")
Back to Journals » OncoTargets and Therapy » Volume 15
Role of Reactivating Mutant p53 Protein in Suppressing Growth and Metastasis of Triple-Negative Breast Cancer
Authors Berke TP, Slight SH, Hyder SM
Received 1 October 2021
Accepted for publication 21 December 2021
Published 8 January 2022 Volume 2022:15 Pages 23—30
DOI https://doi.org/10.2147/OTT.S342292
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Alberto Bongiovanni
Taylor P Berke,1 Simon H Slight,1,2 Salman M Hyder1,2
1Dalton Cardiovascular Research Center, University of Missouri, Columbia, MO, 65211, USA; 2Department of Biomedical Sciences, University of Missouri, Columbia, MO, 65211, USA
Correspondence: Salman M Hyder
Dalton Cardiovascular Research Center, University of Missouri, 134 Research Park Drive, Columbia, MO, 65211, USA
Tel +1 573-882-1261
Fax +1 573-884-4232
Email [email protected]
Abstract: Tumor suppressor p53 protein (p53) plays a vital role throughout the body to conserve DNA stability and prevent cancer. Normally, wild-type p53 protein (wtp53) is either degraded or bound to a negative regulator and is inactive. When damage to DNA occurs within a cell, p53 protein is induced and causes cell cycle arrest. This gives cells a chance to repair, but if damage is too severe, cells undergo apoptosis and are rejected. Mutations in the p53 gene (mtp53) are associated with a variety of cancers and occur in 70– 80% of cases of triple-negative breast cancer (TNBC). Importantly, many mutations occur in the DNA binding domain of p53 gene and the altered mutant p53 protein (mtp53) is subsequently not degraded. High levels of mtp53 protein accumulate within the cell, leading to the development of tumors. Therefore, converting mtp53 protein back into its functional wild-type conformation is a promising means by which to prevent or reverse tumor development. Herein we will briefly examine how tumor suppressor wtp53 exerts its effects, the mechanisms involved in protecting cells that undergo DNA damage and ways in which wtp53 prevents tumorigenesis. Using TNBC as an example, we will describe the use of specific compounds to reactivate mtp53 protein function by reconfiguring its structure and outline the potential benefits of mtp53 protein reactivation. We will also briefly discuss current clinical trials aimed at reactivating mtp53 protein in order to cure certain cancers. Finally, we make the recommendation that greater emphasis should be placed on testing naturally occurring compounds that are generally non-toxic to re-activate mtp53 protein and control progression of TNBC.
Keywords: p53, mutations, tumor suppressor, therapeutics
Introduction
Breast cancer is the most prevalent form of cancer in women, and each year about 200,000 new cases are diagnosed in the United States.1 Almost 40,000 women succumb to the disease annually, usually following metastasis or the emergence of drug-resistant tumor cells.1,2 A particularly aggressive subtype of the disease, known as triple-negative breast cancer (TNBC), accounts for approximately 10–20% of all breast cancer cases worldwide.2,3 TNBC is characterized by a failure to express estrogen and progesterone receptors, as well as human epidermal growth factor receptor 2 (HER-2/neu), three receptors that are commonly targeted with chemotherapeutic agents. The lack of hormone receptors limits the possibility of targeted therapy,2,3 consequently, TNBCs, rich in stem cells, typically metastasize and are associated with poor patient prognosis and death.2–4 It is therefore essential that advanced methods of treatment are developed to combat this especially deadly type of cancer.
Approximately 80% of TNBCs express an inactive, mutant form of the p53 tumor suppressor protein (mtp53), resulting in rapid tumor growth and metastasis.5 The prevalence of mutant p53 gene (mtp53) expression in TNBC and its severe effect on disease progression, highlight the importance of developing means by which to reactivate its functional wild-type conformation of p53 protein in TNBC patients.6
p53 Tumor Suppressor: Structure and Function
The tumor suppressor p53 gene (p53) is located on chromosome 17, and a total of 13 exons and 11 introns are involved in encoding the full length p53 protein. The N-terminus contains two linked transactivation domains (TAD1 and TAD2), which mediate the transcriptional activity of wtp53 protein.6,7 Studies show that variation within codon 47 near the N-terminus is associated with decreased apoptotic efficiency.7 Following the transactivation domains, is a proline rich region with PXXP repeats, where P is proline and X is an amino acid other than proline.6,7 This polyproline region is responsible for suppressing tumor growth and serves some apoptotic functions.7 Variation within codon 72 of exon 4, specifically a G to C transversion, results in a change from arginine to proline. This polymorphism has been found to induce tube-like structures involved in tissue angiogenesis, as well as increased expression of C-X-C Motif Chemokine Receptor 4 (CXCR4) and Cluster of Differentiation 44 (CD44), both of which are associated with increased tumor progression and poor patient prognosis.7 The DNA binding domain of the wtp53 protein is located near the C terminus and contains a central β-sandwich scaffold, a loop-sheet-helix structure, and two large loops. This region has been found to be highly conserved from an evolutionary standpoint and is the site of about 80% of all p53 mutations. The DNA binding domain is followed by a linker region and an oligomerization domain (OD), necessary for the tetramerization of p53 that occurs during DNA binding.6
Wild-type p53 (wtp53) protein is a key regulator of cellular homeostasis that is commonly referred to as the “guardian of the genome.8–10 wtp53 promotes cell cycle arrest and apoptosis, and also inhibits VEGF-dependent angiogenesis, thereby countering rapid tumor growth, metastasis, and possible drug resistance.6,11,12 Upon induction of apoptosis, wtp53 may be rapidly localized to the mitochondria in order to induce mitochondrial outer membrane permeabilization (MOMP), leading to the release of pro-apoptotic factors from the intermembrane space.10 wtp53 also plays an important role in the DNA damage response, senescence, DNA repair, cell migration and autophagy.10 It furthermore inhibits the self-renewal properties of cancer stem cells (CSCs) and opposes the epithelial to mesenchymal transition, a series of rapid changes in phenotype, such as downregulation of cell-cell adhesion structures, that are vital to metastasis.13–15
In response to cellular stress, wtp53 protein acts as a tetrameric transcription factor that binds to specific response elements and activates target genes such as Cyclin Dependent Kinase Inhibitor 1A (CDKN1A) and Serine/threonine kinase 11 (STK11).6,7,16 This activation is achieved in three steps. First, wtp53 is stabilized through events that disrupt its interaction with Mouse double minute 2 homolog (MDM2), a negative regulator that plays a role in a ubiquitin-mediated degradation of wtp53 protein. Following stabilization, functionally active wtp53 protein binds to its DNA response elements;10 however, the prevalence of altered amino acids in p53 protein resulting from mutations within the DNA binding domain of p53 prevents proper p53/DNA association (Figure 1). Successful binding and interaction of wtp53 protein with transcription factors facilitates activation or repression of the appropriate target genes, a process that is disrupted when defective mtp53 protein is present. It is possible that wtp53 protein undergoes posttranslational modifications that may influence its binding to specific promoters through a process referred to as promoter selection.10
 |
Figure 1 Schematic structure of p53 showing the different domains. Mutations frequently occur within the DNA-binding domain. Commonly mutated codons/residues are shown in red. |
As we have seen, most p53 gene mutations are localized to the DNA binding domain, with the majority being missense mutations within exons five through eight.5,6,10 Such mutations generally result in a gain of oncogenic functions, such as the promotion of invasion and metastasis, resistance to apoptosis, lack of cell cycle control, and genome instability.10,15 Alterations in the function of transcriptional regulators, or upregulation of chromatin regulatory genes, such as genome-wide increases in histone methylation, may lead to modified gene expression and cancer cell growth.6 An inability to activate tumor suppressor genes is associated with tumorigenesis, aggressive tumor type, and poor patient prognosis.7,11 In many cases, this failure to activate tumor suppressor genes is also correlated with promotion of invasion, angiogenesis, and, ultimately, drug resistance.11 Furthermore, mtp53 protein is not recognized and is therefore no longer regulated by the MDM2 negative feedback loop, leading to mtp53 protein accumulation.12 Studies show that the mtp53 protein conformation is stabilized through interactions with proteins such as heat shock protein 90 (HSP90), or as a result of post-translational modifications, such as phosphorylation at the Ser20 and Thr180 residues. In addition, its failure to induce MDM2 activation may also play a role in the stabilization of mtp53 protein.16 Typically, MDM2 functions as a ubiquitin ligase of wtp53 protein, downregulating protein levels in the absence of a stress stimulus.6,17,18 Upon activation of MDM2, wtp53 protein is exported from the nucleus to the cytoplasm for degradation and further transcriptional activity is inhibited in a feedback manner.6 Under conditions of cellular stress however, wtp53 protein is activated and accumulates within the nucleus, initiating an appropriate gene response. Inactivation of MDM2 facilitates p53-induced apoptosis as a cellular response to stress,10 The prevalence of mutations in the p53 gene, and the significant downstream consequences of such mutations, emphasizes the potential importance of developing ways of targeting p53 protein as a means of combating TNBCs, for which there are currently few treatment options. Mutations within the p53 gene should be clonal and therefore present in all malignant cancer cells,10 suggesting that treatment strategies involving p53 reactivation might be applicable to a range of cancers (Figure 2).
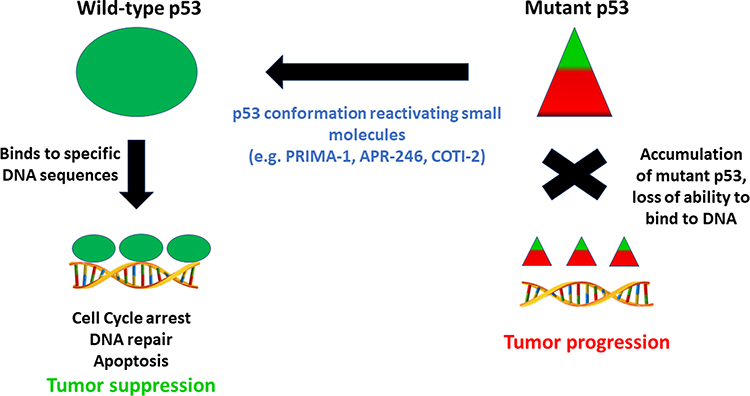 |
Figure 2 Strategy for targeting conformationally altered mutant p53 by small molecule drugs to regain anti-tumor functions. Triangles depict altered shape of wild-type p53 protein which accumulates in cells. |
PRIMA-1/APR-246
PRIMA-1 (p53 reactivation and induction of mass apoptosis) was identified as a non-toxic, low-molecular weight compound that causes mtp53 protein to undergo conformational change that facilitates its binding to DNA in a sequence-specific manner in order to induce apoptosis.16,20 PRIMA-1 was shown to preferentially induce cell cycle arrest and apoptosis in cells expressing different mutant forms of p53 protein, including the ones resulting from hot-spot mutants R273, R175, R282, R248, and G245 in the p53 gene.20 PRIMA-1 has also been found to cause covalent modifications to proteins, such as alkylation of thiol groups of cysteine residues on the surface of the p53 protein.7 So far, PRIMA-1 has been tested on 34 human tumor cell lines of various tissue types and, according to the National Cancer Institute, was found to preferentially inhibit the growth of cancer cell lines carrying p53 mutations. Drug effectiveness strongly correlated with levels of intracellular mtp53 protein levels.16 Upon further investigation, the effects of PRIMA-1 were found to be enhanced by the addition of a methyl group, which yields PRIMA-1Met, also known as APR-246.12,16
APR-246 is both a more soluble and a more active form of PRIMA-1.11 Upon administration, APR-246 is converted to methylene quinuclidinone (MQ). As an α, β-unsaturated carbonyl compound, MQ acts as a Michael acceptor and, similarly to PRIMA-1, has the capability to covalently bind to cysteine residues of the mtp53 protein in order to reactivate the wild-type conformation through the alkylation of thiol groups.6,7,11 Much of the functionality of APR-246 can be attributed to its ability to reactivate mtp53 protein by modifying the DNA-binding domain, promoting the capacity of reconfigured p53 to arrest the cell cycle and induce apoptosis.6,14,15 It is believed that alkylation of these cysteine residues may prevent aggregation caused by the thiol groups, possibly increasing the fraction of the protein able to bind to DNA and regulate gene transcription.7
APR-246 is believed to target a pocket between Loop L1 and beta-sheet S3 located in the core domain of the p53 protein; more specifically, it targets Cys124, located at the center of the pocket, as this residue is the most solvent accessible.6,19 Although Cys124 is not a DNA binding residue, this cysteine residue is responsible for interactions with DNA via Loop 1, anchoring p53 to DNA.11,14 In addition to Cys124, Cys277 has been identified as a binding target that is essential to MQ-mediated thermostabilization of the wtp53 protein core domain.12
Phosphorylation levels of the Ser15 residue, measured using both conformation-specific antibodies and fluorescent staining, have been used as a marker by which to monitor the shift of mtp53 protein towards the wild-type conformation.11 Reduced staining of antibody HO3.5, which detects mtp53 protein, and increased staining of antibody PAb1620, which detects the wtp53 protein conformation, indicate a shift back towards the correctly folded protein.6,16 In addition to these findings, APR-246 was found to reactivate R175H and R273H, both of which represent a substitution of histidine for arginine at positions 175 and 273, respectively, and are the most common forms of mutation detected (Figure 1). While these are two of the highest frequency mtp53 proteins detected, each mutation causes distinct downstream effects. Proteins with the R175H mutation are classified as structural mutants, due to the mutation occurring outside the DNA binding domain and, through an altered 3-dimensional structure, preventing the protein from binding to its response elements. Proteins containing the R273H mutation, on the other hand, are referred to as contact mutants. These mutations occur within the DNA binding domain of p53 gene, preventing protein resulting from mutated p53 to interact with DNA.6,7,14 The ability of APR-246 to reactivate proteins with both types of mutations in the mtp53 protein suggests great promise for its clinical efficacy.
The capacity of APR-246 to induce apoptosis was also measured by monitoring its effect on caspase activity. Following administration of APR-246, caspase activation was demonstrated through cleavage of poly ADP-ribose polymerase (PARP) by caspase-3.11 Caspase-3 is a frequently activated death protease responsible for activating cleavage of specific key proteins and is essential for both the dismantling of the cell and formation of apoptotic bodies.21–23 Concomitant administration of caspase inhibitors and APR-246 results in a reduction in apoptosis, further confirming the relationship between APR-246 and caspase activity.11
APR-246 cytotoxicity is both dose- and time-dependent and also depends on factors such as cellular confluence, hypoxia, cell-cycle progression, and different sensitivities of individual cell lines.11 Both p53 null cells and wtp53-containing cells exhibit reduced cytotoxic effects in response to APR-246, demonstrating a specificity of the drug for cells containing mtp53 protein.11,16 Following APR-246 treatment, an increase in the level of reactive oxygen species (ROS) within p53 null cells, as well as a decrease in the cellular content of glutathione (GSH) was ultimately the cause of cytotoxic effects in these cells. This was confirmed using buthionine-sulfoximine (BSO), an irreversible inhibitor of γ-glutamyl cysteine-synthase (γ-GCS). BSO was found to increase antitumor activities of PRIMA-1 regardless of the cell’s p53 status.11,12 Animals treated with doses of APR-246 between 20 and 400 mg/kg/day maintained body weight and did not exhibit changes in behavior, whether the drug was administered intravenously, intraperitoneally or by intra-tumoral injection.11 We are therefore confident that APR-246 could not only be an effective treatment for TNBC, but would also represent a non-toxic alternative to the highly toxic, non-targeted chemotherapy regimens in current use.
Additional Impacts of APR-246
Numerous studies have shed light on the possible impact of APR-246 in addition to its ability to reactivate mtp53 protein back into the functional wild-type conformation. The effect of APR-246 on cell redox balance may be the most important. As mentioned previously, APR-246 increases levels of ROS while also decreasing intracellular levels of GSH. It is believed that GSH depletion occurs via MQ binding to thiol groups of GSH.12 Regardless of p53 status, this induced oxidative stress typically leads to autophagy.10–12 In both wtp53 protein expressing cells and p53 null cells, a relationship has been found between Solute Carrier Family 7 Member 11 (SLC7A11) expression and potential resistance to APR-246. SLC7A11 is a key component in system xC, responsible for importing cystine for the formation of GSH. Levels of GSH remain high in cells with increased expression of SLC7A11, potentially enabling APR-246 resistance. APR-246 may also affect thioredoxin reductase (TXNRD1), another key regulator of cellular redox balance. Knocking down of this gene in mtp53-containing cell lines partially inhibited APR-246-induced cell death, most likely due to decreased production of ROS in a p53-dependent fashion. These results may explain effects observed in p53 null cells.12
In its effect on cell redox balance, APR-246 may also play a role in the unfolded protein response (UPR). The UPR is typically activated in response to an accumulation of unfolded proteins within the endoplasmic reticulum (ER), causing ER stress. Cells will initially attempt to restore normal cellular function by degrading these misfolded proteins; however, if these mechanisms are overridden apoptosis may occur. Following APR-246 treatment, an upregulation of heat shock proteins HSP70 and HSP90 and induction of numerous genes linked to the UPR leads to a significant increase in cytotoxic effects in p53 null cells. This may also be accompanied by an increase in p73 expression, resulting in an enhancement of UPR markers.11,12
Clinical Trials Utilizing APR-246
Currently, APR-246 is the only mtp53 protein-reactivating small molecule drug that has reached advanced stages of clinical trials, classifying it as a first-in-class drug.12 Approximately ten years after PRIMA-1 was first isolated, its derivative, APR-246, was first studied in Phase I and II clinical trials focusing on hematological malignancies and prostate cancers.22 The preliminary goal of these clinical trials was to determine the drug’s maximum tolerated dose (MTD), its overall safety, dose-limiting toxicities (DLTs), and pharmacokinetics (PK). Patients with a life-expectancy exceeding two months were selected for this clinical trial. p53 status was not a determining factor, but the mtp53 protein was observed in about twenty-five percent of patients. Following administration of APR-246 for four consecutive days,12,22 the MTD was determined to be 60 mg/kg. Overall, the drug was well tolerated with only mild side-effects compared with commonly used toxic chemotherapeutic agents. Favorable pharmacokinetics were demonstrated, including minor interindividual variations that were not dose- or time-dependent. Tumor cells exhibited cell-cycle arrest, increased apoptosis, and upregulation of p53 target genes. Of six patients who presented with circulating malignant cells, cell cycle changes, including complete arrest, were observed in four individuals. In addition, the p53 downstream targets BCL2 Associated X, Apoptosis Regulator (Bax), p53 upregulated modulator of apoptosis (PUMA), and phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), were upregulated, as was Decoy receptor 2 (DcR2), a senescence marker.22
Future Direction and Possibility of Combinational Therapy
The current success of APR-246 in clinical trials raises the possibility that therapy utilizing this small-molecule drug in combination with other agents will be clinically beneficial for treating TNBC. An example of one such therapy consists of a combination of APR-246 and a monoclonal antibody (2aG4), which targets exposed phosphatidylserine residues located on tumor blood vessels. 2aG4 blocks blood vessel function and reduces angiogenesis, both of which are vital for tumor progression. Combination treatment using these two agents typically results in ischemia, hemorrhagic necrosis, and, ultimately, cell death in both hormone-dependent breast cancer and TNBC.23,24 Importantly, a combination of APR-246 and 2aG4 appears to be non-toxic in tumor-bearing animal models,23,34 which nevertheless exhibit suppression of tumor growth and in some cases, complete tumor eradication. There is evidence that a combination of the two is more effective than either agent administered alone. Furthermore, by reducing tumor blood vessel density, an important factor in metastasis, APR-246/2aG4 combination treatment may prevent the growth of secondary malignancies.23,34
Other Small Molecule Drugs That Act on mtp53 Protein
Combinational therapies using APR-246 show promise in the fight against TNBC. However, other small molecule drugs that reactivate mtp53 are also of interest. COTI-2, a third-generation thiosemicarbazone, has been shown to exert antitumor activity in a variety of cancer types, including MDA-MB-231 TNBC cells, both in vitro and in vivo in an animal xenograft model.25 Compared with currently used chemotherapeutic agents and other targeted therapies, COTI-2 has been shown to be more active and well tolerated in animal models. It is believed that, like APR-246, the primary mechanism by which COTI-2 acts, is through reactivation of mtp53 protein, thereby restoring its DNA-binding properties and the ability of converted p53 to induce expression of its target genes.26,27 Studies show, however, that COTI-2 may possess anticancer properties that are independent of p53 status, including an ability to inhibit the target of rapamycin (mTOR) pathway and activate AMP-activated protein kinase (AMPK).26 Another small molecule drug, NSC2287, Reactivation of p53 and Induction of Tumor cell Apoptosis (RITA), also binds to mtp53 protein and restores its proper transcriptional functions.28,29 RITA induces apoptosis in cells that express either mtp53 protein or wtp53, as well as in p53-null cells, a finding that demonstrates that RITA may be able to function both dependently and independently of p53 status. Studies show that RITA may induce apoptosis independently of p53 status by activating caspases through p38 and the stress-activated protein kinase/Jun-amino-terminal kinase SAPK/JNK.30 Following recognition of its anticancer properties, RITA was further studied in a drug development program initiated by the Developmental Therapeutics Program (DTP) at NCI. RITA was administered to rat, dog, and monkey models in doses comparable to those shown to be effective in mouse xenografts.31,32 Unfortunately, in all three animal models RITA caused pulmonary edema, and the program was terminated prematurely. However, studies are ongoing aimed at reducing nonspecific toxicities with analogues, with the hope that RITA might be developed sufficiently to be ultimately safe for clinical use. Since mtp53 protein suppresses normal p73 activity, other drugs, such as Reactivation of Transcriptional Reporter Activity (RETRA), focus on suppressing mtp53 activity to facilitate regular p73 functions.33 Many other drugs that target mtp53 protein and are undergoing clinical development, including those that restore the wild-type conformation of the protein to induce apoptosis/cell cycle arrest, are described in another elegant review article.34
Conclusions
In recent years, there has been a concerted effort to target mutant p53 protein with a view to restoring normal p53 suppressor activity and reducing cancer. APR-246 and COTI-2 are two examples of small molecule drugs that are either undergoing or will soon be tested in clinical trials.35 Rationale structure-activity-based drug design, gene- and cell-based technologies are becoming popular. Although Adenoviral gene therapy and clustered regularly interspaced short palindromic repeats-CRISPR-associated (CRISPR/Cas) gene editing-based therapies are advancing, these technologies have not yet been applied to the targeting of mtp53 protein. Such studies are, however, imminent. Since many small molecule compounds, including APR-246 and COTI-2 are easy to develop therapeutically, these drugs represent a promising way forward for controlling tumor progression. PRIMA-1 was shown to prevent breast cancer in an animal model.36 Newer drugs such as APR-246 and COTI-2 are undergoing clinical trials to establish their effectiveness against different types of cancer. Such agents may eventually be developed for use in a preventative manner, which would indeed represent a major advancement in the field. Since most cases of TNBC are associated with mtp53, new drugs that target the mutant protein are particularly important to women afflicted with this aggressive form of breast cancer, for whom there are no targeted therapies. Several plant-derived nutraceuticals, such as luteolin and curcumin, have been shown to possess anti-tumor properties. It would therefore be extremely beneficial to test such compounds, which are typically non-toxic, for their ability to reactivate mtp53 protein. Naturally occurring agents such as the aforementioned will likely provide a safe and effective means of treating patients with TNBC. It is possible that more potent analogues of such compounds could be developed that will offer non-toxic alternatives to the highly toxic drug regimens now currently used.
Acknowledgments
We would like to thank the many students and technicians involved in the studies reported from the Hyder laboratory. Dr. Yayun Liang deserves special recognition for her expertise and for participation in many experiments in our laboratory. Studies related to p53 were supported by grants from the National Institutes of Health and the Department of Defense, as well as awards from the College of Veterinary Medicine at the University of Missouri. Taylor Berke is an Honors student at MU and a Cherng Scholar in the School of Biological Sciences at the University of Missouri-Columbia. SMH is the Zalk Missouri Professor of Tumor Angiogenesis.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. A cancer statistics. CA Cancer J Clin. 2020;70:7–30. doi:10.3322/caac.21590
2. Aysola K, Desai A, Welch C, et al. Triple negative breast cancer – an overview. Hered Genet. 2013;2013(Suppl 2):001. doi:10.4172/2161-1041.S2-001
3. Wahba HA, El-Hadaad HA. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med. 2015;12:106–116. doi:10.7497/j.issn.2095-3941.2015.0030
4. Park SY, Choi JH, Nam JS. Targeting cancer stem cells in triple-negative breast cancer. Cancers. 2019;11:965. doi:10.3390/cancers11070965
5. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi:10.1038/nature11412
6. Duffy MJ, Synnot NC, O’Grady S, Crown J. Targeting p53 for the treatment of cancer. Semin Cancer Biol. 2020;S1044–579X(20):30160–30167.
7. Kaur RP, Vasudeva K, Kumar R, Munshi A. Role of p53 gene in breast cancer: focus on mutation spectrum and therapeutic strategies. Curr Pharm Des. 2018;24:3566–3575. doi:10.2174/1381612824666180926095709
8. Lane DP. Cancer. P53, guardian of the genome. Nature. 1992;358:15–16. doi:10.1038/358015a0
9. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. doi:10.1016/S0092-8674(00)81871-1
10. Zilfou JT, Lowe SW. Tumor suppressive functions of p53. Cold Spring Harb Perspect Biol. 2009;1(5):00a001883. doi:10.1101/cshperspect.a001883
11. Perdrix A, Najem A, Saussez S, et al. PRIMA-1 and PRIMA-1Met (APR-246): from mutant/wild type p53 reactivation to unexpected mechanisms underlying their potent anti-tumor effect in combinatorial therapies. Cancers. 2017;9:172–188. doi:10.3390/cancers9120172
12. Menichini P, Monti P, Speciale A, et al. Antitumor effects of PRIMA-1 and PRIMA-1Met (APR246) in hematological malignancies: still a mutant p53-dependent affair? Cells. 2021;10(1):98–112. doi:10.3390/cells10010098
13. Klymkowsky MW, Savagner P. Epithelial-mesenchymal transition. Am J Pathol. 2009;174(5):1588–1593. doi:10.2353/ajpath.2009.080545
14. Omar SI, Tuszynski J. The molecular mechanism of action of methylene quinuclidinone and its effects on the structure of p53 mutants. Oncotarget. 2018;9:37137–37156. doi:10.18632/oncotarget.26440
15. Chen J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb Perspect Med. 2016;6:a026104. doi:10.1101/cshperspect.a026104
16. Duffy MJ, Synnott NC, Crown J. Mutant p53 in breast cancer: potential as a therapeutic target and biomarker. Breast Cancer Res Treat. 2018;170:213–219. doi:10.1007/s10549-018-4753-7
17. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi:10.1038/387299a0
18. Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi:10.1016/S1044-579X(02)00099-8
19. Wassman CD, Baronio R, Demir O, et al. Computational identification of a transiently open L2/S3 pocket for reactivation of mutant p53. Nat Commun. 2013;4:1407. doi:10.1038/ncomms2361
20. Bykov VJN, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–288. doi:10.1038/nm0302-282
21. Liang Y, Besch-Williford C, Cook MT, et al. APR-246 alone and in combination with a phosphatidylserine-targeting antibody inhibits lung metastasis of human triple-negative breast cancer cells in nude mice. Breast Cancer. 2019;11:249–259. doi:10.2147/BCTT.S208706
22. Lehmann S, Bykov VJN, Ali D, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30:3633–3639. doi:10.1200/JCO.2011.40.7783
23. Liang Y, Mafuvadze B, Besch-Williford C, Hyder SM. A combination of p53-activating APR-246 and phosphatidylserine-targeting antibody potently inhibits tumor development in hormone-dependent mutant p53-expressing breast cancer xenografts. Breast Cancer. 2018;10:53–67. doi:10.2147/BCTT.S156285
24. Liang Y, Besch-Williford C, Mafuvadze B, Brekken RA, Hyder S. Combined treatment with p53-activating drug APR-246 and a phosphatidylserine-targeting antibody, 2aG4, inhibits growth of human triple-negative breast cancer xenografts. Cancer Rep Rev. 2020;4:1–10. doi:10.15761/CRR.1000205
25. Salim KY, Maleki Vareki S, Danter WR, Koropatnick J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget. 2016;7(27):41363–41379. doi:10.18632/oncotarget.9133
26. Synnott NC, O’Connell D, Crown J, Duffy MJ. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res Treat. 2020;179(1):47–56. doi:10.1007/s10549-019-05435-1
27. Green JA, Von Euler M, Abrahmsen LB. Restoration of conformation of mutant p53. Ann Oncol. 2018;29:1325–1328. doi:10.1093/annonc/mdy057
28. Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53–HDM-2 interaction and activates p53 function in tumors. Nat Med. 2004;10:1321–1328. doi:10.1038/nm1146
29. Doggrell SA. RITA - a small-molecule anticancer drug that targets p53. Expert Opin Investig Drugs. 2005;14:739–742. doi:10.1517/13543784.14.6.739
30. Weilbacher A, Gutekunst M, Oren M, Aulitzky WE, van der Kuip H. RITA can induce cell death in p53-defective cells independently of p53 function via activation of JNK/SAPK and p38. Cell Death Dis. 2014;5:e1318. doi:10.1038/cddis.2014.284
31. Rivera MI, Stinson SF, Vistica DT, Jorden JL, Kenney S, Sausville EA. Selective toxicity of the tricyclic thiophene NSC 652287 in renal carcinoma cell lines. Biochem Pharmacol. 1999;57:1283–1295. doi:10.1016/S0006-2952(99)00046-5
32. Peyser BD, Hermone A, Salamoun JM, et al. Specific RITA modification produces hyperselective cytotoxicity while maintaining in vivo antitumor efficacy. Mol Cancer Ther. 2019;18:1765–1774. doi:10.1158/1535-7163.MCT-19-0185
33. Kravchenko JE, Ilyinskaya GV, Komarov PG, et al. Small-molecule RETRA suppresses mutant p53 bearing cancer cells through a p73-dependent salvage pathway. Proc Natl Acad Sci USA. 2008;105:6302–6307. doi:10.1073/pnas.0802091105
34. Zawacka-Pankau J, Selivanova G. Pharmacological reactivation of p53 as a strategy to treat cancer. J Intern Med. 2015;277:248–259. doi:10.1111/joim.12336
35. Sallman DA, DeZern AE, Guillermo Garcia-Manero DP, et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J Clin Oncol. 2021;39:1584–1594. doi:10.1200/JCO.20.02341
36. Benakanakere I, Besch-Williford C, Ellersieck MR, Hyder SM. Regression of progestin-accelerated 7,12-dimethylbenz[a]anthracene-induced mammary tumors in Sprague-Dawley rats by p53 reactivation and induction of massive apoptosis: a pilot study. Endocr Relat Cancer. 2009;16(1):85–98. doi:10.1677/ERC-08-0069
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.