil-derived neurotoxin; TXB2, thromboxane B2. |
Discussion
The present study investigated the role of TARC in adult asthma in association with various clinical and inflammatory mediators. Significantly higher levels of TARC were found in the sera of adult asthmatic patients than in HCs; additionally, allergic asthmatic patients had significantly higher TARC levels. Moreover, the serum TARC level could discriminate allergic asthmatics from HCs with negative correlations to FEV1% and MMEF%. The TARC-high group had significantly higher serum-free and total IgE levels. Significant correlations were found between serum TARC levels and serum levels of free and total IgE/TXB2/15-HETE/EDN. The serum TARC level could predict the phenotype of type 2-high asthma by ROC analysis. In addition, the present study demonstrated a close relationship between total IgE/free IgE levels and 15-HETE production. TXB2 could favor Th2-cell differentiation via blocking the secretion of interferon-γ in vitro.21 Taken together, increased levels of TARC may induce the release of inflammatory mediators from mast cells, maintaining Th2/eosinophilic responses in asthmatic airways, and thus, contributing to the development of the phenotype of allergic asthma.
Allergic asthma is characterized by childhood-onset asthma, positive skin prick test results, and high serum IgE levels.22 The present study demonstrated that patients with allergic asthma were younger than those with nonallergic asthma and had significantly higher serum levels of free and total IgE. Allergic asthma is mediated by IgE-dependent type 1 hypersensitivity reactions, in which mast cells and basophils are activated by IgE in the asthmatic airway.23 IgE antibody (mainly produced by B cells) binds to its receptor (FcεR1) on mast cells and basophils in order to release various mediators and cytokines, leading to inflammatory responses.23 Released lipid mediators (including cysteinyl LTs, TXA2, and PGs [specially PGD2, PGF2α]) as well as histamine derived from various immune cells could increase vascular permeability and mucus production, and induce bronchoconstriction, enhancing airway inflammation.24 FcεRI-expressing mast cells were accumulated in smooth muscle, bronchial epithelium, and alveolar parenchyma in patients with severe allergic asthma.25,26 Blockage of the infiltration of mast cells and their receptors by using mast cell-deficient mice showed reduced AHR in a mouse model of allergic asthma.27–29 Omalizumab, an anti-IgE antibody, which binds to circulating free IgE in sera has been used to control asthma symptoms in patients with severe allergic asthma.30 It could also reduce FcεRI expression on mast cells, followed by suppression of inflammatory signaling cascades.30 The present study showed increased TARC levels in allergic asthmatics with positive correlations to serum-free IgE/total IgE/EDN levels (type 2 markers). These results suggest that high serum TARC levels are considered the potential target of omalizumab treatment, although further investigations are needed to validate changes in serum TARC levels after omalizumab treatment.
The present study demonstrated that serum TARC level had a positive correlation with serum EDN level, but negative correlations with lung function parameters (FEV1% and MMEF%). Increased serum EDN level (released from activated eosinophils) has been reported as a useful serum biomarker for the degree of eosinophilic inflammation in asthmatic patients.31,32 Furthermore, profibrotic cytokines, including periostin and transforming growth factor beta 1 (released from eosinophils), and close interplays between eosinophils and epithelial cells, could lead to bronchoconstriction, further activation of eosinophils and epithelial cells, and airway remodeling, resulting in lung function declines.7,33 Taken together, TARC-mediated airway dysfunction may be a consequence of activated eosinophils, and eosinophil-epithelial interactions as well as Th2-eosinophil interactions in the asthmatic airways.
Eicosanoids are the most prevalent lipid mediators which regulate the inflammatory process by triggering pro-inflammatory messengers, signaling pathways, and immune cell activation in the pathogenesis of asthma.34 Arachidonic acid (AA), which is released from membrane phospholipids by cytosolic phospholipase A2, is converted into prostanoids (PGs and TXs) by cyclooxygenases (COX) into LTs and lipoxins by lipoxygenases (LOXs), or into epoxyeicosatrienoic and hydroxyoctadecanoic acids via epoxygenases.35,36 AA will be transformed into 5-HETE in the presence of the 5-LOX enzyme and quickly generated into LTA4, LTB4, LTC4, and finally into LTE4 in the cysteinyl LT synthetic pathway.37 However, 15-LOX, which is derived from macrophages, eosinophils, and bronchial epithelial cells in response toward the inflammatory milieu, triggered the transformation of LTA4 into 15-HETE.22,38 By measurement of sub-basement membrane thickness, 15-HETE and its counterpart (15-LOX) would be elevated in severe eosinophilic asthmatics compared to severe non-eosinophilic asthmatics, which was shown to be associated with airway fibrosis,39 and pro-inflammatory effects on tissue remodeling.40,41 However, there is a conflicting result showing that 15-HETE has anti-inflammatory effects through suppressing inflammatory cell migration and activation as well as inflammatory effects through adhering to molecules. The high concentrations of LTE4 and PGF2α released from mast cells could increase inflammatory and contractile responses in asthmatic patients. The present study demonstrated that the production of 15-HETE is related to serum free and total IgE levels, which were similar whether or not asthmatics were maintaining additional leukotriene receptor antagonists or ICS-LABA. These findings suggest that 15-HETE may exert its harmful effects through releasing mediators from activated mast cells.42 In addition, TXA2 derived from the COX-pathway is not a stable form; thus, it is hydrolyzed into TXB2.22,38 TXB2 is known as the inhibitive factors that favor Th2-cell differentiation through blocking Th1 cytokine (interferon-γ) secretion in vitro.21 Taken together, it is conceivable that the increased level of TARC may reflect the dysregulation of AA metabolism, perpetuating eosinophilic airway inflammation and remodeling in asthmatic airways, where LT modifiers or ICS+LABA could not suppress this TARC-induced inflammatory response.
This study has some limitations. First, it is an observational single-center study with limited sample size; further multicenter studies are needed to confirm our results. Second, no direct associations were found between the severity of asthma and serum TARC levels/sputum eosinophils/TEC. Longitudinal follow-up studies are necessary to clarify the changes in serum TARC levels after anti-inflammatory treatments or type 2 biologics in asthmatic patients. Third, our investigation could not clarify the mechanisms on how TARC induces eosinophil activation or dysregulation of inflammatory lipid mediators in vivo and in vitro. Despite these limitations, we first demonstrated two TARC-induced airway inflammatory mechanisms (the TARC-Th2 cell trafficking-eosinophil degranulation-epithelial damage axis and the TARC-IgE-mast cell activation-eicosanoid productions-lung function decline axis) in patients with allergic asthma in the real-world clinical setting.
In conclusion, these findings suggest that TARC may contribute to develop the phenotype of allergic asthma through the Th2-driven activation of eosinophils and mast cells, followed by release of inflammatory lipid mediators. Further studies are needed to evaluate whether biologics (anti-IgE antibody or other type 2 biologics) could overcome this inflammatory signaling cascade.
Acknowledgment
We would like to express our gratitude to Dr Joo-Youn Cho, for her support in the performance of LC-MS/MS analysis for inflammatory lipid mediators.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by the Basic Science Research Program of the National Research Foundation (NRF) grant funded by the Korea government (NRF-2021R1A2B5B01001767) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) grant funded by the Ministry of Health and Welfare, Republic of Korea (grant no. HR16C0001).
Disclosure
The authors report no conflicts of interest for this work and that there are no financial or other issues that might lead to conflicts of interest.
References
1. Global strategy for asthma management and prevention. 2020. Available from: http://ginasthma.org/gina-reports/.
2. Lee E, Kim A, Ye YM, Choi SE, Park HS. Increasing prevalence and mortality of asthma with age in Korea, 2002–2015: a nationwide, population-based study. Allergy Asthma Immunol Res. 2020;12(3):467–484.
3. Caminati M, Morais Almeida M, Bleecker E, et al. Biologics and global burden of asthma: a worldwide portrait and a call for action. World Allergy Organ J. 2021;14:2.
4. Kuruvilla ME, Lee FEH, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy. 2019;56(2):219–233.
5. Weinberger SE, Cockrill BA, Mandel J. Asthma. In: Weinberger SE, Cockrill BA, Mandel J, editors. Principles of Pulmonary Medicine (Seventh Edition). Philadelphia: Elsevier; 2019:75–92.
6. Ying S, Meng Q, Zeibecoglou K, et al. Eosinophil chemotactic chemokines (Eotaxin, Eotaxin-2, RANTES, monocyte chemoattractant protein-3 (MCP-3), and MCP-4), and C-C chemokine receptor 3 expression in bronchial biopsies from atopic and nonatopic (Intrinsic) asthmatics. J Immunol Res. 1999;163(11):6321–6329.
7. Agrawal DK, Shao Z. Pathogenesis of allergic airway inflammation. Curr Allergy Asthma Rep. 2010;10(1):39–48.
8. Subramanian H, Gupta K, Ali H. Roles of Mas-related G protein–coupled receptor X2 on mast cell–mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J Allergy Clin Immunol. 2016;138(3):700–710.
9. Tiotiu A. Biomarkers in asthma: state of the art. Asthma Res Pract. 2018;4:10.
10. Umeda M, Origuchi T, Kawashiri S, et al. Thymus and activation-regulated chemokine as a biomarker for IgG4-related disease. Sci Rep. 2020;10(1):6010.
11. Imai T, Baba M, Nishimura M, Kakizaki M, Takagi S, Yoshie O. The T cell-directed CC chemokine TARC is a highly specific biological ligand for CC chemokine receptor 4. J Biol Chem. 1997;272(23):15036–15042.
12. Kataoka Y. Thymus and activation-regulated chemokine as a clinical biomarker in atopic dermatitis. J Dermatol. 2014;41(3):221–229.
13. Sekiya T, Yamada H, Yamaguchi M, et al. Increased levels of a Th2-type CC chemokine thymus and activation-regulated chemokine (TARC) in serum and induced sputum of asthmatics. Allergy. 2002;57(2):173–177.
14. Kawasaki S, Takizawa H, Yoneyama H, et al. Intervention of thymus and activation-regulated chemokine attenuates the development of allergic airway inflammation and hyperresponsiveness in mice. J Immunol. 2001;166(3):2055–2062.
15. Pham DL, Kim SH, Losol P, et al. Association of autophagy related gene polymorphisms with neutrophilic airway inflammation in adult asthma. Korean J Intern Med. 2016;31(2):375–385.
16. ATS/ERS. ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005. Am J Respir Crit Care Med. 2005;171(8):912–930.
17. Morris JF. Spirometry in the evaluation of pulmonary function. West J Med. 1976;125(2):110–118.
18. Woo SD, Yang EM, Jang J, et al. Serum-free immunoglobulin E: a useful biomarker of atopy and type 2 asthma in adults with asthma. Ann Allergy Asthma Immunol. 2021;127(1):109–115.
19. Holguin F, Cardet JC, Chung KF, et al. Management of severe asthma: a European Respiratory Society/American Thoracic Society guideline. Eur Respir J. 2020;55(1):1900588.
20. Luu QQ, Cao TBT, Shin YS, Yang EM, Moon JY, Park HS. Sputum ANA serves as a biomarker for severe asthma. Allergy. 2021. doi:10.1111/all.15086
21. Li H, Edin ML, Gruzdev A, et al. Regulation of T helper cell subsets by cyclooxygenases and their metabolites. Prostaglandins Other Lipid Mediat. 2013;104-105:74–83.
22. Debeuf N, Lambrecht BN. Eicosanoid control over antigen presenting cells in asthma. Front Immunol. 2018;9. doi:10.3389/fimmu.2018.02006(2006).
23. Froidure A, Mouthuy J, Durham SR, Chanez P, Sibille Y, Pilette C. Asthma phenotypes and IgE responses. Eur Respir J. 2016;47(1):304–319.
24. Broide DH. Molecular and cellular mechanisms of allergic disease. J Allergy Clin Immunol. 2001;108(2 Suppl):S65–71.
25. Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346(22):1699–1705.
26. Amin K, Janson C, Boman G, Venge P. The extracellular deposition of mast cell products is increased in hypertrophic airways smooth muscles in allergic asthma but not in nonallergic asthma. Allergy. 2005;60(10):1241–1247.
27. Williams CMM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med. 2000;192(3):455–462.
28. Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Investig. 2006;116(6):1633–1641.
29. Mayr SI, Zuberi RI, Zhang M, et al. IgE-dependent mast cell activation potentiates airway responses in murine asthma models. J Immunol Res. 2002;169(4):2061–2068.
30. Lee Y, Quoc QL, Park HS. Biomarkers for severe asthma: lessons from longitudinal cohort studies. Allergy Asthma Immunol Res. 2021;13(3):375–389.
31. Lee Y, Lee JH, Yang EM, et al. Serum levels of eosinophil-derived neurotoxin: a biomarker for asthma severity in adult asthmatics. Allergy Asthma Immunol Res. 2019;11(3):394–405.
32. An J, Lee JH, Sim JH, et al. Serum eosinophil-derived neurotoxin better reflect asthma control status than blood eosinophil counts. J Allergy Clin Immunol Pract. 2020;8(8):2681–2688.
33. Vignola AM, Kips J, Bousquet J. Tissue remodeling as a feature of persistent asthma. J Allergy Clin Immunol. 2000;105(6 Pt 1):1041–1053.
34. Sanak M. Eicosanoid mediators in the airway inflammation of asthmatic patients: what is new? Allergy Asthma Immunol Res. 2016;8(6):481–490.
35. Kytikova O, Novgorodtseva T, Denisenko Y, Antonyuk M, Gvozdenko T. Pro-resolving lipid mediators in the pathophysiology of asthma. Medicina (Kaunas). 2019;55(6):284.
36. Chen LC, Tseng HM, Kuo ML, et al. Levels of 15-HETE and TXB2 in exhaled breath condensates as markers for diagnosis of childhood asthma and its therapeutic outcome. Pediatr Allergy Immunol. 2021;32(8):1673.
37. Celejewska Wójcik N, Kania A, Górka K, et al. Eicosanoids and eosinophilic inflammation of airways in stable COPD. Int J Chron Obstruct Pulmon Dis. 2021;16:1415–1424.
38. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15(8):511–523.
39. Chu HW, Balzar S, Westcott JY, et al. Expression and activation of 15-lipoxygenase pathway in severe asthma: relationship to eosinophilic phenotype and collagen deposition. Clin Exp Allergy. 2002;32(11):1558–1565.
40. Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8(5):349–361.
41. Bocan TMA, Rosebury WS, Mueller SB, et al. A specific 15-lipoxygenase inhibitor limits the progression and monocyte–macrophage enrichment of hypercholesterolemia-induced atherosclerosis in the rabbit. Atherosclerosis. 1998;136(2):203–216.
42. Foster HR, Fuerst E, Branchett W, Lee TH, Cousins DJ, Woszczek G. Leukotriene E4 is a full functional agonist for human cysteinyl leukotriene type 1 receptor-dependent gene expression. Sci Rep. 2016;6(1):20461.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.