")
Back to Journals » Journal of Pain Research » Volume 13
Role of the CXCR4/ALK5/Smad3 Signaling Pathway in Cancer-Induced Bone Pain
Authors Peng C , Chen XT, Xu H, Chen LP, Shen W
Received 14 June 2020
Accepted for publication 15 September 2020
Published 14 October 2020 Volume 2020:13 Pages 2567—2576
DOI https://doi.org/10.2147/JPR.S260508
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor E Alfonso Romero-Sandoval
Chong Peng,1,* Xue-Tai Chen,1,* Heng Xu,2 Li-Ping Chen,2 Wen Shen1,2
1Jiangsu Province Key Laboratory of Anesthesiology and Jiangsu Province Key Laboratory of Anesthesia and Analgesia Application Technology, Xuzhou Medical University, Xuzhou, Jiangsu 221002, People’s Republic of China; 2Department of Pain Medicine, Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu 221002, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wen Shen
Department of Pain Medicine, The Affiliated Hospital of Xuzhou Medical University, 99 Huaihai West Road, Xuzhou 221002, People’s Republic of China
Tel +86 13815311266
Email [email protected]
Purpose: The chemokine receptor, CXCR4, and the transforming growth factor-beta receptor, ALK5, both contribute to various processes associated with the sensation of pain. However, the relationship between CXCR4 and ALK5 and the possible mechanisms promoted by ALK5 in the development of pain have not been evaluated.
Materials and Methods: Tumor cell implantation (TCI) technology was used to generate a model of cancer-induced bone pain (CIBP) in rats; intrathecal (i.t.) injections of small interfering (si) RNAs targeting CXCR4 and the ALK5-specific inhibitor, RepSox, were performed. Behavioral outcomes, Western blotting, and immunofluorescence techniques were used to evaluate the expression of the aforementioned specific target proteins in the CIBP model.
Results: The results revealed that i.t. administration of siRNAs targeting CXCR4 resulted in significant reductions in both mechanical and thermal hyperalgesia in rats with CIBP and likewise significantly reduced the expression of ALK5 in the spinal cord. Similarly, i.t. administration of RepSox also resulted in significant reductions in mechanical and thermal hyperalgesia in rats with CIBP together with diminished levels of spinal p-Smad3.
Conclusion: Taken together, our results suggest that CXCR4 expression in the spinal cord may be a critical mediator of CIBP via its capacity to activate ALK5 and downstream signaling pathways.
Keywords: chemokines, neoplasms, sensation, fluorescent antibody technique, cancer pain
Introduction
Cancer-induced bone pain (CIBP) is a complex and multi-faceted response that includes both inflammatory and neuropathic components associated with tumor growth, bone destruction, and other factors.2–4 CXCR4 is a receptor for CXCL12 (stromal-derived factor-1, SDF-1) and is a member of the seven-transmembrane G protein-coupled receptor (GPCR) family. Previous studies have revealed that CXCL12 binding to CXCR4 can activate intracellular signal transduction pathways and is involved in the development of CIBP.5–7 Likewise, ALK5 (S165 phosphorylated-transforming growth factor-beta (TGFβ) receptor I)-mediated signaling pathway plays an important role in the development of inflammatory,8,9 neuropathic,8,10 and cancer-associated pain.11 Earlier studies suggested that TGFβ interactions with TGF beta receptor II resulted in the activation of ALK5,12–14 thereby identifying a peripheral mechanism underlying the development of pain.15,16 Smad3 is a downstream target and transmits signals from ALK5; its activation can promote both canonical and non-canonical pathways of signal transduction. As part of the canonical pathway, ALK5 directly activates the C-terminal Ser423 + 425 site of Smad3, while the non-canonical pathway completes the activation by targeting amino acids at the link regions, including Ser204, Ser208, and Ser213.8,17 Administration of ALK5-specific inhibitors, including SB-431,542 and the Smad3-specific inhibitor, SIS3, can promote the alleviation of pain.11,16 However, few studies have addressed questions regarding the role of ALK5/Smad3 signaling with respect to pain in the central nervous system; this issue has led to controversy with respect to our understanding of TGFβ as a pain-associated mediator. In contrast to its role in the periphery, TGFβ has been reported to promote analgesia in the central nervous system.18–20 Furthermore, G protein-coupled receptors can transactivate the TGFβ receptor I.21–24
In this study, we employed a rat model of CIBP to examine the potential relationship between CIBP and signaling via CXCR4, ALK5, and Smad3 in spinal cord tissue.
Materials and Methods
Animals
Specific pathogen-free Sprague–Dawley (SD) female rats (Experimental Animal Center, Jinan, China) weighing 180–200 g were maintained at ~21 ± 0.5°C with alternating lights simulating day and night. All rats were acclimated to these conditions for 3 days prior to the start of the experiment.
All experimental protocols and animal handling procedures were approved by the Animal Protection and Use Committee of Xuzhou Medical University. The protocols are consistent with the National institute of Health Guide for the Care and Use of Laboratory Animals and the International Association for the Study of Pain’s guidelines for pain research.26
Cell Preparation
Walker 256 breast cancer cells (Institute for Biomedical Research of Shanghai, Shanghai, China) were prepared in SD rats. Tumor cells (2 × 107 cells/mL, 0.5 mL) were injected into the abdominal cavities of SD rats weighing 60–80 g. After 6–7 days, ascitic fluid was extracted aseptically and centrifuged three times at 1000 rpm; the cells in the pellet were washed five times with sterile physiological saline (NS). The pellet was resuspended in NS and adjusted to the appropriate concentration at 1 × 105 cells/μL. The cell suspension was held on ice until use for injections into rats. For sham controls, sterile NS alone was used for injections. The use of cell lines was approved by the ethics committee of Xuzhou Medical University.
Model of Bone Cancer Pain
The female SD rats were anesthetized by intraperitoneal injection of sodium pentobarbital (50mg/kg) and fixed in a supine position. After disinfection with 75% alcohol, an 0.5-cm incision was made in the skin along the long axis of the upper third of the right tibia. The muscles were separated by blunt dissection to expose the tibial plateau. The needle of a 5 mL syringe was used to drill a hole in the bone surface and a 25-μL micro-injector was used to generate a small cavity in the medullary cavity of the tibia through that was large enough to permit fresh blood to be withdrawn. Five μL of a Walker 256 cell suspension at 2 × 104/μL was slowly injected into the cavity;27,28 rats in the sham group were injected with an equal volume of sterile saline. After the injection, the needle was left in place for approximately 60 s, after which the microsyringe was removed. The pinhole was closed with dental glass ionomer cement and erythromycin ointment was applied to the incision. Hematoxylin staining of the tibia was used for evaluation of bone destruction (Supplementary Figure S1)
Administration of Study Drugs
Repsox was synthesized by Selleck Chemicals (Shanghai, China). After inducing anesthesia with sodium pentobarbital (50mg/kg), the rats were placed in a prone position. A 40 U insulin syringe was used to administer Repsox (10 µL at a concentration of 10 µg/µL in dimethyl sulfoxide [DMSO]) to rats in the experimental group; those in the control group received 10 µL of DMSO alone. The needle was into the gap between vertebrae L4–L5 until the characteristic tail flick was observed; drug was then injected slowly while making certain that another tail flick was observed while the injection was taking place. The tip of the needle was removed and the wound was disinfected with iodine before returning the rat to a clean cage; wounds were monitored carefully. The treatment time points are as indicated in the individual Figure Legends.
Administration of Small Interfering RNA (siRNA) Targeting CXCR4
Small interfering RNA (siRNA) targeting CXCR4 was obtained from Sangon Biotech with the sequence based on a GenBank search (Accession No. RS5761). The CXCR4 siRNA sequences used in this study included 5′-GACUGGUACUUUGGGAAAUTT-3′ and 5′-AUUUCCCAAAGUACCAGUCTT-3′. Scrambled sequences (Scr) were introduced as negative controls, including 5′-GUAGCAGGGCAUGUAUUUATT-3′ and 5′-UAAAUACAUGCCCUGCUACTT-3′ was designed as a negative control. The siRNA was mixed with polyethyleneimine (PEI; Sigma-Aldrich, St Louis, MO, USA) to increase cell membrane penetration;17 1 μg siRNA in 0.18 μL PEI was administered by intrathecal injection. CXCR4 knockdown was evaluated by Western blotting (Supplementary Figure S2).
Behavioral Testing
Assessment of Mechanical Allodynia
A polymethyl methacrylate (PMMA) box of dimension 26 × 20 × 14 cm was placed on a metal mesh. After the rats were permitted to acclimate for 30 min in the PMMA box, a von Frey filament was used to vertically stimulate the middle part of the hind paw of the rat.29,30 Behaviors including raising a foot or lameness were considered as positive reactions; the absence of either of these responses was considered as negative reactions. Each rat was evaluated five times. The number of the filament that generated a positive reaction in three or more independent trails was up; the number of the next filament was down.31
Assessment of Thermal Hyperalgesia
Rats were placed on a glass plate and portions of the posterior toes were irradiated with a thermal radiation meter. The time from the start of irradiation to the contraction withdrawal reaction was identified as the paw withdrawal latency.32 Before beginning the experiment, the pain threshold of the plantar base was measured. Repeated measurements were separated by 5 min intervals so that the pain experienced will have dissipated. Each rat was evaluated five times; the average paw withdrawal latency was defined as the thermal pain latency.
Western Blot Analysis
L4–L5 spinal cord lumbosacral segments were removed for evaluation. Pre-cooled lysis buffer with a protease inhibitor cocktail was added to each spinal cord tissue specimen. After homogenization, the sample was centrifuged at 12,000 rpm for 15 min at 4°C; the supernatants were then transferred to a clean centrifuge tube. Protein quantification was performed using the bicinchoninic acid (BCA) method and diluted to 8 μg protein per μL. Loading buffer (2% sodium dodecyl sulfate, 100 mM dithiothreitol, 10% glycerol, and 0.02% bromophenol blue) was added and all samples were denatured in a boiling water bath for 15 min. Forty μg of protein was loaded and electrophoresed on a 10% SDS polyacrylamide gel at 100 V. The proteins were subjected to wet transfer to a PVDF membrane at a constant current of 300 mA. Membranes were blocked with 3% bovine serum albumin at room temperature for 2 h. Primary antibodies included rabbit anti-CXCR4 (1:500 dilution; NB100-74,396, Novus Biologicals, Littleton, CO, USA), rabbit anti-phospho-TGF β Receptor I (Ser165, 1:1000 dilution; AF8080, Affinity), rabbit anti-Smad3 (1:1000 dilution; ab40854, Abcam, UK), rabbit anti-phospho-Smad3 (Ser423 + Ser425, 1:1000 dilution, ab52903, Abcam), rabbit anti-phospho-Smad3 (Ser208, 1:1000 dilution; AF3365, Affinity) and rabbit anti-GAPDH (1:5000 dilution; CW0101, China). Membranes were incubated with primary antibodies overnight at 4°C. The membranes were washed three times, 5 min per wash, with Tris-buffered saline with Tween-20 (TBST buffer) and then incubated with horseradish peroxidase-labeled goat anti-rabbit Ig (1:5000) for 2 h at room temperature. Membranes were then rinsed six times with TBST, 5 min per wash, followed by evaluation using the UVITEC gel camera. Protein band densities were determined and normalized to GAPDH. The fold change observed in specimens from the control group was set to 1.0 for relative quantification.
Immunofluorescence
Rats under deep anesthesia with pentobarbital sodium were rapidly perfused with normal saline (300 mL) followed by a 4% paraformaldehyde solution (300 mL). After perfusion, the L4–L5 segments of the spinal cord were excised and were labeled to distinguish between the left and right sides. The specimens were placed in 4% paraformaldehyde solution and stored overnight at 4°C. On the following day, specimens were transferred to a 30% sucrose solution, frozen, and dehydrated. Spinal tissues were serially sliced (25 μm per section) while in a frozen state. The sections were rinsed three times with 0.01 M PBS for 5 min, followed by blocking solution containing 10% serum. After blocking at room temperature for 2 h, primary antibodies were added, including rabbit anti-CXCR4 (1:200 dilution; NB100-74,396, Novus Biologicals, Littleton, CO, USA), anti-TGF-beta RI/ALK-5 (1:200 dilution, NBP2-58,478, Novus Biologicals, Littleton, CO, USA), rabbit anti-phospho-Smad3 (Ser423 + Ser425, 1:200 dilution; ab52903, Abcam), rabbit anti-phospho-Smad3 (Ser208, 1:1000 dilution; AF3365, Affinity), mouse anti-NeuN (neuronal nuclear marker, 1:1000 dilution, MAB377, Millipore), goat anti-GFAP (astrocyte marker, 1:1000 dilution, ab53554, Abcam), and goat anti-Iba-1 (microglia marker, 1:1000 dilution; ab5076, Abcam). Sections were then washed three times for 10 min each in 0.01 M PBS containing 0.3% Triton X-100 and incubated for 2 h at room temperature with the corresponding Alexa Fluor® 405, 488, and 594-conjugated secondary antibodies (1:1000 dilution; Invitrogen, Carlsbad, CA, USA) overnight at 4°C. The relative mean fluorescence intensity (MFI) for ALK5 immunofluorescence in the entire superficial dorsal horn, including 16 spinal cord sections from four rats (4 sections from each rat from each group) was measured using ImageJ software.1,25 The fold change associated with the control group was set at 1.0 for quantification.
Statistical Analysis
All data were presented as mean ± standard error of the mean. Differences in behavior group were evaluated using a two-way analysis of variance (ANOVA) with repeated measures; multiple group means were compared using the Bonferroni post hoc test. One-way ANOVA was used to evaluate the results of Western blot and immunofluorescence; multiple group means were compared using the Bonferroni post hoc test. Algorithms in GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA) were used for all analyses.
Results
Upregulation of CXCR4, ALK5, and p-Smad3 in Spinal Nerve Cells After TCI
Compared to results obtained from rats in the sham-injection control group, expression of CXCR4, ALK5, and P-Smad3 gradually increased over time in rats subjected to TCI; peak expression was detected at T14 (day 14 after injection of the tumor cells; Figure 1A–H). These results indicate that CXCR4, ALK5, and P-Smad3 were all upregulated in response to TCI. The results of immunofluorescence revealed that, at T14, the fluorescence intensity of the target proteins CXCR4, ALK5, and P-Smad3 were all enhanced; increased expression was observed primarily in the shallow layer of the dorsal horn of the spinal cord (layers I and II; Figure 1I). These results indicate that the upregulated expression of CXCR4, ALK5, and P-Smad3 was detected mainly in the superficial layer of the dorsal horn of the spinal cord.
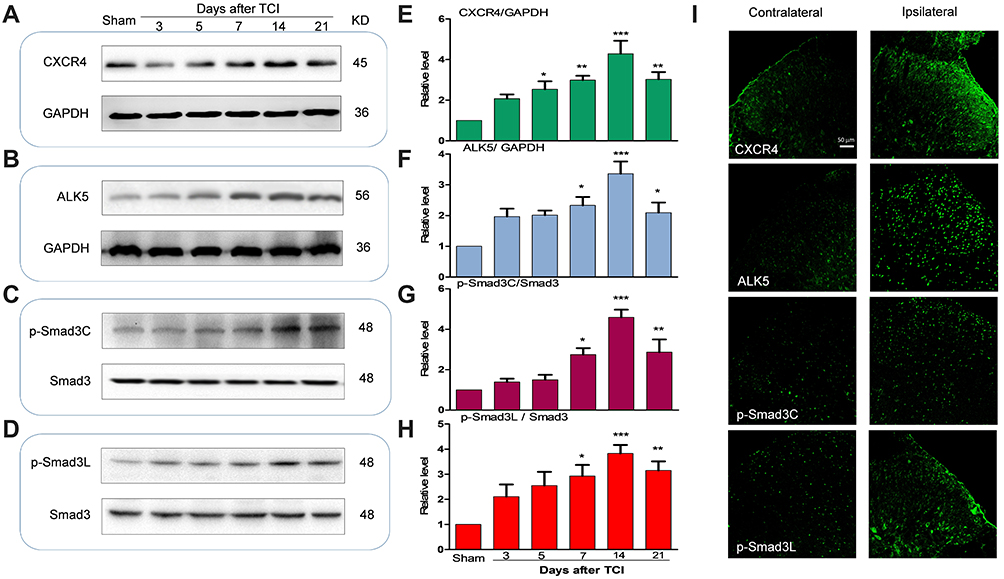 |
Figure 1 Tumor cell implantation (TCI) increased the expression of CXCR4, ALK5 and P -Smad3 in the spinal dorsal horn of rats. (A–H) Compared with the sham-control group, expression of CXCR4, ALK5 and P-Smad3 in the rats receiving tumor cell injections (TCI) gradually increased over time; peak expression was detected at day 14 (*P < 0.05, **P < 0.01, ***P < 0.001, n = 4 in each group). (I) Increased expression was detected in the shallow layer of the dorsal horn of the spinal cord. Scale bar, 50 μm. |
CXCR4 Co-Localizes with ALK5 in Spinal Nerve Cells After TCI
To evaluate this association further, we examined the expression of CXCR4 and ALK5 by confocal immunofluorescence staining. Our results revealed that, up to T14, ALK5 and CXCR4 were co-localized in the spinal cord (Figure 2A). Similar to results obtained in our previous experiments focused on CXCR4 spinal cord localization, ALK5 was also co-localized with markers specific for neurons, astrocytes, and microglia in the spinal dorsal horn (Figure 2B–D). The percentage of ALK5+CXCR4+-positive numbers was analyzed by Image J Software (Supplementary Figure S3).
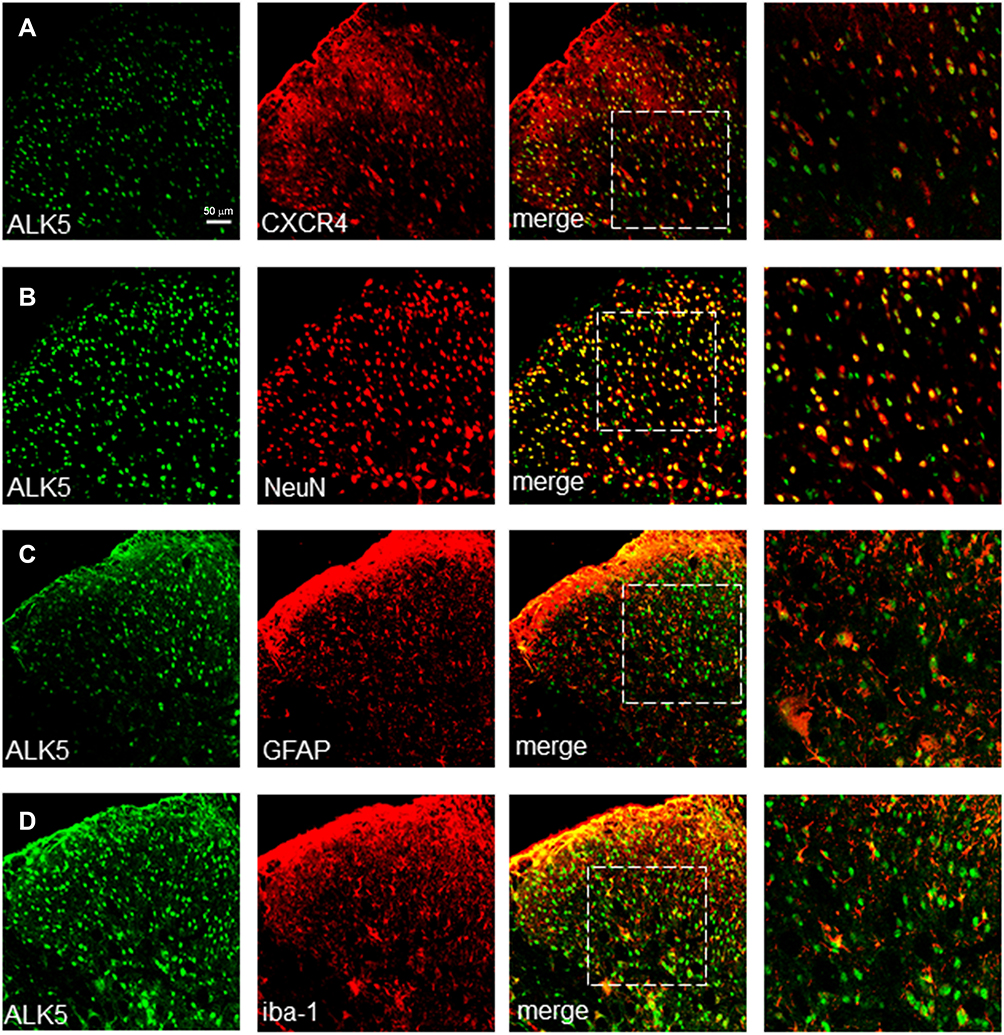 |
Figure 2 Shown are sections of the spinal cord (L4 - L5) at day 14 (A) Up-regulated expression of both ALK5 and CXCR4. (B–D) Upregulated expression of ALK5 corresponded to markers of neurons in the dorsal horn of the spinal cord and likewise with astrocytes and microglia. Scale bar, 50 μm. |
Role of CXCR4 in Nociceptive Behaviors and ALK5 Activation in Rats with CIBP
Behavioral results revealed that rats undergoing TCI gradually responded with pain-associated behavior, manifested as a decrease in paw mechanical withdrawal threshold (a finding that represents mechanical allodynia) and paw withdrawal tendency (representing thermal hyperalgesia). These behavioral responses were observed on post-operative days 5 to 14. Interestingly, i.t. injection of CXCR4-targeted siRNA (5 μg per injection, once daily for 3 consecutive days on post-operative days 12, 13, and 14) reversed the observed mechanical allodynia and thermal hyperalgesia (Figure 3A and B). Administration of scrambled siRNA had no impact on pain hypersensitivity for the entire duration of the experiment. These results suggest that knockdown of CXCR4 expression in cells in the spinal cord has an anti-nociceptive effect with respect to processes associated with CIBP.
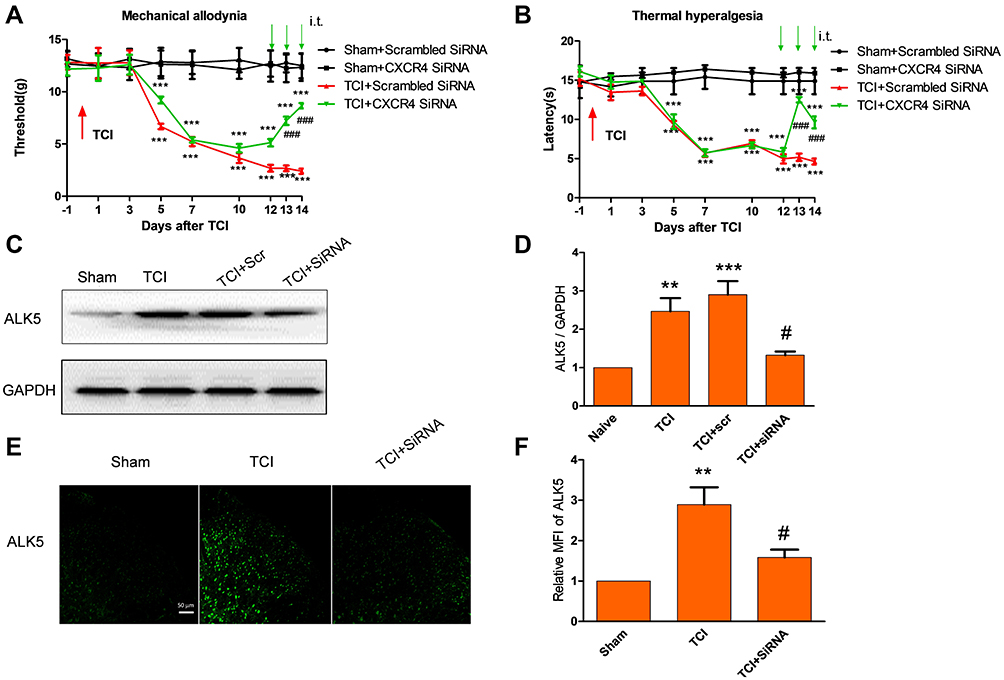 |
Figure 3 CXCR4-targeted siRNA attenuated TCI-induced bone pain and up-regulation of ALK5 expression. (A and B) Compared with the sham group treated with scrambled (control) siRNA, the groups treated with TCI + scrambled siRNAs and TCI + CXCR4-targeted siRNAs revealed significant decreases in both mechanical pain and thermal pain threshold (**P < 0.01, ***P < 0.001). Compared with the TCI + scrambled siRNA group, the TCI + CXCR4-targeted siRNA group revealed significant decreases in both mechanical pain and thermal pain thresholds at both 10 days and14 days (###P < 0.001; n = 8). (C and D) Western blot analysis reveals that, compared with the TCI + scrambled siRNA group, the expression of ALK5 was significantly decreased in the the TCI + CXCR4-targeted siRNA group (#P < 0.05; n = 4). (E and F) Immunofluorescence analysis revealed that, compared with results from the TCI group, relative ALK5 MFI observed in specimens from the TCI + siRNA group was significantly decreased (#P < 0.05; n = 4). Scale bar, 50 μm. |
We then examined the impact of CXCR4 on signaling via the neuronal ALK5 pathway in spinal cord nerve cells. Results of both Western blot and immunofluorescence indicated that repeated administration of CXCR4-targeted siRNA (5 μg per injection, once per day on 3 consecutive days, on days 12, 13 and 14 days after TCI) significantly suppressed the upregulated expression of ALK5 (Figure 3C–F). These results indicate that ALK5 may be a functional target of CXCR4 in spinal cord nerve cells after TCI and in association with CIBP.
Role of ALK5 in Nociceptive Behaviors and Smad3 Activation in Rats with CIBP
We explored the impact of the ALK5-specific inhibitor, RepSox, on hypersensitivity associated with CIBP. Results of behavioral tests revealed that repetitive administration of RepSox (10 μg per i.t. injection, once daily for 3 consecutive days, on days 5, 6, and 7 after TCI) resulted in significantly delayed progression of mechanical allodynia and thermal hyperalgesia (Figure 4A and B). These results suggested that the spinal ALK5/Smad3 signal transduction pathway may play a role in the pathogenesis of CIBP.
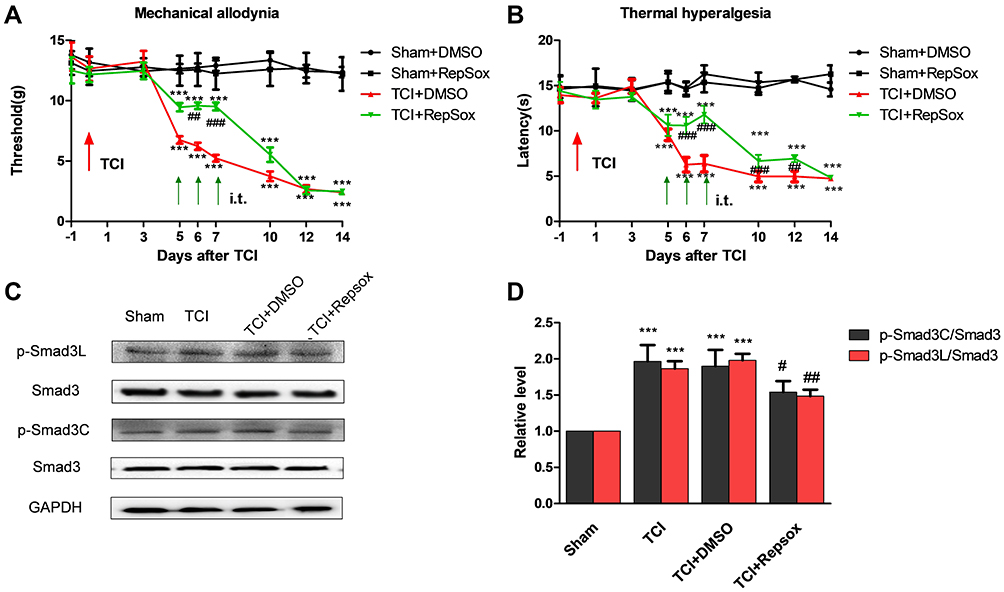 |
Figure 4 ALK5 specific-inhibitor Repsox attenuates TCI-induced cancer pain and p-Smad3 up-regulation. (A and B) Compared results observed from rats in the sham + DMSO group, mechanical pain and the thermal pain threshold were significantly decreased in the TCI + DMSO group and the TCI + RepSox group (***P < 0.001). Compared with the TCI + DMSO group, the TCI + RepSox group revealed significant increases in the mechanical pain threshold on days 6 and 7 (##P < 0.01 and ###P < 0.001); similarly, the thermal pain threshold increased significantly at T6 and T7 (###P < 0.001; n = 8). Behavioral evaluations were performed at 8 h after each intrathecal injection. (C and D) Western blots revealed that administration of RepSox resulted in reduced levels of activation of P-Smad3. Compared with the sham group, expression of P-Smad3L and P-Smad3C was significantly increased in both the TCI group and the TCI + DMSO group (***P < 0.001). Compared with results from the TCI group, the expression of P-Smad3L was significantly decreased in the TCI + RepSox group (##P < 0.01); the expression of P-Smad3C in the TCI + RepSox group was also significantly decreased (#P < 0.05; n = 4). Spinal tissues at L4 – L5 were collected 12 hours after the final injection. |
We then examined whether activation of ALK5 promotes Smad3 activation in spinal cord nerve cells in response to CIBP. The results of Western blotting results revealed that administration of RepSox (10 μg per i.t. injection on days 5, 6, and 7) inhibited activation of P-Smad3C in both the canonical and non-canonical signaling pathways (Figure 4C and D). These results suggest that the activation of Smad3 in the non-canonical pathway is also dependent on ALK5.
Discussion
The results of our study include several interesting observations. First, using a CIBP model, expression of both ALK5 and P-Smad3 were upregulated in a manner similar to that observed for CXCR4; ALK5 and P-Smad3 were also detected in the shallow layer of the dorsal horn of the spinal cord. We also found that ALK5 co-localized with CXCR4 at this locale. Second, we found that repeated i.t. administration of CXCR4-targeting siRNAs or an ALK5-specific inhibitor suppressed TCI-induced nociceptive behaviors. Taken together, our results suggest that ALK5 may be a downstream target of CXCR4; we also found that both the canonical and non-canonical Smad3 activation pathways were regulated by ALK5.
At this time, it is not fully clear whether TGFβ receptors are involved in the development of CIBP; our current understanding of the role of this cytokine in modulating pain responses in the peripheral nervous system appears to be inconsistent with this hypothesis. Neuronal cell culture studies have revealed that blockade of the TGFβ signaling pathway serves to reduce neuronal cell excitability.11,33 Levels of these mediators in the dorsal root ganglion in animal models of pain have revealed similar results, specifically, that TGFβ signaling results in significant increases in the pain threshold in relevant animal models.11,33,34 We can conclude from these studies that TGFβ is clearly involved in modulating pain responses in the peripheral nervous system.
Nonetheless, i.t. injection of exogenous TGFβ did not induce pain but instead alleviated pain of multiple relevant animal models.11,19,35,36 These findings reveal that, contrary to findings generated in the peripheral nervous system, TGFβ may play an analgesic role in the central nervous system. These findings may relate directly to the physiological function of ALK5, which forms in response to binding interactions of TGFβ and TGFβRII which then activates the TGFβRI GS region Ser165.12,13,37 As a classical receptor for TGFβ, its role in animal models of pain appears to be consistent with that of TGFβ. However, recent research has suggested that other interpretations might be considered. For example, the application of SIS3 (Smad3-specific inhibitor) can substantially reduce pain in experimental animals and likewise reduces neuronal excitability in cell culture studies.11,33,38 Smad3 is a downstream target of ALK5 and may be a final target in the TGFβ-TGFβRII-ALK5-Smad3 signaling pathway.9,39 If ALK5/Smad3 can be activated only by TGFβ, ALK5/Smad3 should theoretically be involved in analgesia at the central level; however, our findings are not consistent with this conclusion. One key to solving this problem may be to determine whether ALK5 is involved in the modulation of pain at the central level and likewise to determine whether the non-canonical Smad3 signaling pathway is also dependent on ALK5 activation. In this study, using a CIBP model, we obtained experimental results that reveal that ALK5 is involved in modulating pain at the spinal level and that both the canonical and non-canonical Smad3 signaling pathways are dependent on ALK5. However, there are cytokine mediators other than TGFβ that promote activation of the ALK5/Smad3 signaling pathway as well.
As such, further research is needed in order to answer at least two questions: (1) Under conditions associated with CIBP, which mediator or mediators serve to activate ALK5/Smad3 signaling pathway? (2) How is Smad3 activated by ALK5?
Fortunately, previous research on related subjects can suggest some clues. Previous studies carried out to assess pain suggest the involvement of the CXCL12/CXCR4 signaling axis. Furthermore, previous studies have documented increased concentrations of both CXCL12 and the TGFβ- superfamily member Activin A in sera from patients with metastatic bone cancer.40,41 As such, there appears to be some association between these two very large receptor families. Furthermore, other studies have revealed that GPCR can transactivate the TGFβ receptor.21–24 Signaling via the CXCL12/CXCR4 axis also resulted in the activation of Smad3 via MAPK.42 The results presented here revealed that ALK5 may be among the downstream targets of CXCR4 and may be capable of CXCR4 transactivating ALK5. However, whether the activation of Smad3 in the non-canonical pathway depends on MAPK also remains unclear; further experiments will be needed in order to clarify this point.
This research is associated with several limitations; among these, we have not applied patch-clamp recording technology, and the behavioral analysis presented is not fully comprehensive. However, these results represent a basis for future experimentation. We note that central mechanisms associated with CIBP and the involvement of ALK5 in this process will require ongoing evaluation; additional research will be needed in order to clarify the specific and unique mechanisms underlying CIBP.
Conclusion
The results from our study reveal CXCR4-mediated transactivation of the ALK5/Smad3 signaling pathway, as depicted in the schematic in Figure 5. Blockade of this pathway resulted in significant reductions in pain-associated behavior in a rat model of CIBP. Taken together, our results suggest that the CXCR4/ALK5/Smad3 signaling pathway may be an important therapeutic target and a new focus for intervention in patients with CIBP.
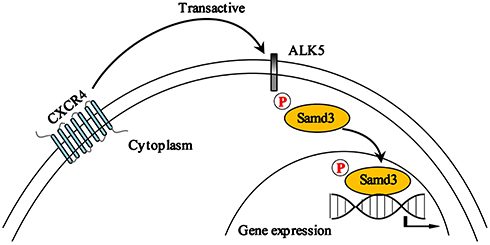 |
Figure 5 Schematic depicting the mechanism underlying CXCR4/ALK5/Smad3 signaling in rats with CIBP. In the lumbar spinal cord, CXCR4-mediated transactivation results in increased activity of the ALK5/Smad3 signaling pathway which contributes to development of CIBP in rats. |
Funding
This study was supported by the National Nature Science Foundation of China (81571066 to Wen Shen), the Key Project of the Natural Science Foundation of Jiangsu Education Department (16KJA320002 to Wen Shen).
Disclosure
The authors declare that they have no competing interests for this work.
References
1. Guirado R, Carceller H, Castillo-Gómez E, Castrén E, Nacher J. Automated analysis of images for molecular quantification in immunohistochemistry. Heliyon. 2018;4(6):e00669. doi:10.1016/j.heliyon.2018.e00669
2. Park SH, Keller ET, Shiozawa Y. Bone marrow microenvironment as a regulator and therapeutic target for prostate cancer bone metastasis. Calcif Tissue Int. 2018;102(2):152–162. doi:10.1007/s00223-017-0350-8
3. Tsuzuki S, Park SH, Eber MR, Peters CM, Shiozawa Y. Skeletal complications in cancer patients with bone metastases. Int J Urol. 2016;23(10):825–832. doi:10.1111/iju.13170
4. Vignani F, Bertaglia V, Buttigliero C, Tucci M, Scagliotti GV, Di Maio M. Skeletal metastases and impact of anticancer and bone-targeted agents in patients with castration-resistant prostate cancer. Cancer Treat Rev. 2016;44:61–73. doi:10.1016/j.ctrv.2016.02.002
5. Hu XM, Zhang H, Xu H, et al. Chemokine receptor CXCR4 regulates CaMKII/CREB pathway in spinal neurons that underlies cancer-induced bone pain. Sci Rep. 2017;7(1):4005. doi:10.1038/s41598-017-04198-3
6. Xu T, Zhang XL, Ou-Yang HD, et al. Epigenetic upregulation of CXCL12 expression mediates antitubulin chemotherapeutics-induced neuropathic pain. Pain. 2017;158(4):637–648. doi:10.1097/j.pain.0000000000000805
7. Shen W, Hu XM, Liu YN, et al. CXCL12 in astrocytes contributes to cancer induced bone pain through CXCR4-mediated neuronal sensitization and glial activation in rat spinal cord. J Neuroinflammation. 2014;11(1):75. doi:10.1186/1742-2094-11-75
8. Muñoz-Félix JM, González-Núñez M, López-Novoa JM. ALK1-Smad1/5 signaling pathway in fibrosis development: friend or foe? Cytokine Growth Factor Rev. 2013;24(6):523–537. doi:10.1016/j.cytogfr.2013.08.002
9. Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP. Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab. 2009;29(6):1084–1098. doi:10.1038/jcbfm.2009.32
10. Wu L, Huang X, Li L, Huang H, Xu R, Luyten W. Insights on biology and pathology of HIF-1α/-2α, TGFα/BMP, Wnt/β-catenin, and NF-ĸB pathways in osteoarthritis. Curr Pharm Des. 2012;18(22):3293–3312. doi:10.2174/1381612811209023293
11. Liu L, Zhu Y, Noë M, Li Q, Pasricha PJ. Neuronal transforming growth factor beta signaling via SMAD3 contributes to pain in animal models of chronic pancreatitis. Gastroenterology. 2018;154(8):2252–2265.e2. doi:10.1053/j.gastro.2018.02.030
12. Fabregat I, Fernando J, Mainez J, Sancho P. TGF-beta signaling in cancer treatment. Curr Pharm Des. 2014;20(17):2934–2947. doi:10.2174/13816128113199990591
13. Massagué J. TGFβ signaling: receptors, transducers, and Mad proteins. Cell. 1996;85(7):947–950. doi:10.1016/S0092-8674(00)81296-9
14. Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi:10.1016/S0092-8674(03)00432-X
15. Utreras E, Keller J, Terse A, Prochazkova M, Iadarola MJ, Kulkarni AB. Transforming growth factor-β1 regulates Cdk5 activity in primary sensory neurons. J Biol Chem. 2012;287(20):16917–16929. doi:10.1074/jbc.M111.329979
16. Xu Q, Zhang XM, Duan KZ, et al. Peripheral TGF-beta1 signaling is a critical event in bone cancer-induced hyperalgesia in rodents. J Neurosci. 2013;33(49):19099–19111. doi:10.1523/JNEUROSCI.4852-12.2013
17. Pineda-Farias JB, Barragán-Iglesias P, Loeza-Alcocer E, et al. Role of anoctamin-1 and bestrophin-1 in spinal nerve ligation-induced neuropathic pain in rats. Mol Pain. 2015;11:41. doi:10.1186/s12990-015-0042-1
18. Chen G, Park CK, Xie RG, Ji RR. Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-beta secretion. J Clin Invest. 2015;125(8):3226–3240. doi:10.1172/JCI80883
19. Chen NF, Chen WF, Sung CS, et al. Contributions of p38 and ERK to the antinociceptive effects of TGF-beta1 in chronic constriction injury-induced neuropathic rats. J Headache Pain. 2016;17(1):72. doi:10.1186/s10194-016-0665-2
20. Chen NF, Huang SY, Chen WF, et al. TGF-β1 attenuates spinal neuroinflammation and the excitatory amino acid system in rats with neuropathic pain. J Pain. 2013;14(12):1671–1685.
21. Burch ML, Osman N, Getachew R. G protein coupled receptor transactivation: extending the paradigm to include serine/threonine kinase receptors. Int J Biochem Cell Biol. 2012;44(5):722–727. doi:10.1016/j.biocel.2012.01.018
22. Dayati P, Rezaei HB, Sharifat N, Kamato D, Little PJ. G protein-coupled receptors can transduce signals through carboxy terminal and linker region phosphorylation of Smad transcription factors. Life Sci. 2018;199:10–15. doi:10.1016/j.lfs.2018.03.004
23. Kamato D, Thach L, Getachew R, et al. Protease activated receptor-1 mediated dual kinase receptor transactivation stimulates the expression of glycosaminoglycan synthesizing genes. Cell Signal. 2016;28(1):110–119. doi:10.1016/j.cellsig.2015.11.003
24. Wojciech S, Ahmad R, Belaid-Choucair Z, et al. The orphan GPR50 receptor promotes constitutive TGFβ receptor signaling and protects against cancer development. Nat Commun. 2018;9(1):1216. doi:10.1038/s41467-018-03609-x
25. Zhuang TT, Pan C, Chen JJ, et al. Chronic asthma-induced behavioral and hippocampal neuronal morphological changes are concurrent with BDNF, cofilin1 and Cdc42/RhoA alterations in immature mice. Brain Res Bull. 2018;143:194–206. doi:10.1016/j.brainresbull.2018.09.006
26. Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16(2):109–110. doi:10.1016/0304-3959(83)90201-4
27. Appel CK, Gallego-Pedersen S, Andersen L, et al. The Src family kinase inhibitor dasatinib delays pain-related behaviour and conserves bone in a rat model of cancer-induced bone pain. Sci Rep. 2017;7(1):4792.
28. Buehlmann D, Ielacqua GD, Xandry J, Rudin M. Prospective administration of anti-nerve growth factor treatment effectively suppresses functional connectivity alterations after cancer-induced bone pain in mice. Pain. 2019;160(1):151–159. doi:10.1097/j.pain.0000000000001388
29. Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi:10.1016/0165-0270(94)90144-9
30. Martin K, Parvin S, Garraway SM. Peripheral inflammation accelerates the onset of mechanical hypersensitivity after spinal cord injury and engages TNFα signaling mechanisms. J Neurotrauma. 2018;36(12):2000–2010. doi:10.1089/neu.2018.5953
31. Bevilaqua-Grossi D, Zanin M, Benedetti C, Florencio L, Oliveira A. Thermal and mechanical pain sensitization in patients with osteoarthritis of the knee. Physiother Theory Pract. 2019;35(2):139–147. doi:10.1080/09593985.2018.1441930
32. Galbraith JA, Mrosko BJ, Myers RR. A system to measure thermal nociception. J Neurosci Methods. 1993;49(1–2):63–68. doi:10.1016/0165-0270(93)90109-5
33. Zhang X, Zheng H, Zhu HY, et al. Acute effects of transforming growth factor-beta1 on neuronal excitability and involvement in the pain of rats with chronic pancreatitis. J Neurogastroenterol Motil. 2016;22(2):333–343. doi:10.5056/jnm15127
34. Mika J, Zychowska M, Popiolek-Barczyk K, Rojewska E, Przewlocka B. Importance of glial activation in neuropathic pain. Eur J Pharmacol. 2013;716(1–3):106–119. doi:10.1016/j.ejphar.2013.01.072
35. Azizzadeh F, Mahmoodi J, Sadigh-Eteghad S, Farajdokht F, Mohaddes G. Ghrelin exerts analgesic effects through modulation of IL-10 and TGF-beta levels in a rat model of inflammatory pain. Iran Biomed J. 2017;21(2):114–119. doi:10.18869/acadpub.ibj.21.2.114
36. Xie L, Tintani F, Wang X, et al. Systemic neutralization of TGF-beta attenuates osteoarthritis. Ann N Y Acad Sci. 2016;1376(1):53–64. doi:10.1111/nyas.13000
37. Souchelnytskyi S, Ten Dijke P, Miyazono K, Heldin CH. Phosphorylation of Ser165 in TGF-beta type I receptor modulates TGF-beta1-induced cellular responses. EMBO J. 1996;15(22):6231–6240. doi:10.1002/j.1460-2075.1996.tb01013.x
38. Tanabe K, Kojima A, Tachi J, Nakashima D, Kozawa O, Iida H. Limitation by Rho-kinase and Rac of transforming growth factor-beta-induced interleukin-6 release from astrocytes. Neurosci Lett. 2019;696:191–196. doi:10.1016/j.neulet.2018.12.040
39. Lin PS, Cheng RH, Chang MC, et al. TGF-β1 stimulates cyclooxygenase-2 expression and PGE2 production of human dental pulp cells: role of ALK5/Smad2 and MEK/ERK signal transduction pathways. J Formos Med Assoc. 2017;116(10):748–754. doi:10.1016/j.jfma.2017.07.008
40. Hofbauer LC, Rachner TD, Coleman RE, Jakob F. Endocrine aspects of bone metastases. Lancet Diabetes Endocrino. 2014;2(6):500–512. doi:10.1016/S2213-8587(13)70203-1
41. Vallet S, Mukherjee S, Vaghela N, et al. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc Natl Acad Sci U S A. 2010;107(11):5124–5129. doi:10.1073/pnas.0911929107
42. Lin CH, Shih CH, Lin YC, Yang YL, Chen BC. MEKK1, JNK, and SMAD3 mediate CXCL12-stimulated connective tissue growth factor expression in human lung fibroblasts. J Biomed Sci. 2018;25(1):19. doi:10.1186/s12929-018-0421-9
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.