")
Back to Journals » Infection and Drug Resistance » Volume 8
Role of small colony variants in persistence of Pseudomonas aeruginosa infections in cystic fibrosis lungs
Authors Malone J
Received 3 May 2015
Accepted for publication 5 June 2015
Published 29 July 2015 Volume 2015:8 Pages 237—247
DOI https://doi.org/10.2147/IDR.S68214
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Suresh Antony
Jacob G Malone1,2
1John Innes Centre, Norwich, UK; 2School of Biological Sciences, University of East Anglia, Norwich, UK
Abstract: Pseudomonas aeruginosa is an opportunistic pathogen that predominates during the later stages of cystic fibrosis (CF) lung infections. Over many years of chronic lung colonization, P. aeruginosa undergoes extensive adaptation to the lung environment, evolving both toward a persistent, low virulence state and simultaneously diversifying to produce a number of phenotypically distinct morphs. These lung-adapted P. aeruginosa strains include the small colony variants (SCVs), small, autoaggregative isolates that show enhanced biofilm formation, strong attachment to surfaces, and increased production of exopolysaccharides. Their appearance in the sputum of CF patients correlates with increased resistance to antibiotics, poor lung function, and prolonged persistence of infection, increasing their relevance as a subject for clinical investigation. The evolution of SCVs in the CF lung is associated with overproduction of the ubiquitous bacterial signaling molecule cyclic-di-GMP, with increased cyclic-di-GMP levels shown to be responsible for the SCV phenotype in a number of different CF lung isolates. Here, we review the current state of research in clinical P. aeruginosa SCVs. We will discuss the phenotypic characteristics underpinning the SCV morphotype, the clinical implications of lung colonization with SCVs, and the molecular basis and clinical evolution of the SCV phenotype in the CF lung environment.
Keywords: small colony variants, cystic fibrosis, cyclic-di-GMP, Pseudomonas aeruginosa, RsmA, antibiotics
Introduction
Cystic fibrosis (CF) is a recessively inherited genetic disease in which the cystic fibrosis transmembrane conductance regulator (CFTR) gene is mutated, leading either to partial or complete loss-of-function. CFTR encodes a chloride ion channel, and the loss of ion transport across epithelial cell membranes leads to osmotic imbalance, and consequently to the buildup of sticky mucus in the lower respiratory tract.1 CF lungs are highly prone to microbial infection, with chronic infection beginning in infancy for the overwhelming majority of cases and continuing throughout the patient’s life. Pseudomonas aeruginosa is an opportunistic Gram-negative pathogen and the predominant infective species isolated from late-stage CF lung infections.2,3 Once established in the lungs of CF patients, P. aeruginosa infections are extremely difficult to completely eradicate, and the aggravation and progressive tissue degradation associated with repeated infectious relapses lead to morbidity and eventually death.3 P. aeruginosa lung infections typically progress clonally from infection with a single environmentally acquired genotype4,5 and, over the course of long-term chronic CF infections, undergo extensive genetic and phenotypic adaptation to the lung environment.6,7
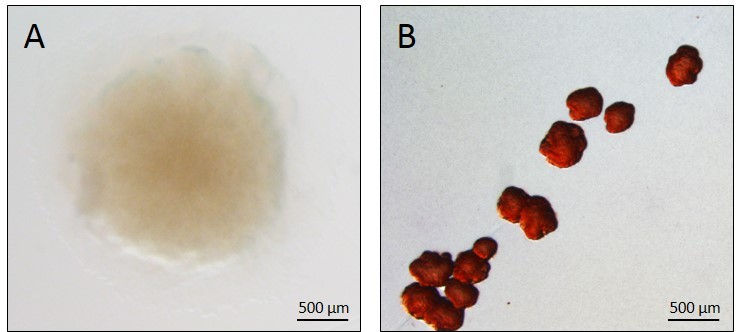
There are two main consequences of this adaptation: first, the progressive transition toward a persistent, low virulence state, and second, a related diversification into a number of different phenotypes.6,7 These include mucoid cells, which overproduce alginate and form distinctive slimy colonies,8 and small colony variants (SCVs, Figure 1), which are typically slow-growing isolates that show strong attachment to surfaces, autoaggregation, enhanced exopolysaccharide production, and biofilm formation.9,10 In recent years, several studies have established a causal link between P. aeruginosa SCVs and persistence of infection in animal models,11–13 supporting the hypothesis that the SCV phenotype confers a fitness advantage under chronic infection conditions, and thus plays an important role in the pathogenesis of P. aeruginosa lung infections. P. aeruginosa SCVs also emerge in other situations that favor chronic infections, including in mechanically ventilated patients or in those suffering from chronic obstructive pulmonary disease.14,15 These studies suggest that persistent P. aeruginosa morphotypes such as SCV represent genetic adaptations to the hostile milieu in the patient, with characteristics including resistance to phagocytosis,12 antimicrobial resistance due to slow growth or increased persister cell populations,16,17 and reduced virulence18 potentially contributing to selection.
 | Figure 1 The Pseudomonas aeruginosa SCV phenotype. |
In this review, we will summarize the current state of research into clinical P. aeruginosa SCVs. We will discuss the initial discovery and characterization of SCVs in the sputum of CF patients, the phenotypic characteristics associated with the SCV morphotype, and the clinical implications of lung colonization with SCVs. Finally, we will address the molecular basis of the SCV phenotype and possible evolutionary routes to SCV generation, with an emphasis on SCV evolution in the lung environment.
Discovery and initial characterization of SCVs in the CF lung
Phenotypic variation between different P. aeruginosa isolates in CF sputum has been recognized for many years. While the alginate-overproducing mucoid phenotype8 has until recently garnered the most attention, it has long been recognized that SCVs (previously known as dwarf colonies) also arise in the chronically infected respiratory tract of CF patients.19 For example, Thomassen et al20 examined P. aeruginosa isolates from 286 CF patients over a 3-month period in 1976–1977 and determined the frequency and distribution of different morphotypes. In this study, dwarf colonies were identified in the sputum of 14% of patients, although these colonies were always found in conjunction with other morphotypes.20 The first clinical study to specifically focus on the distribution and phenotypic characteristics of P. aeruginosa SCV isolates from CF patients took place in Germany from 1996 to 1998. Häussler et al17 tested the sputum of 86 CF patients for P. aeruginosa SCVs, identifying them in 33 patients. Over the course of this study, around 3.0% of the total P. aeruginosa isolates collected were classed as SCVs. These colonies were characterized as small (1–3 mm in diameter), slow growing, and more resistant to several different classes of antibiotics than subsequent planktonic revertants.17 Häussler et al17 went on to characterize some of their SCV isolates, reporting that these strains were hyperpiliated and autoaggregative. The SCV morphs tested showed increased twitching motility and a marked ability to form biofilms and to attach to human pneumocytes.10 Simultaneously with these clinical SCV studies, the molecular biology of SCV formation was being investigated by two independent groups. Drenkard and Ausubel21 showed that P. aeruginosa SCV formation could be induced under laboratory conditions by the addition of kanamycin and linked this phenotype to the putative cyclic-di-GMP (cdG) phosphodiesterase gene pvrR. At the same time, D’Argenio et al22 identified the WspR diguanylate cyclase (DGC) and showed that it was responsible for SCV formation in the laboratory strain PA01.
Phenotypic characteristics of the SCV morphotype
In agreement with the early clinical research, clinical and laboratory-derived SCVs generally display increased resistance to a broad range of antibiotics.17,21,23 Exposure to subinhibitory antibiotic concentrations has been shown to induce attachment and SCV formation in vitro, possibly as part of a defense response.21,24 SCVs are also usually nonmotile, with flagellar motility absent in most SCVs characterized to date.12,23,25 Hyperpiliation has been reported for some strains, with these isolates displaying increased twitching motility as a result,10 although the distribution of this phenotype is unknown. Probably the most prominent phenotype associated with SCV formation is the significantly enhanced production of one or more exopolysaccharide molecules.9,12,25 P. aeruginosa produces at least three exopolysaccharides. The most noticeable of these in clinical CF isolates is alginate, the overproduction of which produces the slimy, mucoid phenotype.8 The role of alginate in SCV formation is unclear, although SCV strains have been identified that overproduce this molecule.26 Also produced are the glucose-rich exopolysaccharide Pel, and Psl, which contains glucose, mannose, and rhamnose.27 Both Pel and Psl overproduction have been explicitly linked to SCV formation, and deletion of both pel and psl operons reverts an SCV to a smooth phenotype.12,25 Many of the other phenotypes associated with SCV, including their enhanced biofilm formation and surface attachment,10 small size (stemming from autoaggregation),17 resistance to phagocytosis,12 and infectious persistence,11–13 have been linked to exopolysaccharide overproduction in these morphs.
A striking characteristic of P. aeruginosa in the CF lung is its extraordinary degree of phenotypic diversity. For example, based on a year-long study of sputum samples from ten CF patients infected with the same initial P. aeruginosa strain, Mowat et al28 examined the phenotypic characteristics of 1,720 individual isolates. The P. aeruginosa populations in each individual sputum sample in this study showed extensive phenotypic diversity, with the majority of diversity occurring within sputum samples rather than between patients. In agreement with this, Workentine et al29 examined a more extensive series of phenotypes for 169 clonal isolates from a single chronically colonized patient. Once again, the researchers observed a very high degree of phenotypic variation. Interestingly, every isolate in this study presented a different phenotypic profile from the others, with very little correlation seen between the majority of tested phenotypes.29
The implications of these findings for SCVs in the lung environment are that apart from the core phenotypic characteristics by which SCVs are identified in the clinic, a high degree of variation might be expected for every other phenotype. Examination of the phenotypes of clinical SCVs has largely confirmed this hypothesis. For example, Häussler et al17 reported reduced growth rates for their clinical SCV isolates, an observation that has since been repeated for other SCV genotypes growing in liquid media.23 However, work from other researchers has shown that this does not apply to all strains or under all growth conditions.30 Similarly, siderophore production has been reported as being downregulated in some SCVs,30 but as being upregulated in others.12,23,31 Cytotoxicity/virulence is another potentially plastic phenotype in SCVs. Long-term lung colonization is generally associated with a loss of cytotoxicity, and downregulation of the type III secretion system (T3SS).32 Reduced cytotoxicity is a recognized characteristic of rsmA- strains,13 which form SCVs under laboratory conditions.33 However, a transcriptional analysis of the well-studied strain SCV20265 showed significant upregulation of T3SS genes compared to its clonal predecessor.31 This increased expression agreed with the results of an earlier proteomic analysis of the same strain34 and corresponded to significantly increased cytotoxicity in a murine infection model.31 Given that SCVs generally arise after significant periods of lung colonization, many of the phenotypes associated with long-term, chronic lung infection (loss of virulence, reduced quorum sensing, hypermutability, etc18,23,35–37) might be expected to correlate with SCV. However, as the studies above indicate, many exceptions exist, and phenotypic variation between SCV morphotypes is likely to be extensive.28,29
The clinical implications of SCV lung infection
At least two independent reports have identified a strong relationship between the presence of SCVs in the CF lung and poor clinical outcomes. The 1999 Häussler et al17 study of SCVs in CF sputum examined a total of 86 patients with chronic P. aeruginosa lung infections. While only 3% of all colonies recovered during the study were classed as SCVs, those patients from whom SCVs were isolated (33 out of 86) displayed poorer lung function than those who did not display SCV colonization. The forced expiratory volume in the first second (FEV1) score for these patients was 56%, which was significantly lower than the FEV1 of SCV-negative patients (80%). More recently, Schneider et al38 looked for P. aeruginosa and Staphylococcus aureus SCVs in sputum samples from 98 CF patients. Over a 3-month period, 9.2% of patients in this study were colonized with P. aeruginosa SCVs.38 In agreement with the findings of Häussler et al,17 patients having SCVs had poorer lung function, with lower blood oxygen levels and a significantly lower FEV1 score (39%) than the non-SCV patients (65.5%). The body mass of SCV-colonized patients was also significantly lower (mean 14.0% underweight compared with 2.7% for non-SCV patients).38
While it is difficult to confidently infer a causal relationship between the SCV phenotype and poor clinical prognosis based on correlation studies alone, there is substantial evidence supporting a role for SCVs in persistence in a clinical setting. Three independent research groups, using different animal infection models and genetically distinct SCV strains, have established causal relationships between the SCV phenotype and persistence of infection.11–13 Furthermore, many of the phenotypes associated with SCV have been directly linked to advantageous survival traits during chronic infection. For example, Malone et al12 showed that extracellular polysaccharides (EPS) overproduction rendered P. aeruginosa SCVs highly resistant to macrophage phagocytosis. As mentioned previously, SCVs generally show enhanced antibiotic resistance compared to wild-type P. aeruginosa.17,21,23 While a causal relationship again remains to be established, this antibiotic resistance may contribute to SCV survival in the CF lung during antibiotic chemotherapy.
Fully defining the relationship between SCVs and persistence in the CF lung is likely to remain a challenge for the foreseeable future. While correlations between clinical SCV colonization and poor lung function are clear, and SCVs contribute to infectious persistence in laboratory and animal studies, it is currently unknown whether SCVs trigger poor clinical outcomes or just preferentially associate with deteriorating lungs. Short of invasive or postmortem investigations of CF lungs, an interesting recent advance that may provide an answer is the ex vivo porcine lung model developed by Harrison et al.39 This system has been successfully tested with P. aeruginosa quorum sensing mutants and promises to shed further light onto microbial colonization of the lung environment.
The molecular bases of the SCV phenotype: cyclic-di-GMP and the GacAS/RsmAZY pathway
Following early indications from two independent research labs,21,22 strong evidence has accumulated for a causal link between the SCV phenotype and the bacterial second messenger cdG.12,25,40–42 cdG is a ubiquitous bacterial signaling molecule that controls a wide range of cellular processes involved in the transition between motile, virulent, and sessile biofilm forming lifestyles.43,44 The cyclic dinucleotide is produced from two molecules of GTP (guanosine triphosphate) by DGCs and degraded to pGpG (5′-phosphoguanylyl-(3′-5′)-guanosineguanosine) by phosphodiesterases;45 enzymes containing the conserved GGDEF and EAL/HD-GYP domains, respectively.46–49 In general, cdG production is associated with community behavior phenotypes such as EPS production and biofilm formation, while low cdG levels lead to enhanced motility, virulence, and a single-celled lifestyle43 (Figure 2). cdG signal transduction is a highly complex process, with many bacterial species containing dozens of different cdG-signaling proteins.43 P. aeruginosa is no exception, with 33 predicted GGDEF and 17 EAL domain-containing proteins.50,51 These cdG metabolic enzymes control the intracellular level of cdG and hence regulate the expression of various phenotypic outputs. In P. aeruginosa, this includes exopolysaccharide production,52–54 production and deployment of proteinaceous adhesins,55,56 siderophore production,12 rhamnolipid biosynthesis,57 and virulence and cytotoxicity systems,51,58–60 as well as the assembly, function, and control of type IV pili61,62 and the bacterial flagellum.54,63–65
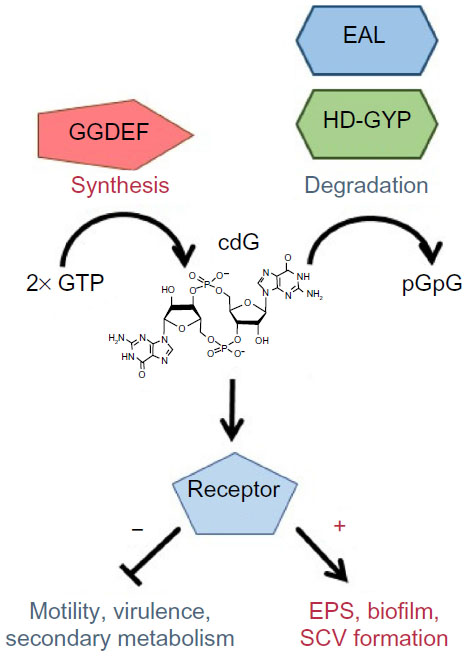 | Figure 2 cdG signaling. |
cdG affects cell behavior by controlling phenotypic outputs at every regulatory level, from gene expression through to allosteric modulation of phenotypic outputs.43 These effects occur through binding to specific effector proteins, whose activity is altered upon interaction with cdG. Consistent with the high complexity of cdG signal transduction, binding is highly promiscuous and occurs with a wide range of different protein folds.66,67 For example, the PilZ domain is a dedicated cdG binding protein, with examples like Alg44 in P. aeruginosa, which is a regulator of alginate biosynthesis.52 Furthermore, not all GGDEF, HD-GYP, and EAL domain proteins have cdG metabolic activity. A substantial minority have degenerate active sites and instead function by binding cdG, via protein–protein interactions or through other alternative mechanisms.66,67 For example, the P. aeruginosa EPS synthase component PelD binds to cdG, at a conserved site on its degenerate GGDEF domain, and induces a conformational change in the Pel synthase machinery that leads to activation and EPS production.68 cdG also binds to a number of different transcriptional regulators. In P. aeruginosa, this includes the flagellar and EPS-gene master regulator FleQ, which controls pel and psl EPS operon transcription.54,69 The metabolism of cdG is under extensive spatial and temporal control in P. aeruginosa. Expression and translation of cdG genes is regulated by various cellular inputs, including sigma factors, cell cycle control, quorum sensing, and translational regulation.43,60,70 A further, significant level of control exists at the posttranslational level, with the majority of GGDEF, EAL, and HD-GYP domain proteins also containing diverse sensory inputs, including PAS, GAF, and response-regulator receiver domains.43
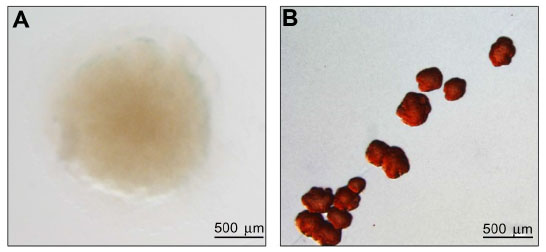
The role of cdG in P. aeruginosa SCV formation is now well-established. For example, a strong SCV phenotype can be triggered under laboratory conditions by overexpressing a DGC gene in trans,44 a phenotype that can be reversed by disrupting the pel and psl EPS synthase operons (Figure 3).12 The intracellular level of cdG has been shown to be elevated in several clinical and laboratory-derived SCVs.12,25,42 Furthermore, mutations in several P. aeruginosa cdG signaling pathways have been shown to induce SCV phenotypes in vitro. In each case, elevated intracellular levels of cdG have been linked to overproduction of either exopolysaccharides9,25,41 or fimbrial adhesins,40,71 and consequently to SCV formation.
 | Figure 3 cdG and EPS define the SCV phenotype. |
As well as cdG signaling pathways, other genetic loci have been implicated in persistence of infection and/or SCV formation in P. aeruginosa. A prominent example is the GacAS/RsmAZY signaling system (Figure 4), an important regulator of the switch between chronic and acute infectious lifestyles in many Gram-negative bacterial species, including P. aeruginosa.70,72 RsmA is a small, translational regulator that specifically recognizes and binds to GGA sequences in the 5′ leader region of target mRNAs. RsmA reciprocally controls the translation of mRNAs associated with Type III/Type VI secretion and Psl exopolysaccharide synthesis,33,70 leading to the suppression of biofilm-associated phenotypes and promoting secondary metabolism, motility, and virulence. Deletion of the rsmA gene in P. aeruginosa PA01 induces an SCV phenotype as a consequence of increased psl mRNA translation,33 and has been shown to increase infectious persistence in a murine lung model.13
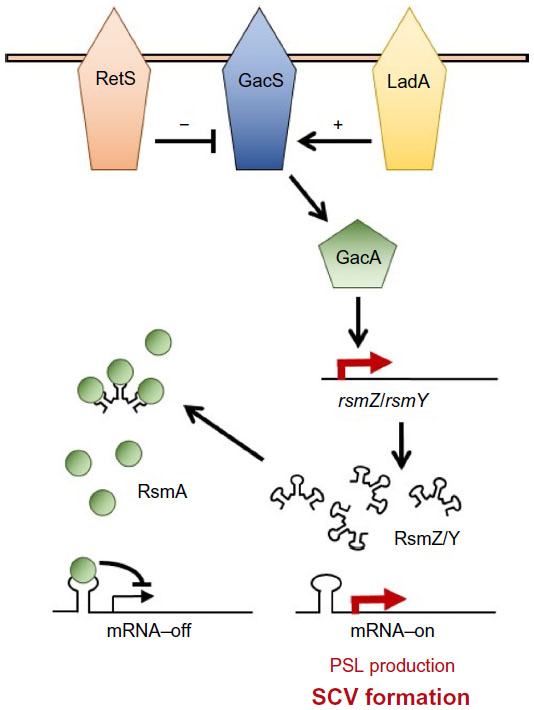 | Figure 4 The GacAS/RsmAZY signaling network. |
In Pseudomonas spp. the activity of RsmA is controlled by the GacAS two-component signal transduction system. GacS is a transmembrane histidine protein kinase and activates its cognate response regulator GacA by phosphotransfer. Phosphorylated GacA promotes transcription of the antagonistic sRNAs, RsmY and RsmZ.73 RsmY/RsmZ contain multiple GGA trinucleotides in the exposed stem-loops of their predicted secondary structures and inhibit RsmA activity by competing for RsmA binding, thus titrating the translational regulator away from its target mRNAs (Figure 4). The GacAS system is itself controlled by the two-component signal hybrid proteins LadS and RetS. RetS is an antagonist of GacS activity, and thus suppresses RsmZ/Y levels. Rather than operating via a conventional histidine protein kinase phosphotransfer mechanism, RetS forms heterodimers with GacS, and thus blocks GacS autophosphorylation phosphotransfer to GacA.74 The LadS protein positively controls rsmZ/Y expression, and thus works to suppress RsmA activity. Although the mechanism of LadS function is currently uncharacterized, it may work by counteracting RetS activity (Figure 4).72 Signal transduction through the Gac/Rsm pathway is highly complex, and affects the production of dozens of different proteins.70 In addition to direct RsmA translational regulation, the Gac/Rsm system indirectly affects numerous additional cellular behaviors via the regulation of signaling pathways, including both cdG metabolism and quorum sensing.60,70 Similar to cdG, mutations in quorum sensing genes are strongly associated with persistence during chronic CF lung infections.6,75
Evolution of SCVs in the CF lung
Having colonized the respiratory passage and lungs of a CF patient, in most cases, a P. aeruginosa infection persists throughout the patient’s lifetime. Patients are typically colonized by a single environmentally acquired genotype4,5 (although some genotypes such as the Liverpool epidemic strain are infectious and can be transmitted from patient to patient76). P. aeruginosa CF lung infections generally alternate between periods of chronic, largely asymptomatic colonization and relapses into aggravated infection.1 Over several years, the progressive tissue degradation that accompanies these repeated infectious relapses leads to lung failure and premature death.3 As stated above, P. aeruginosa strains undergo extensive adaptation to the lung environment throughout the period of chronic lung infection,6,7 with multiple distinct phenotypes including SCVs arising in the lung population.7,10,77 Despite generally originating from a single clonal genotype, significant genetic variation exists between individual sputum isolates sampled at any one time,28,29 providing an explanation for the long-observed phenotypic heterogeneity in CF P. aeruginosa isolates.19,20
Perhaps unsurprisingly, the most commonly identified SCV-inducing mutations are loss-of-function mutations in repressor proteins that control the activity of DGCs (Table 1). A well-studied example is the Wsp pathway,41 which contains a methyl-accepting chemotaxis receptor (WspA) and a DGC response regulator (WspR). WspR overproduction/activation induces an SCV phenotype,22,41 displaying strong attachment and increased expression of the pel and psl exopolysaccharide operons. Under laboratory conditions, the principle route to SCV evolution is via loss-of-function mutations in the methylesterase gene wspF.25,41 Without WspF, WspA and hence WspR are constitutively activated, leading to cdG synthesis and SCV formation through EPS overproduction (Figure 5).41,78 There is evidence that wspF loss-of-function also represents a route to clinical SCV evolution. In 2006, Smith et al6 carried out a longitudinal study of lung-adapted P. aeruginosa isolates and identified several different backgrounds in which the wspF gene was mutated. This study also identified a number of mutations in the transcriptional regulator fleQ. Deletion of fleQ induces an autoaggregative phenotype in PA01, with many of the characteristics of SCV colonies.54 While the morphologies of the lung isolates in the Smith et al6 study were not characterized in detail, these data nonetheless implicate both wspF and fleQ as potential mutagenic targets for SCV generation in the CF lung (Figure 5).
 | Table 1 Mutational targets for Pseudomonas aeruginosa SCV formation |
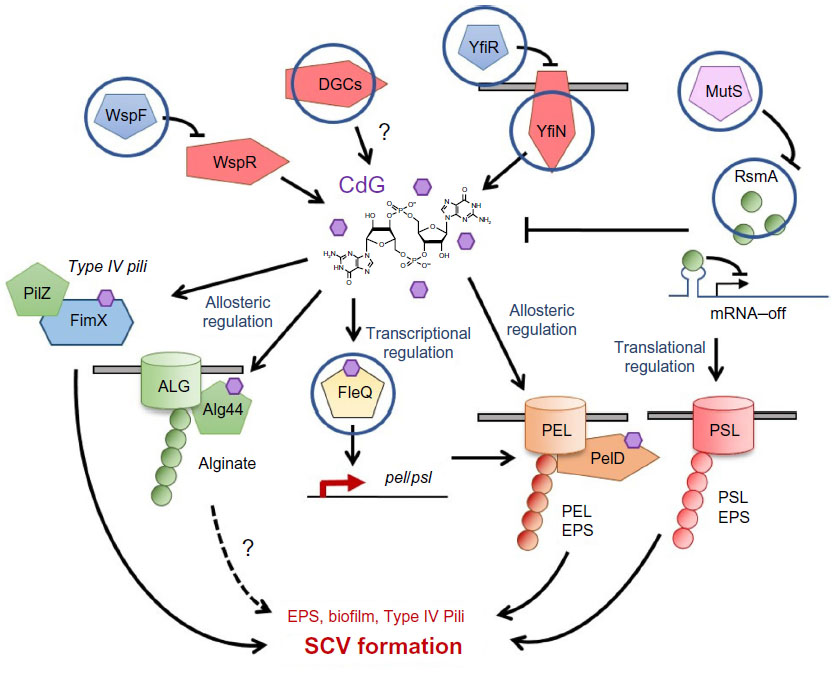 | Figure 5 Mutational routes to SCV formation. |
Blanka et al42 recently identified an interesting additional mechanism for SCV evolution in the CF lung isolate SCV20265. Comparison of the SCV20265 genome with those of several laboratory-derived revertants identified a causal mutation for the SCV20265 aggregative phenotype. This study showed that a point mutation in the 5′ untranslated region of accBC, a gene cluster responsible for fatty acid biosynthesis, leads to mRNA stabilization and a consequent increase in the proportion of short-chain fatty acids in the plasma membrane. In turn, this change in membrane composition triggered activation of the Wsp system and cdG overproduction via WspR.42 Intriguingly, the genome of SCV20265 was also shown to contain a second, additive mutation in the methylesterase gene wspF, shown in previous studies to induce an SCV phenotype.6,25 These findings suggest that both regulatory and genetic inputs combine to control cdG production, and hence SCV evolution, in the CF lung.42
The yfiBNR signaling operon12 represents a further, well-characterized genetic target for clinically-arising SCVs. YfiN is a membrane-bound DGC whose activity is normally allosterically repressed by the soluble periplasmic repressor YfiR. Release of YfiR repression, either through a loss-of-function mutation in yfiR or an “escape” mutation in yfiN, leads to DGC activation, cdG overproduction, and SCV formation under laboratory conditions.12,26 As with wspF and fleQ, activating mutations arise in the yfi locus (in both yfiR and yfiN) during long-term CF lung infections, driving SCV formation in vivo (Figure 5).26 Interestingly, further examination of the CF isolates included in this study identified mutational “scars” in the yfi genes of two independent clinical CF lines. These strains contained both activating and loss-of-function mutations in the yfiN DGC gene, supporting the idea that YfiN activity is under both positive and negative selection in vivo, with Yfi-SCVs acting as an environmental pool for the generation of new smooth morphotypes in the complex and heterogeneous fitness landscape of the CF lung.26
Another class of mutations associated with the clinical emergence of the SCV phenotype are those in mutS and other mismatch repair genes. These hypermutatable strains arise with high frequency in the course of chronic CF lung infection.37,79 Mena et al35 showed that a P. aeruginosa mismatch-repair mutation increased the long-term persistence of oropharyngeal colonization in CF mice, and that SCV mutants readily emerged over time in this background. The phenomenon of SCV morphotypes arising in hypermutatable P. aeruginosa backgrounds has been independently observed for another ΔmutS mutant strain,80 which was also shown to produce a large number of mutations in the lasR quorum sensing locus.36 The work of Mena et al35 strongly suggests that mismatch repair mutants are selected in chronic P. aeruginosa lungs due to their increased persistence. As a possible explanation for this, hypermutatable strains have been linked to the RsmA signaling network in Erwinia carotovora,81 placing RsmA downstream of mutS and suggesting that the persistence of these strains may result (at least in part) from reduced levels of RsmA (Figure 5). Another possibility is simply that the increased rate of mutation in these strains facilitates the emergence of mutations in the Gac/Rsm or cdG signaling pathways. Genetic adaptation to the CF lung has been shown to be catalyzed by the initial acquisition of mismatch repair mutations, with subsequent mutations arising much more rapidly than in nonmutator lines.82 Mutants in the cdG signaling genes fleQ and wspF were identified in this study, although the isolate library used was the same as described in the earlier work of Smith et al,6 where the wspF and fleQ mutations were initially identified.
Large-scale DNA inversions have also been associated with the generation of morphological diversity, and hence potentially SCV generation, in the lung environment. In a study of CF lung isolates, Römling et al83 showed that 50% of tested P. aeruginosa genomes contained large chromosomal inversions. These inversions were exclusively detected in CF lung isolates, suggesting that they are selected during adaptation to chronic lung infection.83 Chromosomal inversions have been linked to the mobile element IS6100 and are proposed to represent a source of phenotypic variation,84 similar to that emerging from the loss of mismatch repair described above.80 Furthermore, reversible genomic inversion has been shown to induce an SCV phenotype in S. aureus.85
Recently, the advent of next-generation sequencing has enabled the molecular basis of SCV formation to be examined much more closely. The complete genome sequences of at least two clinically-evolved SCVs have been published, allowing candidate causal mutations for generation of the SCV phenotype to be investigated. For SCV20265,86 the mutational routes to SCV are known,42 while for the urethral catheter SCV MH27,87 the underlying genetics of the SCV phenotype has yet to be established. As sequencing technology continues to improve and becomes ever more accessible, it is likely that the coming years will see a much more complete examination of the mutational and regulatory routes to SCV generation in the CF lung environment.
Summary
P. aeruginosa SCVs are frequently isolated from the lungs of CF patients. The appearance of these distinctive, small, autoaggregative colonies strongly correlates with both deteriorating lung function and associated poor clinical prognosis. Whether this connection is causal or simply correlative is not yet clear. Nonetheless, the importance of the SCV phenotype in persistence of P. aeruginosa infection is beyond doubt, with SCVs conferring both increased antibiotic tolerance and resistance to immune phagocytosis. Hopefully, an increased awareness of the importance of SCVs in chronic CF infection, alongside recent improvements in both diagnostic and analytical tools, will allow us to make headway in the treatment of these unusual morphotypes.
Acknowledgment
The author would like to thank Sebastian Pfeilmeier for the photographs used in Figures 1 and 3.
Disclosure
The author reports no conflicts of interest in this work.
References
O’Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891–1904. | |
Harrison F. Microbial ecology of the cystic fibrosis lung. Microbiology. 2007;153:917–923. | |
Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15:194–222. | |
Struelens MJ, Schwam V, Deplano A, Baran D. Genome macrorestriction analysis of diversity and variability of Pseudomonas aeruginosa strains infecting cystic fibrosis patients. J Clin Microbiol. 1993;31:2320–2326. | |
Breitenstein S, Walter S, Bosshammer J, Römling U, Tummler B. Direct sputum analysis of Pseudomonas aeruginosa macrorestriction fragment genotypes in patients with cystic fibrosis. Med Microbiol Immunol. 1997;186:93–99. | |
Smith EE, Buckley DG, Wu Z, et al. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A. 2006;103:8487–8492. | |
Huse HK, Kwon T, Zlosnik JE, et al. Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. MBio. 2010; 1(4):e00199. | |
Govan JR, Deretic V. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev. 1996;60:539–574. | |
Kirisits MJ, Prost L, Starkey M, Parsek MR. Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl Environ Microbiol. 2005;71:4809–4821. | |
Häussler S, Ziegler I, Löttel A, et al. Highly adherent small-colony variants of Pseudomonas aeruginosa in cystic fibrosis lung infection. J Med Microbiol. 2003;52:295–301. | |
Byrd MS, Pang B, Hong W, et al. Direct evaluation of Pseudomonas aeruginosa biofilm mediators in a chronic infection model. Infect Immun. 2011;79:3087–3095. | |
Malone JG, Jaeger T, Spangler C, et al. YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog. 2010;6:e1000804. | |
Mulcahy H, O’Callaghan J, O’Grady EP, et al. Pseudomonas aeruginosa RsmA plays an important role during murine infection by influencing colonization, virulence, persistence, and pulmonary inflammation. Infect Immun. 2008;76:632–638. | |
Reinhardt A, Köhler T, Wood P, et al. Development and persistence of antimicrobial resistance in Pseudomonas aeruginosa: a longitudinal observation in mechanically ventilated patients. Antimicrob Agents Chemother. 2007;51:1341–1350. | |
Murphy TF, Brauer AL, Sethi S, et al. Haemophilus haemolyticus: a human respiratory tract commensal to be distinguished from Haemophilus influenzae. J Infect Dis. 2007;195:81–89. | |
Spoering AL, Lewis K. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J Bacteriol. 2001;183:6746–6751. | |
Häussler S, Tummler B, Weissbrodt H, Rohde M, Steinmetz I. Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin Infect Dis. 1999;29:621–625. | |
Burke V, Robinson JO, Richardson CJ, Bundell CS. Longitudinal studies of virulence factors of Pseudomonas aeruginosa in cystic fibrosis. Pathology. 1991;23:145–148. | |
Zierdt CH, Schmidt PJ. Dissociation in Pseudomonas aeruginosa. J Bacteriol. 1964;87:1003–1010. | |
Thomassen MJ, Demko CA, Boxerbaum B, Stern RC, Kuchenbrod PJ. Multiple isolates of Pseudomonas aeruginosa with differing antimicrobial susceptibility patterns from patients with cystic fibrosis. J Infect Dis. 1979;140:873–880. | |
Drenkard E, Ausubel FM. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature. 2002;416:740–743. | |
D’Argenio DA, Calfee MW, Rainey PB, Pesci EC. Autolysis and autoaggregation in Pseudomonas aeruginosa colony morphology mutants. J Bacteriol. 2002;184:6481–6489. | |
Wei Q, Tarighi S, Dötsch A, et al. Phenotypic and genome-wide analysis of an antibiotic-resistant small colony variant (SCV) of Pseudomonas aeruginosa. PLoS One. 2011;6:e29276. | |
Hoffman LR, D’Argenio DA, MacCoss MJ, et al. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature. 2005;436:1171–1175. | |
Starkey M, Hickman JH, Ma L, et al. Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol. 2009;191:3492–3503. | |
Malone JG, Jaeger T, Manfredi P, et al. The YfiBNR signal transduction mechanism reveals novel targets for the evolution of persistent Pseudomonas aeruginosa in cystic fibrosis airways. PLoS Pathog. 2012;8:e1002760. | |
Friedman L, Kolter R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol. 2004;186:4457–4465. | |
Mowat E, Paterson S, Fothergill JL, et al. Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am J Respir Crit Care Med. 2011;183:1674–1679. | |
Workentine ML, Sibley CD, Glezerson B, et al. Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS One. 2013;8:e60225. | |
Sabra W, Haddad AM, Zeng AP. Comparative physiological study of the wild type and the small colony variant of Pseudomonas aeruginosa 20265 under controlled growth conditions. World J Microbiol Biotechnol. 2014;30:1027–1036. | |
von Gotz F, Häussler S, Jordan D, et al. Expression analysis of a highly adherent and cytotoxic small colony variant of Pseudomonas aeruginosa isolated from a lung of a patient with cystic fibrosis. J Bacteriol. 2004;186:3837–3847. | |
Roy-Burman A, Savel RH, Racine S, et al. Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J Infect Dis. 2001;183:1767–1774. | |
Irie Y, Starkey M, Edwards AN, et al. Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol Microbiol. 2010;78:158–172. | |
Wehmhoner D, Häussler S, Tümmler B, et al. Inter- and intraclonal diversity of the Pseudomonas aeruginosa proteome manifests within the secretome. J Bacteriol. 2003;185:5807–5814. | |
Mena A, Maciá MD, Borrell N, et al. Inactivation of the mismatch repair system in Pseudomonas aeruginosa attenuates virulence but favors persistence of oropharyngeal colonization in cystic fibrosis mice. J Bacteriol. 2007;189:3665–3668. | |
Lujan AM, Moyano AJ, Segura I, Argarana CE, Smania AM. Quorum-sensing-deficient (lasR) mutants emerge at high frequency from a Pseudomonas aeruginosa mutS strain. Microbiology. 2007;153:225–237. | |
Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. | |
Schneider M, Mühlemann K, Droz S, et al. Clinical characteristics associated with isolation of small-colony variants of Staphylococcus aureus and Pseudomonas aeruginosa from respiratory secretions of patients with cystic fibrosis. J Clin Microbiol. 2008;46:1832–1834. | |
Harrison F, Muruli A, Higgins S, Diggle SP. Development of an ex vivo porcine lung model for studying growth, virulence, and signaling of Pseudomonas aeruginosa. Infect Immun. 2014;82:3312–3323. | |
Meissner A, Wild V, Simm R, et al. Pseudomonas aeruginosa cupA-encoded fimbriae expression is regulated by a GGDEF and EAL domain-dependent modulation of the intracellular level of cyclic diguanylate. Environ Microbiol. 2007;9:2475–2485. | |
Hickman JW, Tifrea DF, Harwood CS. A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci U S A. 2005;102:14422–14427. | |
Blanka A, Düvel J, Dötsch A, et al. Constitutive production of c-di-GMP is associated with mutations in a variant of Pseudomonas aeruginosa with altered membrane composition. Sci Signal. 2015;8:ra36. | |
Hengge R. Principles of c-di-GMP signalling in bacteria. Nat Rev Microbiol. 2009;7:263–273. | |
Jenal U, Malone J. Mechanisms of cyclicdi-GMP signaling in bacteria. Annu Rev Genet. 2006;40:385–407. | |
Ross P, Weinhouse H, Aloni Y, et al. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature. 1987;325:279–281. | |
Schmidt AJ, Ryjenkov DA, Gomelsky M. The ubiquitous protein domain EAL is a cyclic diguanylate-specific phosphodiesterase: enzymatically active and inactive EAL domains. J Bacteriol. 2005;187:4774–4781. | |
Paul R, Weiser S, Amiot NC, et al. Cell cycle-dependent dynamic localization of a bacterial response regulator with a novel di-guanylate cyclase output domain. Genes Dev. 2004;18:715–727. | |
Christen M, Christen B, Folcher M, Schauerte A, Jenal U. Identification and characterization of a cyclic di-GMP-specific phosphodiesterase and its allosteric control by GTP. J Biol Chem. 2005;280:30829–30837. | |
Ryan RP, Fouhy Y, Lucey JF, et al. Cell-cell signaling in Xanthomonas campestris involves an HD-GYP domain protein that functions in cyclic di-GMP turnover. Proc Natl Acad Sci U S A. 2006;103:6712–6717. | |
Galperin MY. A census of membrane-bound and intracellular signal transduction proteins in bacteria: bacterial IQ, extroverts and introverts. BMC Microbiol. 2005;5:35. | |
Kulasakara H, Lee V, Brencic A, et al. Analysis of Pseudomonas aeruginosa diguanylate cyclases and phosphodiesterases reveals a role for bis-(3′–5′)-cyclic-GMP in virulence. Proc Natl Acad Sci U S A. 2006;103:2839–2844. | |
Merighi M, Lee VT, Hyodo M, Hayakawa Y, Lory S. The second messenger bis-(3′–5′)-cyclic-GMP and its PilZ domain-containing receptor Alg44 are required for alginate biosynthesis in Pseudomonas aeruginosa. Mol Microbiol. 2007;65:876–895. | |
Lee SY, Matewish JM, Kessler JL, et al. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol. 2007;65:1474–1484. | |
Hickman JW, Harwood CS. Identification of FleQ from Pseudomonas aeruginosa as a c-di-GMP-responsive transcription factor. Mol Microbiol. 2008;69:376–389. | |
Kulasekara HD, Ventre I, Kulasekara BR, et al. A novel two-component system controls the expression of Pseudomonas aeruginosa fimbrial cup genes. Mol Microbiol. 2005;55:368–380. | |
Borlee BR, Goldman AD, Murakami K, et al. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol Microbiol. 2010;75:827–842. | |
An S, Wu J, Zhang LH. Modulation of Pseudomonas aeruginosa biofilm dispersal by a cyclic-di-GMP phosphodiesterase with a putative hypoxia-sensing domain. Appl Environ Microbiol. 2010;76:8160–8173. | |
Kuchma SL, Connolly JP, O’Toole GA. A three-component regulatory system regulates biofilm maturation and type III secretion in Pseudomonas aeruginosa. J Bacteriol. 2005;187:1441–1454. | |
Moscoso JA, Mikkelsen H, Heeb S, Williams P, Filloux A. The Pseudomonas aeruginosa sensor RetS switches Type III and Type VI secretion via c-di-GMP signalling. Environ Microbiol. 2011;13:3128–3138. | |
Moscoso JA, Jaeger T, Valentini M, et al. The diguanylate cyclase SadC is a central player in Gac/Rsm-mediated biofilm formation in Pseudomonas aeruginosa. J Bacteriol. 2014;196:4081–4088. | |
Kazmierczak BI, Lebron MB, Murray TS. Analysis of FimX, a phosphodiesterase that governs twitching motility in Pseudomonas aeruginosa. Mol Microbiol. 2006;60:1026–1043. | |
Alm RA, Bodero AJ, Free PD, Mattick JS. Identification of a novel gene, pilZ, essential for type 4 fimbrial biogenesis in Pseudomonas aeruginosa. J Bacteriol. 1996;178:46–53. | |
Baraquet C, Harwood CS. Cyclic diguanosine monophosphate represses bacterial flagella synthesis by interacting with the Walker A motif of the enhancer-binding protein FleQ. Proc Natl Acad Sci U S A. 2013;110:18478–18483. | |
Kulasekara BR, Kamischke C, Kulasekara HD, et al. c-di-GMP heterogeneity is generated by the chemotaxis machinery to regulate flagellar motility. Elife. 2013;2:e01402. | |
Kuchma SL, Delalez NJ, Filkins LM, et al. Cyclic Di-GMP-mediated repression of swarming motility by Pseudomonas aeruginosa PA14 requires the MotAB stator. J Bacteriol. 2015;197:420–430. | |
Ryan RP, Tolker-Nielsen T, Dow JM. When the PilZ don’t work: effectors for cyclic di-GMP action in bacteria. Trends Microbiol. 2012;20:235–242. | |
Boyd CD, O’Toole, GA. Second messenger regulation of biofilm formation: breakthroughs in understanding c-di-GMP effector systems. Annu Rev Cell Dev Biol. 2012;28:439–462. | |
Lee VT, Matewish JM, Kessler JL, et al. A cyclic-di-GMP receptor required for bacterial exopolysaccharide production. Mol Microbiol. 2007;65:1474–1484. | |
Baraquet C, Murakami K, Parsek MR, Harwood CS. The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res. 2012;40(15):7207–7218. | |
Brencic A, Lory S. Determination of the regulon and identification of novel mRNA targets of Pseudomonas aeruginosa RsmA. Mol Microbiol. 2009;72:612–632. | |
Häussler S. Biofilm formation by the small colony variant phenotype of Pseudomonas aeruginosa. Environ Microbiol. 2004;6:546–551. | |
Chambers JR, Sauer K. Small RNAs and their role in biofilm formation. Trends Microbiol. 2013;21:39–49. | |
Brencic A, McFarland KA, McManus HR, et al. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol Microbiol. 2009;73:434–445. | |
Goodman AL, Merighi M, Hyodo M, et al. Direct interaction between sensor kinase proteins mediates acute and chronic disease phenotypes in a bacterial pathogen. Genes Dev. 2009;23:249–259. | |
Rau MH, Hansen SK, Johansen HK, et al. Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ Microbiol. 2010;12:1643–1658. | |
Scott FW, Pitt TL. Identification and characterization of transmissible Pseudomonas aeruginosa strains in cystic fibrosis patients in England and Wales. J Med Microbiol. 2004;53:609–615. | |
Hogardt M, Heesemann J. Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int J Med Microbiol. 2010;300:557–562. | |
Bantinaki E, Kassen R, Knight CG, et al. Adaptive divergence in experimental populations of Pseudomonas fluorescens. III. Mutational origins of wrinkly spreader diversity. Genetics. 2007;176:441–453. | |
Hogardt M, Hoboth C, Schmoldt S, et al. Stage-specific adaptation of hypermutable Pseudomonas aeruginosa isolates during chronic pulmonary infection in patients with cystic fibrosis. J Infect Dis. 2007;195:70–80. | |
Smania AM, Segura I, Pezza RJ, et al. Emergence of phenotypic variants upon mismatch repair disruption in Pseudomonas aeruginosa. Microbiology. 2004;150:1327–1338. | |
Mukherjee A, Cui Y, Ma W, Liu Y, Chatterjee AK. hexA of Erwinia carotovora ssp. carotovora strain Ecc71 negatively regulates production of RpoS and rsmB RNA, a global regulator of extracellular proteins, plant virulence and the quorum-sensing signal, N-(3-oxohexanoyl)-L-homoserine lactone. Environ Microbiol. 2000;2:203–215. | |
Mena A, Smith EE, Burns JL, et al. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J Bacteriol. 2008;190:7910–7917. | |
Römling U, Schmidt KD, Tummler B. Large chromosomal inversions occur in Pseudomonas aeruginosa clone C strains isolated from cystic fibrosis patients. FEMS Microbiol Lett. 1997;150:149–156. | |
Kresse AU, Dinesh SD, Larbig K, Römling U. Impact of large chromosomal inversions on the adaptation and evolution of Pseudomonas aeruginosa chronically colonizing cystic fibrosis lungs. Mol Microbiol. 2003;47:145–158. | |
Cui L, Neoh HM, Iwamoto A, Hiramatsu K. Coordinated phenotype switching with large-scale chromosome flip-flop inversion observed in bacteria. Proc Natl Acad Sci U S A. 2012;109:E1647–E1656. | |
Eckweiler D, Bunk B, Sproer C, Overmann J, Häussler S. Complete genome sequence of highly adherent Pseudomonas aeruginosa small-colony variant SCV20265. Genome Announc. 2014;2(1):e01232. | |
Tielen P, Wibberg D, Blom J, et al. Genome sequence of the small-colony variant Pseudomonas aeruginosa MH27, isolated from a chronic urethral catheter infection. Genome Announc. 2014;2(1):e01174. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.