")
Back to Journals » Infection and Drug Resistance » Volume 13
Role of Renin-Angiotensin System in Acute Lung Injury Caused by Viral Infection
Authors Gao YL, Du Y, Zhang C, Cheng C, Yang HY, Jin YF, Duan GC , Chen SY
Received 10 June 2020
Accepted for publication 12 September 2020
Published 21 October 2020 Volume 2020:13 Pages 3715—3725
DOI https://doi.org/10.2147/IDR.S265718
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Yan-Lei Gao,* Yue Du,* Chao Zhang, Cheng Cheng, Hai-Yan Yang, Yue-Fei Jin, Guang-Cai Duan, Shuai-Yin Chen
Department of Epidemiology, College of Public Health, Zhengzhou University, Zhengzhou, Henan, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guang-Cai Duan; Shuai-Yin Chen Email [email protected]
Abstract: The renin-angiotensin system (RAS) is the most important regulatory system of electrolyte homeostasis and blood pressure and acts through angiotensin-converting enzyme (ACE)/angiotensin II (Ang II)/Ang II type 1 (AT1) receptor axis and angiotensin-converting enzyme 2 (ACE2)/angiotensin (1-7)/MAS receptor axis. RAS dysfunction is related to the occurrence and development of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) and causes a serious prognosis and even death. ALI/ARDS can be induced by various ways, one of which is viral infections, such as SARS-CoV, SARS-CoV-2, H5N1, H7N9, and EV71. This article reviews the specific mechanism on how RAS dysfunction affects ALI/ARDs caused by viral infections. SARS-CoV and SARS-CoV-2 enter the host cells by binding with ACE2. H5N1 and H7N9 avian influenza viruses reduce the ACE2 level in the body, and EV71 increases Ang II concentration. Treatment with angiotensin-converting enzyme inhibitor and angiotensin AT1 receptor blocker can alleviate ALI/ARDS symptoms. This review provides suggestions for the treatment of lung injury caused by viral infections.
Keywords: renin angiotensin system, acute lung injury and acute respiratory distress syndrome, viral infection
Introduction
The renin-angiotensin system (RAS) is important because it regulates blood pressure and maintains the water and electrolyte balance and cardiovascular homeostasis in organisms1 and is related to the inflammation, oxidative stress, immunity, apoptosis, fibrosis, and other physiological activities.2
The main components of this system include angiotensinogen (AGT), renin, angiotensin I (Ang I), angiotensin II (Ang II), and various enzymes and receptors, such as angiotensin-converting enzyme (ACE), angiotensin-converting enzyme 2 (ACE2), MAS receptor, Ang II type 1 (AT1) receptor, and Ang II type 2 (AT2) receptor. RAS functions through ACE/Ang II/AT1 and ACE2/Ang I (1-7)/MAS axes with opposite effects. A new branch has recently been discovered. When alanine replaces the aspartate at the amino terminal of Ang I (1-7), Ala-Ang I (1-7) (alamandine) is produced, binds to the new receptor Mas-related G protein-coupled receptor D (MrgD), and protects against hypertension, hypertrophy, lung damage, and fibrosis.3–5 The regulatory mechanisms of RAS are listed in Figure 1. RAS is distributed in many human tissues, including brain, heart, kidney, and lung, which has the largest amount.6–8
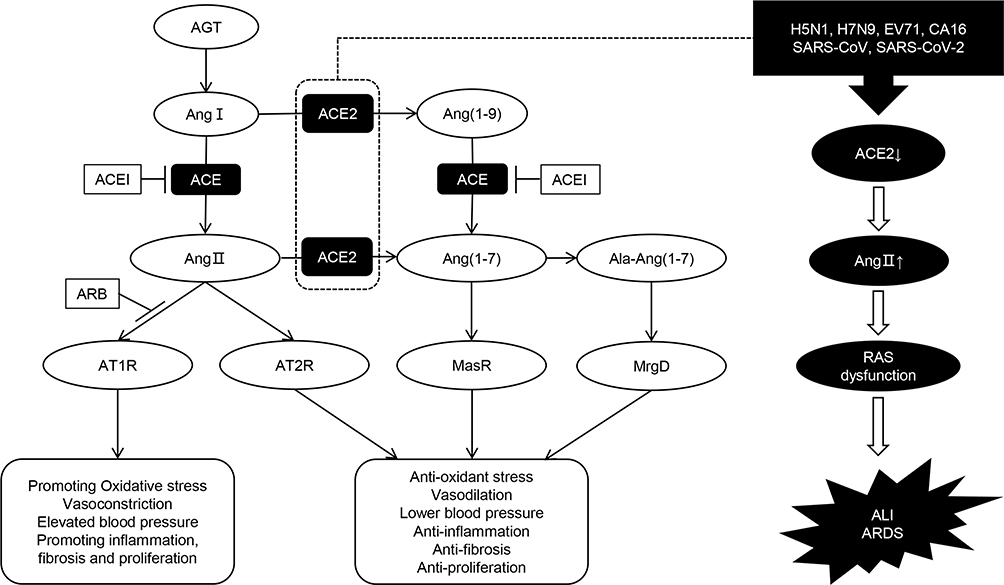 |
Figure 1 Diagram of regulatory mechanism of renin-angiotensin system (RAS). The classical RAS pathway begins with the conversion of angiotensinogen (AGT) to an inactive decapeptide precursor angiotensin I (Ang I) through the action of renin. Ang I is further recognized and cleaved by angiotensin converting enzyme (ACE) to produce active octapeptide angiotensin II (Ang II). Ang II is the key biological peptide of RAS which has various physiological regulatory effects. It acts mainly through the combination of type 1 angiotensin II receptor (AT1R) to constrict blood vessels, raise blood pressure, promote inflammation, and promote fibrosis and apoptosis. It can also bind to AT2, AT3, and AT4 receptors, and plays different physiological roles. In addition, angiotensin converting enzyme 2 (ACE2) can cleave Ang I C-terminal peptide to produce inactive Ang (1-9), then Ang (1-9) can be hydrolyzed by ACE and other peptidases to produce active Ang (1-7). ACE2 can also directly cleave Ang II to form Ang (1-7). Ang (1-7) binds to the Mas receptor (MasR) and exerts physiological functions that are contrary to Ang II, including vasodilation, lower blood pressure, anti-inflammation, anti-oxidative stress, anti-fibrosis, etc. These form two main functional axes of RAS. Ala-Ang (1-7) is produced when the aspartate at the amino terminal of Ang (1-7) is replaced by alanine. It can bind to the new receptor Mas-related G protein-coupled receptor D (MrgD) and protect against hypertension, inflammation, and lung damage. Furthermore, ACEI and ARB can protect against RAS imbalance by inhibiting the conversion of Ang I to Ang II and the binding of Ang II to the AT1R, respectively. We also showed that some viral infections can interfere with the function of RAS, leading to acute lung injury (ALI) and even acute respiratory distress syndrome (ARDS). |
Acute lung injury (ALI) is acute hypoxic respiratory insufficiency caused by non-cardiogenic pathogenic factors and may develop to acute respiratory distress syndrome (ARDS) in severe cases. The 28-day mortality rates of normal and critically ill patients with ARDS are 30% and over 40%, respectively.9–11 One of the important causes of ALI is virus infection. When the virus invades the respiratory tract, the epithelial cells and vascular endothelial cells are first damaged, and protein-rich edema fluid then accumulates in the lung interstitium and alveoli,12 and triggers various immune cells such as macrophages and neutrophils to release a large number of inflammatory factors. When RAS homeostasis is disrupted, a cytokine storm appears and eventually causes ALI or even ARDS.11–13 Some viral infections can also directly regulate the expression of RAS components by accelerating the imbalance of RAS and the occurrence and development of ALI/ARDS. RAS dysfunction induces ALI primarily by triggering a cascade of downstream reactions. Ang II is the main effective molecule of RAS and binds to AT1R to activate the NF-κB, AK2/STATs, and MAPK pathways and triggers inflammation, thus stimulating the production of reactive oxygen species (ROS) and promoting cell apoptosis.14–18 When the concentration of Ang II in the lungs increases, a large number of cytokines, chemokines, and ROS are released and exacerbate the lung damage caused by viral invasion. Pulmonary edema, pulmonary fibrosis, and lung cell apoptosis are related to RAS.19–22 ACE inhibitors (ACEI) and angiotensin II receptor blockers (ARB) can alleviate ALI, improve pulmonary capillary permeability, and reduce apoptosis.14,23 Therefore, RAS imbalance can promote the development of ALI/ARDS.
This article focuses on the role of RAS in ALI/ARDS caused by viral infections, as shown in Figure 1. The three parts aimed to explain RAS's role in the progression of different viral infections causing lung injury.
Role of RAS in ALI Caused by Coronavirus Infections
SARS
In 2003, severe acute respiratory syndrome (SARS) was first reported in China and then spread worldwide.24 By July 31, 2003, SARS had resulted in 8096 reported cases, including 774 deaths in 26 countries.25 A new coronavirus named severe acute respiratory syndrome coronavirus (SARS-CoV) is the pathogen of this epidemic,26,27 and binds to host cells via its surface Spike protein, particularly S1 domain, and then enters the body and causes severe lung injury.28 SARS pathogenesis has been widely studied.
ACE2 protein is the functional receptor of SARS-CoV, and the combination of ACE2 and S protein of SARS-CoV is crucial in lung injury. Li et al28 found that viral genome copies in ACE2-transfected cells increase by more than 100,000-fold in the first 48 hours. ACE2 contributes to the efficiency of SARS-CoV replication. When wild-type (WT) mice were infected with SARS-CoV, the lung ACE level did not change, whereas ACE2 was reduced substantially because of virus–receptor interaction. The type II transmembrane serine protease TMPRSS2 can cleave ACE2 and S protein to promote the S protein for membrane fusion and the entry of SARS-CoV into cells.29 Therefore, the ACE2 concentration decreases in the membrane but increases in the cytoplasm in SARS-CoV infected cells. Meanwhile, Ang II level in lung tissues increases considerably in SARS-CoV-infected mice. ARB treatment for the mice with infected SARS-CoV can remarkably alleviate the pulmonary edema and severe ALI.30 This finding suggests that SARS-CoV-mediated lung failure is caused by Ang II overexpression and excessive activation of the AT1 receptor.
ACE gene polymorphism, which is characterized by the insertion/deletion (I/D) of a 287-bp Alu repeat sequence in intron 16 of the ACE gene, is related to hypoxemia in patients with SARS.31,32 A study reported that patients in the hypoxemic group had a high frequency of D allele and consequently had high serum ACE levels. I/D polymorphism affects the serum and tissue ACE levels in the general population.33 The elevated ACE and decreased ACE2 levels in patients with SARS promote the huge expression of Ang II and AT1 receptor, thereby accelerating the lung injury and even leading to death.31 Furthermore, the innate immunity response is activated, and the expression of tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-8, IL-1β, interferon inducible protein-10 (IP-10), caspase 3 (CASP3), CASP9, and fibroblast growth factor-7 (FGF-7) increase in patients with SARS.34–36 The inflammatory pathways, apoptosis, oxidative stress, and fibrosis are all activated in patients with SARS.35,36
SARS-CoV infects people through the combination of its S protein and lung ACE2, which reduces the ACE2 level in the lungs, increases Ang II expression, and overactivates AT1 receptor. The function of RAS is no longer balanced. Finally, plenty of cytokines, chemokines, and proteases are released, resulting in cytokine storm and eventually severe ALI/ARDS or even death. Figure 2 shows the graphic pathogenesis of SARS-CoV.
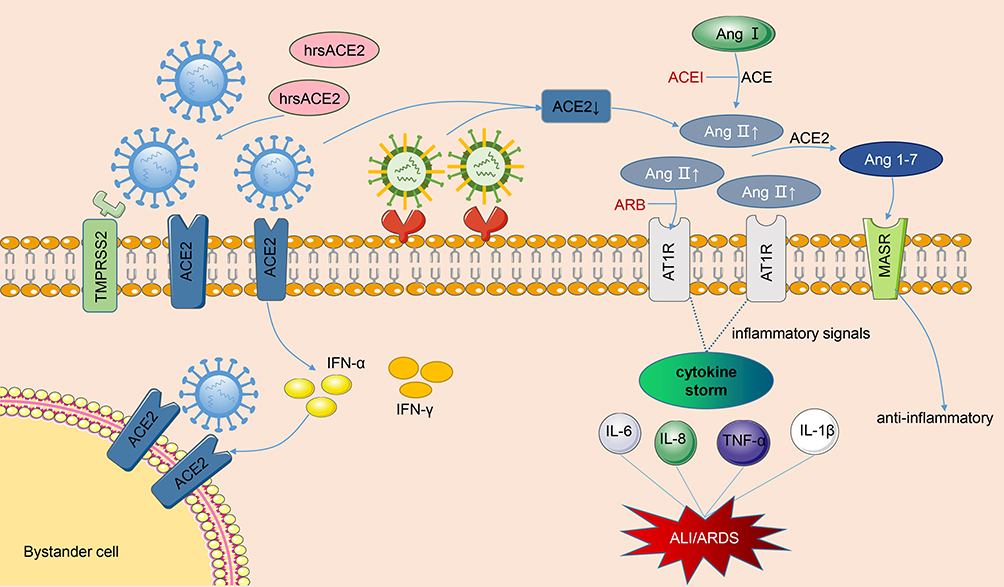 |
Figure 2 Pathogenesis of renin-angiotensin system in acute lung injury caused by viral infections. The SARS-CoV and SARS-CoV-2 bind to membranes angiotensin converting enzyme 2 (ACE2) of and employ type II transmembrane serine protease (TMPRSS2) for Spike protein priming, facilitating the fusion of viral and cellular membranes. After SARS-CoV-2 invades cells, it activates the natural antiviral pathway, prompts the infected cells to secrete interferon, stimulates bystanders to up-regulate ACE2 expression, and creates more targets for the entry of SARS-CoV-2. Occupation of ACE2 by the virus leads to the reduction of cell surface ACE2 levels. As the virus enters the cell, not limited to SARS-CoV and SARS-CoV-2, but also H5N1, H7N9 and EV71, the angiotensin II (Ang II) levels are significantly upregulated, a large amount of Ang II binds to the type 1 angiotensin II receptor (AT1R), then the inflammatory signaling pathways are overactivated, and then secrete various cytokines, resulting in cytokine storm and eventually lung injury. In the meantime, the ACE2 can cleave Ang II into Ang 1-7 which combines with Mas receptor (MasR) to fight against inflammation. ACEI and ARB can help restore the RAS balance after viral infections. Moreover, clinical-grade human soluble ACE2 (hrsACE2) can inhibit the coronavirus invasion by binding to coronavirus outside the cell. The blue virus in the figure represents coronaviruses, and the other one represents other viruses, such as H1N1, H7N9, and EV71. |
SARS-CoV-2
Coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 infection has spread in many countries and regions and is currently a major global health concern. Infected patients mainly show typical influenza-like symptoms. For severe cases, dyspnea or hypoxemia occurs 1 week after the disease onset and further develops into ARDS, septic shock, irreversible metabolic acidosis, and coagulopathy.37
Similar to SARS-CoV, the S protein of SARS-CoV-2 uses the cellular membrane ACE2 for entry, and is followed by employing TMPRSS2 for S protein priming to facilitate the fusion of viral and cellular membranes.38–40 Virus infectivity studies showed that SARS-CoV-2 uses ACE2 as an entry receptor in the ACE2-expressing cells, such as Hela cells in Chinese horseshoe bats, civets, and pigs, but not Hela cells without ACE2. SARS-CoV-2 does not employ other coronavirus receptors such as DPP4 and APN entry host cells.39 Virus entry in the host prompts two possible therapeutic targets for early-stage viral infection, including interference with the SARS-CoV-2 virus binding to the host and inhibition of serine protease TMPRSS2 activity. Another coronavirus, HNL63-CoV, also uses ACE2 protein as a receptor for cell entry, but its S protein has lower affinity for ACE2 than SARS-CoV and SARS-CoV-2. ACE2 is primarily distributed on the endothelial cells and smooth muscle cells of organs and tissues, including the oral and nasal mucosa, lung, small intestine, kidney, heart, and blood vessels; this condition is consistent with the multi-organ failure in patients with COVID-19.40–43 Single-cell sequences showed that the alveolar type II cells (AT2) are the major cell type that express ACE2 in all human lung cells and exhibit viral replication and transmission functions compared with AT2 cells not expressing ACE2. This phenomenon may partly explain the severe alveolar damage of SARS-CoV-2 infection. However, the small sample size and individuals heterogeneity should be considered.44
In experimental models of acid-aspirating ALI, the Ang II level is upregulated markedly in lungs and plasma of mice, but is substantially downregulated by ACE inactivation. The acid-treated ACE2 knockout mice showed evident lung injury compared with wild-type mice, the injection of recombinant human ACE2 protein alleviated pulmonary edema, and the use of specific AT1 and AT2 receptor blockers rescued the ALI in ACE2-knockout mice.45 In other ALI animal models induced by lipopolysaccharide or bleomycin, lung ACE2 level was severely decreased, and Ang II increased accordingly; ACE2 injection can weaken the lung pathological injury,46,47 and RAS imbalance is accompanied by the upregulation of inflammatory injury.47 These studies are limited to animal experimental models but can explain the mechanism of RAS systems to some extent. For the RAS imbalance caused by SARS-CoV and SAR-CoV-2 infections, the “abduction” effect of the virus on ACE2 must be considered.
In engineered extra pulmonary tissues, including engineered human blood vessel organoids and human kidney organoids, SARS-CoV-2 infection is inhibited by clinical-grade human soluble ACE2 (hrsACE2) – a decoy protein that has already been tested in phase I and II clinical trials. Vero E6 cells treated with hrsACE2 can dose-dependently inhibit SARS-CoV-2,48 suggesting that this protein can prevent early coronavirus from entering host cells. Additional clinical trials are needed to clarify the effects of hrsACE2 on RAS and clinical prognosis. Jose, Alex, and their colleagues found that ACE2 is a human interferon-stimulated gene (ISG). Interferon-α (IFN-α) and IFN-γ dose-dependently induce the upregulation of ACE2 in human primary basal epithelial cells. They also leveraged single cell RNA-seq and further screened the cells with enrichment for ACE2 and TMPRSS2 in non-infected controls and those infected with influenza A and B. The results showed that ACE2 is most upregulated in bystander goblet or squamous cells that are not directly infected by virus in the infected samples. This finding suggests that IFN produced by the virus-infected cells stimulates the surrounding uninfected cells and prompts the high ACE2 expression in bystander cells, thus providing additional invasive targets for SARS-CoV-2 infection. Individuals suffering from upper respiratory tract are highly susceptible to SARS-CoV-2.49
In addition to ACE2 as a viral receptor affecting viral entry, SARS-CoV-2 infection causes RAS disorders and systemic inflammatory response. Liu et al50 found that the plasma Ang II level of patients with COVID-19 is substantially higher than that of healthy individuals and is linearly associated to viral load and lung injury, thus providing the contact between tissue ACE2 downregulation and RAS imbalance. A clinical descriptive study showed that cytokine storm syndrome (CSS) appears in patients with COVID-19 and severe pneumonia, and some cases may rapidly progress to ARDS or even multi-organ failure.51 Inflammatory cytokines and chemokines, including IL-6, IL-2, IL-1β, IL-8, IL-17, IFN-γ, TNF-α, and monocyte chemoattractant protein-1 (MCP-1), are substantially upregulated in patients with COVID-19, especially IL-6, the key cytokine that triggers an inflammatory response and is correlated with mortality in these patients.52,53 After the spike protein of SARS-CoV-2 binds to the ACE2 on the cell surface, the level of membrane ACE2 is down-regulated, Ang II cannot convert to Ang1-7 and thus increasingly binds to AT1 receptors. The uncontrolled ACE/Ang II/AT1R axis stimulates downstream inflammatory signaling pathways, resulting in an increased permeability of pulmonary capillaries, injury to lung endothelium and epithelial cells, and ultimately ALI or even ARDS. This finding is consistent with the above experimental model of artificially induced lung injury. In addition to CSS, viral replication in lung epithelial and endothelial cells is also a major cause of lung injury. The pathogenesis of RAS in ALI caused by SARS-CoV-2 is presented in Figure 2.
The association of ACE gene polymorphism with SARS-CoV-2 infection and mortality has been studied. According to the available data, the frequency of D allele is positively correlated with the number of SARS-CoV-2 infection cases in Asians and African Americans.32,54 Owing to the combined effect of D allele and SARS-CoV-2 infection, the high ACE levels and decreased ACE2 levels lead to a high amount of Ang II in plasma and tissues and eventually result in severe lung injury and even death.
The possibility of using RAS inhibitors in COVID-19 treatment has become controversial. Patients with COVID-19 and hypertension, cardiovascular disease (CVD), diabetes, or chronic are advised to suspend the use of ACEIs or ARBs due to the possibility of upregulating ACE2, which may be conducive to viral invasion and may aggravate lung injury.55 On the contrary, other researchers claimed discontinuing ACEIs and ARBs in COVID-19 patients may be harmful due to the superior effect in pneumonia prevention.56–60 Some recent studies support this view, and population-based reports revealed that the use of ACEI and ARB inhibitors in patients with COVID-19 does not aggravate the severity of the disease, increase the risk of infection, improve prognosis, or reduce mortality.61–66 In addition, a systematic review and meta-analysis of related research has reconfirmed this thesis.67–71 Given that hypertension and CVD can increase the severity of the disease, dividing patients without hypertensive and CVD into a non-ACEI/ARB group may underestimate the favorable effects of ACEI/ARB drugs on affected patients. Limited by the retrospective design of most of these investigations, large-scale cohort studies are needed to confirm this view.
On the basis of existing evidence, ACE2 upregulation is limited only in animal experiments with the use of relatively high doses of ARBs and ACEIs. These results are inconsistent, and cannot be extrapolated to humans,72–75 and the expression of ACEI/ARB-derived ACE2 on lungs and the respiratory epithelium remains unclear. Further mechanism studies are needed to illustrate the effects of RAS inhibitors on human ACE2 expression.
Role of RAS in ALI Caused by Avian Influenza Virus Infections
H5N1
Infections caused by avian influenza viruses often lead to severe ALI and can even progress to ARDS. In 1997, the first case of infection with highly pathogenic avian influenza with a high fatality rate of 33.3% was reported in Hong Kong.76 In February 2020, two outbreaks of H5N1 and H5N6 avian influenza occurred in Hunan and Sichuan, China, suggesting that avian influenza virus is still one of the potential pathogens causing the global pandemic of viral infections.
The lung tissues of most dead patients exhibit the clinical features of ARDS and multiple organ dysfunction syndrome.77,78 Some studies suggested that ALI caused by avian influenza virus is related to RAS. Zou et al79 showed that the level of ACE2 protein in lung tissue decreases rapidly after H5N1 virus infection in mice, followed by a substantial increase in plasma Ang II concentration. However, no change is observed for ACE expression level. Compared with WT mice, ACE2-knockout mice infected with H5N1 show more severe lung injusy, and no matter before and after infection, and the injection of ACE2 protein can reduce the pathological damage caused by H5N1 infection. Yan et al80 found that RAS is imbalanced after H5N1 infection, and the plasma Ang II level may be used as one of the clinical indicators to evaluate the fatality of H5N1 infection. With the increase in disease severity, the Ang II concentration in H5N1-infected patients also gradually increases, and the plasma concentration of Ang II is high in dead patients. The viral loads and final virus titers in the lung tissues of WT mice were substantially lower than those in ACE2-knockout mice. When the recombinant hACE2 protein was injected into H5N1-infected mice, the replication of H5N1 in the lungs decreased, and the survival duration was extended. Treatment with AT1 receptor inhibitor losartan can increase the survival rate of mice, but cannot reduce the viral load in lung tissues, suggesting that ACE2 protein may decrease plasma Ang II level by reducing virus replication to restore normal RAS function in ALI caused by H5N1.
The pathogenicity of H5N1 avian influenza is associated with cytokine storm. The levels of intercellular adhesion molecule-1 (ICAM-1), IL-1β, IL-6, IL-8, and TNF-α in the lung tissues and peripheral in the mice of H5N1 virus pneumonia model were substantially higher than those in the control group.81 Other cytokines and chemokines, such as IL-8, MCP-1, and IP-10 were found in the serum of fatal cases infected by H5N1.82 In vitro infection experiments have revealed that H5N1 virus has higher ability than H1N1 in inducing the release of cytokines and chemokines in epithelial cells and macrophages.83
In summary, H5N1 infection dysregulates lung RAS and increases Ang II expression to stimulate the AT1 receptor, thereby inducing the activation of downstream inflammatory signals, forming a cytokine storm, and eventually resulting in ALI, even ARDS (Figure 2).
H7N9
H7N9 avian influenza virus infection was first reported in humans in 2013.84 Since then, H7N9 virus has spread continuously among humans and birds and had resulted in 1568 human infections and 615 deaths as of March 31, 2019.85 H7N9 virus has dual-receptor binding tropism and binds to α-2, 6-linked sialic acid (human receptor), and α-2, 3-linked sialic acid (avian receptor).86,87 Human upper respiratory tract and lung tissues contain α-2,6 glycans, and only lung tissues contain α-2,3 glycans.88 In vitro experiment showed that the virus titer in lungs is higher than that in the trachea under H7N9 infection.89 The epithelial cells of lower respiratory tract and type II alveolar cells are susceptible to the H7N9 virus.89 Meanwhile, ACE2 is mainly expressed in the epithelial cells of lung tissues and type II alveolar cells.
When H7N9 virus infects the human respiratory tract and induces ALI, the ACE2 expression is decreased, whereas the Ang II level is elevated.90 To understand the regulatory mechanism, researchers infected the WT mice and ACE2-knockout mice with H7N9 virus.91 ACE2 expression was substantially down-regulated in the WT mice 3 days after infection, while the expression of ACE was similar between H7N9-infected mice and control mice. In addition, pulmonary edema was aggravated, and the viral titers in the lung increased in the H7N9-infected mice. All ACE2-knockout mice that were infected with H7N9 virus died quickly, and 20% of WT mice survived. Pulmonary edema and lung injury were more severe in ACE2-knockout mice than in WT mice. Therefore, ACE2 plays a protective role against lung injury. In addition, the Ang II levels in plasma and lung tissues of H7N9-infected mice increased substantially. In H7N9-infected patients, the plasma Ang II concentrations increased continuously and reached a peak when the patient was close to death.90 Meanwhile, the plasma Ang II concentrations of the patients who recovered within 28 days were remarkably lower in the second week than that in the first week. Therefore, plasma Ang II level is related to the viral load and the severity of lung injury. Treatment with an AT1 receptor blocker can seriously attenuate the symptoms of lung injury and reduce lung viral titers, whereas treatment with an AT2 receptor blocker does not attenuate the lung edema and lung injury.91 In addition, the levels of IP-10, MIG, IL-6, IL-8, IFN-α, and IFN-γ are remarkably higher in H7N9-infected patients than in healthy subjects.89 Dysfunctions in chemokine and cytokine are responsible for the severe lung injury in human H7N9 infection.
In summary, H7N9 virus enters human lungs by binding to α-2,6-linked sialic acid and α-2,3-linked sialic acid, and then reduces ACE2 expression in lung tissues, and finally increases the plasma Ang II level. Ang II combined with AT1 receptor activates NF-κB and JAK/STATs inflammatory pathways,89,92 and promotes the release of various cytokines and inflammatory mediators (Figure 2). Dysfunctions of RAS and cytokine storm lead to ALI and ARDS in H7N9-infected patients.
Role of RAS in ALI Caused by Another Virus
Hand, Foot, and Mouth Disease
Hand, foot, and mouth disease (HFMD) is an infectious disease mainly caused by coxsackie 16 (CA16) and enterovirus 71 (EV71). Most HFMD cases are wild and limited to fever, oral ulcers, and vesicular rashes on the hands, feet, and buttocks. Some patients may rapidly progress to severe HFMD with serious complications, including brainstem encephalitis, encephalomyelitis, meningitis, neurogenic pulmonary edema (NPE), and cardiopulmonary failure.93 NPE and pulmonary hemorrhage are the main causes of ALI caused by HFMD.
RAS is involved in the systemic inflammatory response induced by HFMD virus infection.94 A clinical research study found that the serum Ang II concentrations of critically ill children with HFMD are substantially higher than those in the mild or severe group; this finding indicates that RAS plays a key role in the occurrence and development of pre-cardiopulmonary failure of HFMD, and its activation may eventually lead to NPE and ALI.95 Time-series analysis showed Ang II level is positively related to noradrenaline (NA) in mild and severe cases. NA secretion is induced by the combination of Ang II and AT1R and can induce the water and sodium storage and associate with pulmonary edema.96 Animal experiments suggested that EV71 infection can induce Ang II and NA expression in the brains, skeletal muscle, and lungs of mice.97 Therefore, RAS participates in the development of HFMD, and the elevated Ang II may be one of the early warning indicators of disease progression into cardiopulmonary failure. In addition, the increased Ang II facilitates its binding to the receptor, activates the downstream signals, increases water and sodium retention, promotes capillary contraction, and increases blood pressure, all of which are important in the development of HFMD to NPE and induce the lung injury.
HFMD viral infections can induce excessive immune response.98–103 During this process, lymphocytes and macrophages are continuously activated and expanded, thus aggravating the injury of lung endothelial cells and epithelial cells and inducing the release of inflammatory cytokines such as IL-1, IL-4, IL-6, IL-10, IL-13, IL-17, TNF-α, and INF-γ and the formation of a cytokine storm (Figure 2). In addition, large amounts of nitric oxide are expressed, thus further diluting blood, destroying blood vessels, and leading to ARDS and cardiopulmonary failure.
Summary and Outlook
Pneumonia caused by viral infection is one of the major threats to human health. RAS plays an important role in lung injury caused by viral infection, and RAS dysfunction is the key to the occurrence and development of ALI/ARDS in patients with viral infections. As a key member of RAS, ACE2 can be used as a receptor for SARS-CoV, SARS-CoV-2, and HCoV-NL63 to assist the viruses to enter the body, decrease the level of ACE2, and cause RAS dysfunction. Viral infections such as H5N1, H7N9, and EV71 can inhibit the expression of ACE2 and increase the level of Ang II after entering the bodies by binding to other receptors, thus forcing the imbalance of RAS. Inflammatory pathways and innate immunity are then activated, and plenty of cytokines, chemokines, and proteases act together to induce ALI or even ARDS. ACEI, ARB, and hrsACE2 may alleviate the severity of ALI by acting on RAS, thereby lowering the release of inflammatory molecules, and inhibiting the viral replication. However, the investigations on ACEI, ARB, and hrsACE2 as treatment for pneumonia caused by viral infection are limited to experimental studies and a few population-based studies.104–106 This must be further confirmed by more basic research and clinical studies. And this review could be a stimulus.
Acknowledgments
The authors would thank all those involved for their contributions to this article.
Disclosure
Yan-Lei Gao and Yue Du are co-first authors; these two authors contributed equally to this article. The authors report no conflicts of interest in this work.
References
1. Menikdiwela KR, Ramalingam L, Rasha F, et al. Autophagy in metabolic syndrome: breaking the wheel by targeting the renin-angiotensin system. Cell Death Dis. 2020;11(2):87. doi:10.1038/s41419-020-2275-9
2. Mizuiri S, Ohashi Y. ACE and ACE2 in kidney disease. World J Nephrol. 2015;4(1):74–82. doi:10.5527/wjn.v4.i1.74
3. Santos RAS, Oudit GY, Verano-Braga T, Canta G, Steckelings UM, Bader M. The renin-angiotensin system: going beyond the classical paradigms. Am J Physiol Heart Circ Physiol. 2019;316(5):H958–H970. doi:10.1152/ajpheart.00723.2018
4. Sarzani R, Giulietti F, Di Pentima C, Filipponi A, Spannella F. Antagonizing the renin-angiotensin-aldosterone system in the era of COVID-19. Intern Emerg Med. 2020;15(5):885–887. doi:10.1007/s11739-020-02365-5
5. Paz Ocaranza M, Riquelme JA, Garcia L, et al. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat Rev Cardiol. 2020;17(2):116–129. doi:10.1038/s41569-019-0244-8
6. Agrawal V, Gupta JK, Qureshi SS, Vishwakarma VK. Role of cardiac renin angiotensin system in ischemia reperfusion injury and preconditioning of heart. Indian Heart J. 2016;68(6):856–861. doi:10.1016/j.ihj.2016.06.010
7. Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical renin-angiotensin system in kidney physiology. Compr Physiol. 2014;4(3):1201–1228.
8. Abdul-Hafez A, Mohamed T, Omar H, Shemis M, Uhal BD. The renin angiotensin system in liver and lung: impact and therapeutic potential in organ fibrosis. J Lung Pulm Respir Res. 2018;5(1).
9. Maca J, Jor O, Holub M, et al. Past and present ARDS mortality rates: a systematic review. Respir Care. 2017;62(1):113–122. doi:10.4187/respcare.04716
10. Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800. doi:10.1001/jama.2016.0291
11. Butt Y, Kurdowska A, Allen TC. Acute lung injury: a clinical and molecular review. Arch Pathol Lab Med. 2016;140(4):345–350. doi:10.5858/arpa.2015-0519-RA
12. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):562–572. doi:10.1056/NEJMra1608077
13. Ling Y, Wei J, Wang A, Jin H. Advances in the pathogenesis of acute lung injury and acute respiratory distress syndrome. Carcinogen Teratogen Mutagen. 2017;29(02):151–154.
14. Li C, Bo L, Li P, et al. Losartan, a selective antagonist of AT1 receptor, attenuates seawater inhalation induced lung injury via modulating JAK2/STATs and apoptosis in rat. Pulm Pharmacol Ther. 2017;45:69–79. doi:10.1016/j.pupt.2017.05.002
15. Wang R, Zagariya A, Ibarra-Sunga O, et al. Angiotensin II induces apoptosis in human and rat alveolar epithelial cells. Am J Physiol. 1999;276(5):L885–L889.
16. Zhang M, Gao Y, Zhao W, Yu G, Jin F. ACE-2/ANG1-7 ameliorates ER stress-induced apoptosis in seawater aspiration-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2018;315(6):L1015–L1027. doi:10.1152/ajplung.00163.2018
17. Liu L, Qiu HB, Yang Y, Wang L, Ding HM, Li HP. Losartan, an antagonist of AT1 receptor for angiotensin II, attenuates lipopolysaccharide-induced acute lung injury in rat. Arch Biochem Biophys. 2009;481(1):131–136. doi:10.1016/j.abb.2008.09.019
18. Martins JO, Zanoni FL, Martins DO, et al. Insulin regulates cytokines and intercellular adhesion molecule-1 gene expression through nuclear factor-kappaB activation in LPS-induced acute lung injury in rats. Shock. 2009;31(4):404–409. doi:10.1097/SHK.0b013e318186275e
19. Cao CS, Yin Q, Huang L, Zhan Z, Yang JB, Xiong HW. Effect of angiotensin II on the expression of aquaporin 1 in lung of rats following acute lung injury. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2010;22(7):426–429.
20. Deng J, Wang DX, Deng W, Li CY, Tong J. The effect of endogenous angiotensin II on alveolar fluid clearance in rats with acute lung injury. Can Respir J. 2012;19(5):311–318. doi:10.1155/2012/951025
21. Lee YH, Mungunsukh O, Tutino RL, Marquez AP, Day RM. Angiotensin-II-induced apoptosis requires regulation of nucleolin and Bcl-xL by SHP-2 in primary lung endothelial cells. J Cell Sci. 2010;123(Pt 10):1634–1643. doi:10.1242/jcs.063545
22. Marshall RP, Gohlke P, Chambers RC, et al. Angiotensin II and the fibroproliferative response to acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286(1):L156–L164. doi:10.1152/ajplung.00313.2002
23. Asmussen S, Bartha E, Olah G, et al. The Angiotensin-converting enzyme inhibitor captopril inhibits poly(adp-ribose) polymerase activation and exerts beneficial effects in an ovine model of burn and smoke injury. Shock. 2011;36(4):402–409. doi:10.1097/SHK.0b013e318228f614
24. Kuiken T, Fouchier RA, Schutten M, et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet. 2003;362(9380):263–270. doi:10.1016/S0140-6736(03)13967-0
25. WHO. Summary of probable SARS cases; 2003. Available from: https://www.who.int/csr/sars/country/table2004_04_21/en/.
26. Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1953–1966. doi:10.1056/NEJMoa030781
27. Rota PA, Oberste MS, Monroe SS, et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300(5624):1394–1399. doi:10.1126/science.1085952
28. Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi:10.1038/nature02145
29. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pohlmann S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol. 2014;88(2):1293–1307. doi:10.1128/JVI.02202-13
30. Kuba K, Imai Y, Rao S, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11(8):875–879. doi:10.1038/nm1267
31. Itoyama S, Keicho N, Quy T, et al. ACE1 polymorphism and progression of SARS. Biochem Biophys Res Commun. 2004;323(3):1124–1129. doi:10.1016/j.bbrc.2004.08.208
32. Zheng H, Cao JJ. Angiotensin-converting enzyme gene polymorphism and severe lung injury in patients with coronavirus disease 2019. Am J Pathol. 2020;190(10):2013–2017. doi:10.1016/j.ajpath.2020.07.009
33. Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86(4):1343–1346. doi:10.1172/JCI114844
34. Tang NL, Chan PK, Wong CK, et al. Early enhanced expression of interferon-inducible protein-10 (CXCL-10) and other chemokines predicts adverse outcome in severe acute respiratory syndrome. Clin Chem. 2005;51(12):2333–2340. doi:10.1373/clinchem.2005.054460
35. Gu H, Li J. The introduction of cytokines in SARS and their elimination and antagonism. Chin J Dialysis Artif Organs. 2003;02:1–9.
36. Kong SL, Chui P, Lim B, Salto-Tellez M. Elucidating the molecular physiopathology of acute respiratory distress syndrome in severe acute respiratory syndrome patients. Virus Res. 2009;145(2):260–269. doi:10.1016/j.virusres.2009.07.014
37. Zhan J, Liu Q, Zhang Y, et al. The preliminary understanding of the new coronavirus 2019-nCoV. Environ Chem. 2020;39(02):283–291.
38. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280 e278. doi:10.1016/j.cell.2020.02.052
39. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020.
40. Xu X, Chen P, Wang J, et al. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci China Life Sci. 2020.
41. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562–569. doi:10.1038/s41564-020-0688-y
42. Li J, Liu W. Puzzle of highly pathogenic human coronaviruses (2019-nCoV). Protein Cell. 2020.
43. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS. J Virol. 2020;94(7). doi:10.1128/JVI.00127-20
44. Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. Am J Respir Crit Care Med. 2009;202(5):756–759. doi:10.1164/rccm.202001-0179LE
45. Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436(7047):112–116. doi:10.1038/nature03712
46. Li X, Molina-Molina M, Abdul-Hafez A, Uhal V, Xaubet A, Uhal BD. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295(1):L178–L185. doi:10.1152/ajplung.00009.2008
47. Ye R, Liu Z. ACE2 exhibits protective effects against LPS-induced acute lung injury in mice by inhibiting the LPS-TLR4 pathway. Exp Mol Pathol. 2020;113:104350. doi:10.1016/j.yexmp.2019.104350
48. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020;181(4):905–913.e7. doi:10.1016/j.cell.2020.04.004
49. Ziegler CGK, Allon SJ, Nyquist SK, et al. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell. 2020;181(5):1016–1035.e19. doi:10.1016/j.cell.2020.04.035
50. Liu Y, Yang Y, Zhang C, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63(3):364–374. doi:10.1007/s11427-020-1643-8
51. Chen N, Zhou M, Dong X, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. 2020;395(10223):507–513. doi:10.1016/S0140-6736(20)30211-7
52. Imai Y, Kuba K, Neely GG, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. 2008;133(2):235–249. doi:10.1016/j.cell.2008.02.043
53. Zhou F, Yu T, Du R, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi:10.1016/S0140-6736(20)30566-3
54. Pati A, Mahto H, Padhi S, Panda AK. ACE deletion allele is associated with susceptibility to SARS-CoV-2 infection and mortality rate: an epidemiological study in the Asian population. Clin Chim Acta. 2020;510:455–458. doi:10.1016/j.cca.2020.08.008
55. Fang L, Karakiulakis G, Roth M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med. 2020;8(4):e21. doi:10.1016/S2213-2600(20)30116-8
56. Rossi GP, Sanga V, Barton M. Potential harmful effects of discontinuing ACE-inhibitors and ARBs in COVID-19 patients. Elife. 2020;9. doi:10.7554/eLife.57278
57. Bian J, Zhao R, Zhai S, Li Z. Letter to the editor: anti-RAS drugs and SARS-CoV-2 infection. Acta Pharm Sin B. 2020;10(7):1251–1252. doi:10.1016/j.apsb.2020.04.013
58. Cappuccio FP, Siani A. Covid-19 and cardiovascular risk: susceptibility to infection to SARS-CoV-2, severity and prognosis of Covid-19 and blockade of the renin-angiotensin-aldosterone system. An evidence-based viewpoint. Nutr Metab Cardiovasc Dis. 2020;30(8):1227–1235. doi:10.1016/j.numecd.2020.05.013
59. Kai H, Kai M. Interactions of coronaviruses with ACE2, angiotensin II, and RAS inhibitors-lessons from available evidence and insights into COVID-19. Hypertens Res. 2020;43(7):648–654. doi:10.1038/s41440-020-0455-8
60. Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N Engl J Med. 2020;382(17):1653–1659. doi:10.1056/NEJMsr2005760
61. Ombarak RA, Zayda MG, Hinenoya A, Yamasaki S. Serotypes, pathogenic potential, and antimicrobial resistance of Escherichia coli isolated from subclinical bovine mastitis milk samples in Egypt. Jpn J Infect Dis. 2019;72(5):337–339. doi:10.7883/yoken.JJID.2018.538
62. Alexandre J, Cracowski JL, Richard V, Bouhanick B; Drugs C-wgotFSoPT. Renin-angiotensin-aldosterone system and COVID-19 infection. Ann Endocrinol (Paris). 2020;81(2–3):63–67. doi:10.1016/j.ando.2020.04.005
63. Lopez-Otero D, Lopez-Pais J, Cacho-Antonio CE, et al. Impact of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers on COVID-19 in a western population. CARDIOVID registry. Rev Esp Cardiol (Engl Ed). 2020. doi:10.1016/j.recesp.2020.05.030
64. Lam KW, Chow KW, Vo J, et al. Continued in-hospital ACE inhibitor and ARB use in hypertensive COVID-19 patients is associated with positive clinical outcomes. J Infect Dis. 2020;222(8):1256–1264. doi:10.1093/infdis/jiaa447
65. Hu J, Zhang X, Zhang X, et al. COVID-19 patients with hypertension have more severity condition, and ACEI/ARB treatment have no influence on the clinical severity and outcome. J Infect. 2020. doi:10.1016/j.jinf.2020.05.056
66. Bean DM, Kraljevic Z, Searle T, et al. Angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers are not associated with severe COVID-19 infection in a multi-site UK acute hospital trust. Eur J Heart Fail. 2020;22(6):967–974. doi:10.1002/ejhf.1924
67. Grover A, Oberoi M. A systematic review and meta-analysis to evaluate the clinical outcomes in COVID-19 patients on angiotensin-converting enzyme inhibitors or angiotensin receptor blockers. Eur Heart J Cardiovasc Pharmacother. 2020. doi:10.1093/ehjcvp/pvaa064
68. Guo X, Zhu Y, Hong Y. Decreased mortality of COVID-19 with renin-angiotensin-aldosterone system inhibitors therapy in patients with hypertension: a meta-analysis. Hypertension. 2020;76(2):e13–e14. doi:10.1161/HYPERTENSIONAHA.120.15572
69. Zhang X, Yu J, Pan LY, Jiang HY. ACEI/ARB use and risk of infection or severity or mortality of COVID-19: A systematic review and meta-analysis. Pharmacol Res. 2020;158:104927. doi:10.1016/j.phrs.2020.104927
70. Sriram K, Insel PA. Risks of ACE inhibitor and ARB usage in COVID-19: evaluating the evidence. Clin Pharmacol Ther. 2020;108(2):236–241. doi:10.1002/cpt.1863
71. Mackey K, King VJ, Gurley S, et al. Risks and impact of angiotensin-converting enzyme inhibitors or angiotensin-receptor blockers on SARS-CoV-2 infection in adults: a living systematic review. Ann Intern Med. 2020;173(3):195–203. doi:10.7326/M20-1515
72. Hamming I, van Goor H, Turner AJ, et al. Differential regulation of renal angiotensin-converting enzyme (ACE) and ACE2 during ACE inhibition and dietary sodium restriction in healthy rats. Exp Physiol. 2008;93(5):631–638. doi:10.1113/expphysiol.2007.041855
73. Ferrario CM, Jessup J, Chappell MC, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005;111(20):2605–2610. doi:10.1161/CIRCULATIONAHA.104.510461
74. Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26(4):
75. Lakshmanan AP, Thandavarayan RA, Watanabe K, et al. Modulation of AT-1R/MAPK cascade by an olmesartan treatment attenuates diabetic nephropathy in streptozotocin-induced diabetic mice. Mol Cell Endocrinol. 2012;348(1):104–111. doi:10.1016/j.mce.2011.07.041
76. Yuen KY, Chan PK, Peiris M, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet. 1998;351(9101):467–471. doi:10.1016/S0140-6736(98)01182-9
77. Liu S, Pang L, Ruan S, Zhang X. Global dynamics of avian influenza epidemic models with psychological effect. Comput Math Methods Med. 2015;2015:913726. doi:10.1155/2015/913726
78. Gao HN, Lu HZ, Cao B, et al. Clinical findings in 111 cases of influenza A (H7N9) virus infection. N Engl J Med. 2013;368(24):2277–2285. doi:10.1056/NEJMoa1305584
79. Zou Z, Yan Y, Shu Y, et al. Angiotensin-converting enzyme 2 protects from lethal avian influenza A H5N1 infections. Nat Commun. 2014;5:3594. doi:10.1038/ncomms4594
80. Yan Y, Liu Q, Li N, et al. Angiotensin II receptor blocker as a novel therapy in acute lung injury induced by avian influenza A H5N1 virus infection in mouse. Sci China Life Sci. 2015;58(2):208–211. doi:10.1007/s11427-015-4814-7
81. Zhao HX Establishment of animal model of highly pathogenic H5N1 avian influenza virus pneumonia and its pathogenesis [phD]. Jilin, China: Jilin University; 2006.
82. de Jong MD, Simmons CP, Thanh TT, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12(10):1203–1207. doi:10.1038/nm1477
83. Chan MC, Cheung CY, Chui WH, et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir Res. 2005;6:135. doi:10.1186/1465-9921-6-135
84. Su S, Gu M, Liu D, et al. Epidemiology, evolution, and pathogenesis of H7N9 influenza viruses in five epidemic waves since 2013 in China. Trends Microbiol. 2017;25(9):713–728. doi:10.1016/j.tim.2017.06.008
85. WHO. Avian influenza A(H7N9) virus-Monthly Risk Assessment Summary; 2019. Available from: https://www.who.int/influenza/human_animal_interface/Influenza_Summary_IRA_HA_interface_09_04_2019.pdf?ua=1.
86. Shi Y, Zhang W, Wang F, et al. Structures and receptor binding of hemagglutinins from human-infecting H7N9 influenza viruses. Science. 2013;342(6155):243–247. doi:10.1126/science.1242917
87. Xu Y, Peng R, Zhang W, et al. Avian-to-human receptor-binding adaptation of avian H7N9 influenza virus hemagglutinin. Cell Rep. 2019;29(8):2217–2228 e2215. doi:10.1016/j.celrep.2019.10.047
88. Chandrasekaran A, Srinivasan A, Raman R, et al. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol. 2008;26(1):107–113. doi:10.1038/nbt1375
89. Zhou J, Wang D, Gao R, et al. Biological features of novel avian influenza A (H7N9) virus. Nature. 2013;499(7459):500–503.
90. Huang F, Guo J, Zou Z, et al. Angiotensin II plasma levels are linked to disease severity and predict fatal outcomes in H7N9-infected patients. Nat Commun. 2014;5:3595. doi:10.1038/ncomms4595
91. Yang P, Gu H, Zhao Z, et al. Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury. Sci Rep. 2014;4:7027. doi:10.1038/srep07027
92. Lei J, Yin X, Shang H, Jiang Y. IP-10 is highly involved in HIV infection. Cytokine. 2019;115:97–103. doi:10.1016/j.cyto.2018.11.018
93. National Health Commission of People’s Republic of China. Guidelines for the diagnosis and treatment of hand foot and mouth disease (2018 edition). Chin J Viral Dis. 2018;8(05):347–352.
94. Kao SJ, Yang FL, Hsu YH, Chen HI. Mechanism of fulminant pulmonary edema caused by enterovirus 71. Clin Infect Dis. 2004;38(12):1784–1788. doi:10.1086/421021
95. Shen HY, Wang L, Xia Q, Xu JX, Xuan AL. Changes and significance of serum angiotensin II level and basic vital signs in children with hand-foot-mouth disease. Maternal Child Health Care China. 2019;34(16):3695–3697.
96. Yamamura H, Kawazoe Y, Miyamoto K, Yamamoto T, Ohta Y, Morimoto T. Effect of norepinephrine dosage on mortality in patients with septic shock. J Intensive Care. 2018;6:12. doi:10.1186/s40560-018-0280-1
97. Zhang C, Chen S, Zhou G, et al. Involvement of the renin-angiotensin system in the progression of severe hand-foot-and-mouth disease. PLoS One. 2018;13(5):e0197861. doi:10.1371/journal.pone.0197861
98. Duan G, Yang H, Shi L, et al. Serum inflammatory cytokine levels correlate with hand-foot-mouth disease severity: a nested serial case-control study. PLoS One. 2014;9(11):e112676. doi:10.1371/journal.pone.0112676
99. Lin TY, Hsia SH, Huang YC, Wu CT, Chang LY. Proinflammatory cytokine reactions in enterovirus 71 infections of the central nervous system. Clin Infect Dis. 2003;36(3):269–274. doi:10.1086/345905
100. Wang SM, Lei HY, Huang KJ, et al. Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: roles of cytokines and cellular immune activation in patients with pulmonary edema. J Infect Dis. 2003;188(4):564–570. doi:10.1086/376998
101. Lin TY, Chang LY, Huang YC, Hsu KH, Chiu CH, Yang KD. Different proinflammatory reactions in fatal and non-fatal enterovirus 71 infections: implications for early recognition and therapy. Acta Paediatr. 2002;91(6):632–635. doi:10.1111/j.1651-2227.2002.tb03292.x
102. Zhu D, Zhao XY, Yao Y, et al. A new factor influencing pathogen detection by molecular assay in children with both mild and severe hand, foot, and mouth disease. Diagn Microbiol Infect Dis. 2013;76(2):162–167. doi:10.1016/j.diagmicrobio.2013.02.011
103. Zheng XQ, Chen XQ, Gao Y, et al. Elevation of human leukocyte antigen-G expression is associated with the severe encephalitis associated with neurogenic pulmonary edema caused by Enterovirus 71. Clin Exp Med. 2014;14(2):161–167.
104. Kim J, Choi SM, Lee J, et al. Effect of renin-angiotensin system blockage in patients with acute respiratory distress syndrome: a retrospective case control study. Korean J Crit Care Med. 2017;32(2):154–163. doi:10.4266/kjccm.2016.00976
105. Spannella F, Giulietti F, Balietti P, et al. Renin-angiotensin system blockers and statins are associated with lower in-hospital mortality in very elderly hypertensives. J Am Med Dir Assoc. 2018;19(4):342–347. doi:10.1016/j.jamda.2017.09.023
106. Henry C, Zaizafoun M, Stock E, Ghamande S, Arroliga AC, White HD. Impact of angiotensin-converting enzyme inhibitors and statins on viral pneumonia. Proc (Bayl Univ Med Cent). 2018;31(4):419–423. doi:10.1080/08998280.2018.1499293
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.