")
Back to Journals » Journal of Inflammation Research » Volume 15
Role of Pyroptosis in Respiratory Diseases and its Therapeutic Potential
Authors Liu J , Fan G, Tao N, Sun T
Received 5 December 2021
Accepted for publication 16 March 2022
Published 28 March 2022 Volume 2022:15 Pages 2033—2050
DOI https://doi.org/10.2147/JIR.S352563
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Jingjing Liu,1– 3 Guoqing Fan,1– 3 Ningning Tao,4 Tieying Sun1,2
1Department of Respiratory Medicine and Critical Care, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 2Graduate School of Peking Union Medical College, Beijing, People’s Republic of China; 3The MOH Key Laboratory of Geriatrics, Beijing Hospital, National Center of Gerontology, Beijing, People’s Republic of China; 4Department of Respiratory Medicine and Critical Care, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, Shandong, People’s Republic of China
Correspondence: Tieying Sun, Department of Respiratory Medicine and Critical Care, Beijing Hospital, Dongcheng District, Beijing, 100730, People’s Republic of China, Tel +86 15153169108, Email [email protected]
Abstract: Pyroptosis is an inflammatory type of regulated cell death that is dependent on inflammasome activation and downstream proteases such as caspase-1 or caspase 4/5/11. The main executors are gasdermins, which have an inherent pore-forming function on the membrane and release inflammatory cytokines, such as interleukin (IL)-1β, IL-18 and high mobility group box 1. Emerging evidence demonstrates that pyroptosis is involved in the pathogenesis of various pulmonary diseases. In this review, we mainly discuss the biological mechanisms of pyroptosis, explore the relationship between pyroptosis and respiratory diseases, and discuss emerging therapeutic strategies for respiratory diseases.
Keywords: pyroptosis, pulmonary disease, caspase-1, caspase-4/5/11, gasdermins
Introduction
Pyroptosis is a newly discovered type of regulated cell death that was first described in Salmonella-infected macrophages and dendritic cells.1 The morphological characteristics of pyroptosis which are distinguishable from necroptosis include cell membrane pore formation, cytomembrane fracture, cellular swelling, chromatin condensation, and increased inflammatory cytokine production. However, in contrast to apoptosis, the integrity of the mitochondria is maintained, and pyroptosis has no effect on the release of cytochrome C.1–3 Pyroptosis was initially reported to occur in innate immune cells, such as macrophages, dendritic cells, and neutrophils. However, recent studies have demonstrated that pyroptosis can also occur in epithelial cells, endothelial cells, keratinocytes, and neurons.4
Pyroptosis was initially described as an inflammatory type of regulated cell death mediated by caspase-1 which belongs to the inflammatory caspase family and is activated by inflammasome.5 An inflammasome is an intracellular multi-protein compound, which mainly includes the nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) and PYHIN protein families.6 The inflammasomes can be activated by pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs).7 Bioactive caspase-1 can process gasderminD (GSDMD) into a GSDMD N-terminal (GSDMD-NT) fragment and cleave pro-IL-1β and pro-IL-18 into mature IL-1β and IL-18, eventually leading to canonical pyroptosis and the release many inflammatory cytokines.7
Lipopolysaccharides (LPS) from gram-negative bacteria can directly bind to caspase-4/5/11 independent of caspase-1 to induce pyroptosis, and this is described as the non-canonical pyroptosis pathway.8 In 2015, it was discovered that GSDMD is an executor of pyroptosis, N terminal of GSDMD can form pores on the plasma membrane, thereby releasing many inflammatory cytokines, pro-fibrotic cytokines, and cell contents.9,10 Notably, other gasdermin family members (such as GSDMA, GSDMB, GSDMC, and GSDME) have been increasingly involved in cell membrane pore formation and pyroptosis induction.11–14 Therefore, the Nomenclature Committee on Cell Death modified the definition of pyroptosis, and it is now described as a cell death dependent on the gasdermin family’s participation in membrane pore formation.15
Pyroptosis is involved in the development of various diseases, such as cancer, and autoimmune, metabolic, and nervous system diseases.16–19 Emerging evidence has confirmed that Dermatophagoides farina 1 (Der f1)-induced pyroptosis in human bronchial epithelia contributes to the development of asthma.20 In addition, inflammasome-mediated pyroptosis participates in the pathogenesis of pulmonary infections, pulmonary fibrosis, and chronic obstructive pulmonary disease (COPD).21–24 Therefore, the role of pyroptosis in pulmonary diseases, particularly, in the development of novel diagnostic and therapeutic methods to manage these diseases, has become a research focus.
This review highlights the signaling pathway and inhibitors involved in pyroptosis and the relationship between pyroptosis and pulmonary diseases. It also discusses potential therapeutic targets related to pyroptosis in pulmonary diseases.
Molecular Mechanisms of Pyroptosis
Components of the Inflammasome
The inflammasome is an intracellular multi-protein compound consisting of a sensor protein (for example, pattern recognition receptors [PRRs]), apoptosis-associated speck like protein containing a caspase recruitment domain (ASC) and a caspase-1 family protease.25 PRRs can identify PAMPs or DAMPs and include the NLR family (NLRP3, and NLR family caspase recruitment domain [CARD] containing proteins, such as NLRC1, NLRC4, and NLRC6) localized in the cytoplasm, which is present within the plasma membrane or lysosomal organelles and PYHIN protein family including absent in melanoma (AIM2).26,27 NLR proteins consist of C-terminal leucine-rich repeats (LRRs), a central NOD/NACHT, and an N-terminal CARD or pyrin domain (PYD). The NOD/NACHT domain is shared by the NLR family and its downstream pathway is activated by adenosine triphosphate (ATP)-dependent oligomerization. LRRs are associated with ligand sensing and autoregulation, and the CARD and PYD domains interact with the downstream signals.28 Of note, NLRP3 has been identified as a crucial NLR family member, which recognizes PAMPs (such as bacteria, viruses, and fungi) or DAMPs (eg ATP and uric acid).26 Other NLR family members include NLRP1, which is activated by the anthrax lethal toxin;29 NLRC4, which is activated by cytosolic bacterial flagellin, such as that of Salmonella;30 and NLRP6, which is activated by microbes, such as Porphyromonas gingivalis.31 AIM2 proteins typically contain a DNA-binding HIN-200 domain and a PYD-signaling domain, which specifically recognize cytoplasmic double-stranded DNA.27 Pyrin is encoded by the MEFV gene, which has four functional domains; PYD, and the zinc finger, coiled coil, and B30.2/SPRY domains. It specifically recognizes RhoA guanosine triphosphatase inactivation induced by pathogens.32 ASC is an essential intermediate protein that combines sensor proteins (NLRs and AIM2) and caspase-1, whereas NLRC4 is an exception. NLRC4 activates caspase-1 by directly recruiting pro-caspase-1.6,33
Canonical inflammasome activation requires two independent steps. Taking the activation of the NLRP3 inflammasome as an example, the first step is priming, which is induced by the Toll-like receptor (TLR)/myeloid differentiation primary response 88 (MyD88)-nuclear factor kappa B (NF-κB) signaling pathway to upregulate the transcription of NLRP3, pro-IL-1β and pro-IL-18.6,7,25,34 The second step involves formation and modification of the NLRP3 inflammasome. There are three classic activation pathways of the NLRP3 inflammasome. (i) potassium efflux,35 which is a mutual pathway for the assembly of the NLRP3 inflammasome which promotes the interaction between NEK7-NLRP3.36,37 (ii) lysosomes damage.35,38 (iii) the generation of mitochondrial reactive oxygen species (ROS),7,35 mitochondrial ROS promotes the dissociation of thioredoxin from thioredoxin-interacting protein (TXNIP). Subsequently, TXNIP can bind to NLRP3 and trigger the activation of the NLRP3 inflammasome by stimulating the recruitment of ASC and caspase-1.39 Pro-caspase-1 is activated by inflammasomes to form bioactive caspase-1. Caspase-1 can cut the precursors of downstream inflammatory cytokines (such as pro IL-1β and pro IL-18) and turn them into bioactive cytokines (IL-1β and IL-18).
Non-canonical NLRP3 inflammasome activation has been recently discovered, and it is associated with potassium efflux.40–42 Potassium efflux via GSDMD pores in caspase-4/5/11-mediated non-classical pyroptosis is essential for NLRP3 inflammasome activation.41 Another study showed that LPS-induced caspase-11 activation triggered cleavage of the pannexin-1 channel, which promoted the release of ATP. ATP triggers P2X7, a purinoreceptor, to form a channel for potassium efflux, and subsequently, the NLRP3 inflammasome is activated.42 Emerging evidence has demonstrated that caspase-8 is associated with NLRP3 inflammasome activation by promoting the formation of ASC and activation of caspase-1, and is also possibly related to potassium efflux.43
Caspase Family Proteases
Caspase-1/4/5 and caspase-1/11 expressed in humans and mice, respectively, are the main caspase family proteases associated with pyroptosis.44 Caspase-1 exists as an inert precursor in the cytoplasm, which can be activated by inflammasomes and can promote apoptosis and mediate the canonical pyroptosis pathway.45 Caspase-1 proteolytically cleaves the precursor pro-IL-1β/pro-IL-18 to generate mature IL-1β/IL-18 and cleaves GSDMD into a 31kD N-terminal and a 22kD C-terminal. GSDMD-NT forms pores on the cell membrane, thus, inducing pyroptosis and releasing IL-1β, IL-18, IL-1α and high mobility group box 1 (HMGB1).46
Caspase-4/5/11 are involved in the non-canonical pyroptosis pathway. CARD in caspase-4/5/11 recognizes LPS lipid A and induces the oligomerization of the caspases. Activated caspase-4/5/11 cleave GSDMD and release the GSDMD-NT fragment to induce cell membrane pore formation, release IL-1α and HMGB1, and lead to pyroptosis.9,44 However, caspase-4/5/11-mediated pyroptosis has no effect on the processing of IL-1β and IL-18. Interestingly, caspase-4/5/11 triggers non-canonical NLRP3 inflammasome activation to promote IL-1β and IL-18 secretion.9
An increasing number of caspase family members have been found to play a role in pyroptosis. Caspase-3 was previously known as an executor of apoptosis, and emerging evidence has demonstrated that activated caspase-3 cleaves GSDME, thereby releasing the GSDME N-terminal and triggering pyroptosis.14,47,48 Caspase-3 is activated by caspase-8 through the death receptor pathway and can also be activated by caspase-9 via the mitochondrial apoptotic pathway.48,49 Various death stimuli or viral infections can result in permeabilization of the mitochondrial outer membrane, leading to the release of cytochrome C, which promotes the activation of caspase-9 which then cleaves caspase-3.48 Emerging evidence suggests that caspase-1/3/7 cleave GSMDE to generate GSDME-N terminal and induce pyroptosis in teleosts, and GSDME is cleaved by caspase-1 with high efficiency and by caspase-3 and 7 with less efficiency.50 Furthermore, caspase-8 is considered a molecular switch for pyroptosis.51–53 Previous studies have demonstrated that caspase-8 triggers the formation of ASC specks, activation of caspase-1, and subsequent activation of the NLRP3 inflammasome, leading to pyroptosis in bone marrow-derived macrophages (BMDMs) and intestinal epithelial cells.51 Sarhan et al found that caspase-8 could cleave both GSDMD and GSDME, resulting in pyroptosis in murine macrophages during Yersinia infections.52 A recent study claimed that caspase-8 directly cleaves GSDMD to trigger pyroptosis, which provides the host defense against Yersinia infections.53 Therefore, caspase-3/7/8 can also mediate pyroptosis.
Gasdermins: Executioners of Pyroptosis
Gasdermins are a family of pore-forming effector proteins that increase membrane permeabilization and induce pyroptosis. The gasdermin family is encoded by six paralogous genes in humans: GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and PJVK, and nine genes in mice: Gsdma 1–3, Gsdmc 1–4, Gsdme and pjvk.10 The gasdermin family is composed of a cytotoxic N-terminal domain and a C-terminal inhibitory domain, and is linked by a long loop in the middle.10 This structure is shared by all gasdermin family members except for PJVK, which has a truncated C-terminal domain.54
GSDMD is an executor of pyroptosis.9 Activated caspase-1/4/5/11 acts on the aspartic acid site on the circle to cleave GSDMD into two fragments: a 31-kDa GSDMD-NT fragment with an inherent pore-forming function and a 22-kDa GSDMD C-terminal (GSDMD-CT) fragment, which has an autoinhibitory role in the GSDMD-NT fragment.9,10 The GSDMD-NT fragment can target, using cardiolipin and phosphoinositides, the membrane to destroy its structure by forming pores with a diameter of 10–14nm and inducing pyroptosis.55 Recent studies found that caspase-8 can cleave GSDMD into GSDMD-NT and GSDMD-CT fragments and induce pyroptosis.43,52,53 The pathway leading to GSDMD activation extends beyond the caspase family, and emerging evidence has demonstrated that neutrophil elastase can cleave GSDMD in active neutrophils.56,57 Although the site cleaved by neutrophil elastase is different from that of caspases, GSDMD still generates a fragment with pore-forming functions.57
An increasing number of gasdermin family members have been found to play a role in pyroptosis. GSDME cleaved by caspase-3 can induce pyroptosis.47,48 Rogers et al revealed that caspase-3 cleaved GSDME at Asp270 and divided into N-terminal and C-terminal fragments. The N-terminus of GSDME has the same function as GSDMD-NT, which has intrinsic pore-forming activity and induces pyroptosis in apoptotic cells, but is not scavenged.48 Wang et al claimed that GSDME cleaved by caspase-3 can switch from apoptosis, induced by tumor necrosis factor alpha (TNF-α) or chemotherapy drugs, to pyroptosis, and GSDME-/- mice are protected from chemotherapy-induced tissue damage.58 A study of pyroptosis in teleosts suggested that GSDME, which is the only gasdermin family member present in teleosts, triggers pyroptosis, and GSDME is cleaved by caspase-1/3/7, all of which occur at the FEVD site in the linker region of GSDME.50
GSDMA acts as an effector of epithelial pyroptosis during Streptococcus pyogenes infections, and the cysteine protease SpeB of Streptococcus pyogenes cleaves GSDMA in the linker region after Gln246 and generates N-terminus of GSDMA, triggering pyroptosis.11
In 2020, Feng et al claimed that GSDMB was cleaved by lymphocyte-derived granzyme A to induce pyroptosis in natural killer cells and cytotoxic T lymphocytes.12 GSDMB is highly expressed in various tissues, especially in the digestive tract epithelia, including derived tumors. GSDMB cleaved by granzyme A-induced pyroptosis promotes tumor clearance in mice, and may enhance anti-tumor immunity.12
Hou et al revealed that the N-terminus of GSDMC also induces pyroptosis, and GSDMC is cleaved by caspase-8.59 Phosphorylated signal transducer and activator of transcription 3 interacts with programmed death ligand 1 and promotes its nuclear translocation, which upregulates GSDMC transcription under hypoxic conditions. GSDMC is cleaved by caspase-8 under TNF-α treatment, thereby switching from apoptosis to pyroptosis and facilitating tumor necrosis.13 Although the N-terminus of GSDMA, GSDMB, GSDMC, and GSDME also drive cells to undergo pyroptosis, GSDMD is still the best characterized executor of pyroptosis.
Canonical (Caspase-1-Mediated) Pyroptosis Pathway
The canonical pyroptosis signaling pathway is triggered by inflammasomes, mainly the NLR family and pyrin protein families. Inflammasomes can be activated by different stimulants, such as PAMPs or DAMPs.7 Caspase-1 is triggered by the inflammasome and pro-IL-1β/pro-IL-18 is converted to IL-1β/IL-18 via caspase-1. Simultaneously, caspase-1 cleaves GSDMD and releases the GSDMD-NT.5 The GSDMD-NT fragment forms pores on the cytomembrane and promotes the secretion of the inflammatory cytokines, IL-1β, IL-18, IL-1α and HMGB1, resulting in plasma membrane rupture, cellular swelling, and eventually cell death (pyroptosis) (Figure 1).9
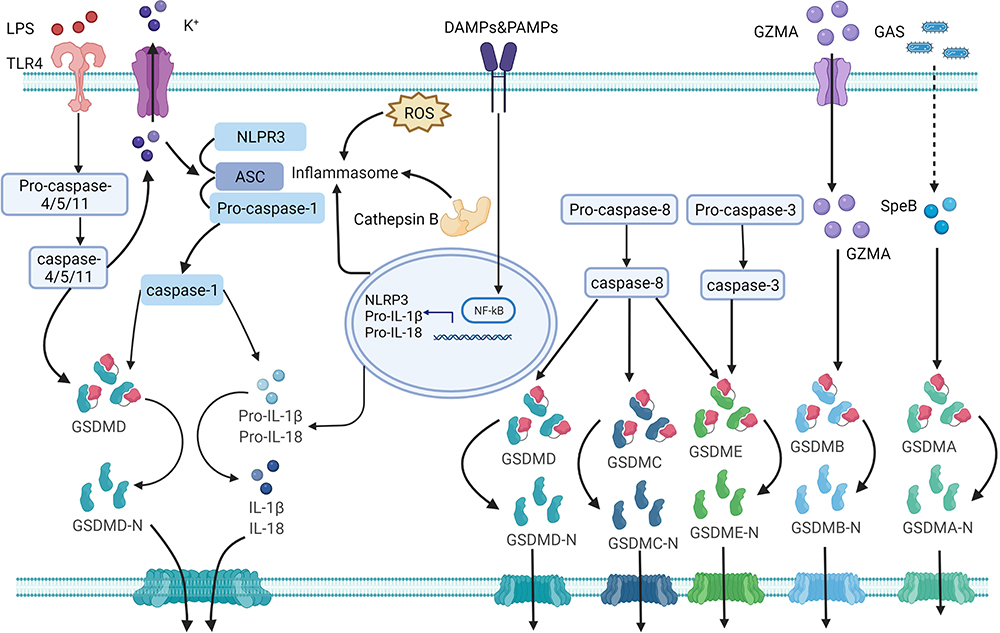 |
Figure 1 The mechanism of pyroptosis. Pyroptosis involves canonical, non-canonical, and other pathways. NLRP3 inflammasome activation requires DAMPs or PAMPs to trigger the NF-κB-mediated upregulation the transcription of of NLRP3, pro-IL-1β and pro-IL-18. The formation and modification of NLRP3 inflammasomes by potassium efflux, the generation of reactive oxygen species (ROS), and cathepsin B released by lysosomal damage. Caspase-1 is triggered by the inflammasome and pro-IL-1β/pro-IL-18 is converted to IL-1β/IL-18 via caspase-1. Simultaneously, caspase-1 cleaves GSDMD and releases the GSDMD-NT. The GSDMD-NT fragment forms pores on the cytomembrane and promotes the secretion of the inflammatory cytokines, IL-1β and IL-18, resulting in plasma membrane rupture, cellular swelling, and eventually pyroptosis. LPS from gram-negative bacteria can access the cell cytoplasm via TLR4 receptor-mediated endocytosis and bind directly to caspase-4/5/11. Activated caspase-4/5/11 cleaves GSDMD into a GSDMD-NT fragment to induce cell membrane pore formation and initiate pyroptosis. This process can also cause NLRP3 activation by potassium efflux via nonselective pores. The cysteine protease (SpeB) from group A Streptococcus (GAP) can cleave GSDMA, triggering pyroptosis. The cleavage of GSDMA by the cysteine protease (SpeB), triggers pyroptosis during Streptococcus pyogenes infections. Active caspase-3 cleave GSDME and release GSDME-NT, thereby triggers pyroptosis. Caspase-8 can cleave GSDMD, GSDME, and GSDMC, to trigger pyroptosis. |
Non-Canonical (Caspase-4/5/11-Mediated) Pyroptosis Pathway
The non-canonical pyroptosis pathway was initially thought to be mediated by caspase-4/5/11. LPS from gram-negative bacteria can access the cell cytoplasm via CD14/TLR4/MD2 receptor-mediated endocytosis or bacterial outer membrane vesicles and bind directly to caspase-4/5/11.40,60 The CARD in caspase-4/5/11 recognizes lipid A, which is the most conserved region in LPS, and induces oligomerization of caspases.9 Activated caspase-4/5/11 cleaves GSDMD into a GSDMD-NT fragment to induce cell membrane pore formation and initiate pyroptosis. However, caspase-4/5/11-mediated pyroptosis cannot release IL-1β and IL-18, in contrast to the caspase-1-mediated classical pyroptosis pathway. Interestingly, caspase-4/5/11 triggers non-canonical NLRP3 inflammasome activation through a cell-intrinsic process to promote IL-1β and IL-18 secretion (Figure 1).41,42,61
Other Pyroptosis Pathways
In 2017, it was discovered that the caspase-3/GSDME pathway induces pyroptosis.48 GSDME, cleaved by caspase-3 during apoptosis, mediates the switch to pyroptosis. Furthermore, caspase-3/GSDME-mediated pyroptosis is involved in chemotherapy-induced tissue damage, providing a new target for the treatment of cancer.58 A recent study revealed that cleavage of GSDME by caspase-1/3/7 is the only pathway that induces pyroptosis in teleosts.50
The mechanism underlying caspase-8-mediated pyroptosis is complex. Caspase-8 induces pyroptosis by activating the NLRP3 inflammasome.51 Another report claims that caspase-8 cleaves GSDMD and releases GSDMD-NT to trigger pyroptosis.53 Interestingly, a recent study has demonstrated that caspase-8 could cleave both GSDMD and GSDME and release their N-terminal fragments, thus inducing pyroptosis.52 The cleavage of GSDMA by the cysteine protease SpeB triggers pyroptosis during Streptococcus pyogenes infections.11 GSDMB cleaved by granzyme A as an effector of pyroptosis mainly occurs in natural killer cells and cytotoxic T lymphocytes and is involved in the pathogenesis of gastrointestinal tumors.12 Furthermore, caspase-8/GSDMC mediates a non-classical pyroptosis pathway in cancer cells, resulting in tumor necrosis (Figure 1).13
Role of Pyroptosis in Respiratory Diseases
Emerging evidence has revealed that pyroptosis is a key process for various diseases, including autoimmune diseases (eg cryptothermal protein-associated cycle syndrome, rheumatoid arthritis, and Crohn’s disease),62,63 nervous system diseases (eg Alzheimer’s disease and Parkinson’s disease),64,65 metabolic diseases (eg gout and type II diabetes),66,67 atherosclerosis,68 and cancer (eg colon cancer, breast cancer and melanoma).69,70 The role of pyroptosis in respiratory diseases has received considerable attention. Pyroptosis is relevant to the pathogenesis of respiratory diseases, whether as a positive or negative regulator. Therefore, we illuminate the role of pyroptosis in several respiratory diseases, including COPD, asthma, acute respiratory distress syndrome (ARDS), pulmonary fibrosis, lung cancer, and pulmonary infection.
COPD
COPD is a heterogeneous disease characterized by irreversible airflow obstruction, involving chronic inflammation of the small airways and lung parenchyma, which causes small airway fibrosis and emphysema.71 COPD is predicted to become the third leading cause of death and the fifth leading cause of economic burdens worldwide.72 Despite intense research efforts focused on the pathogenesis and therapy of COPD, the pathogenesis of COPD has not been fully elucidated, and only a few therapies can lower mortality rates and exacerbations in patients with COPD. Infection, exposure to toxic particles, air pollution and cigarette smoke (CS) are the major causes of COPD.
CS is a major cause of COPD worldwide. Increased inflammasome activation, ASC speck accumulation, and IL-1β expression are observed in the lungs of patients with COPD, as well as in experimental models including murine models of COPD.73–75 Furthermore, the NLRP3 inflammasome was upregulated in an in vitro model of COPD exacerbation, and it may serve as a novel biomarker in the diagnosis of COPD exacerbation and as a new target for therapies.76 However, another report revealed that NLRP3 inflammasome had no correlation with the severity of stable COPD patients, possibly due to the increased expression of the inflammatory and inflammasome inhibitory molecules IL-37 and NALP7.77 These studies indicate that inflammasome activation and expression of IL-1β may play important roles in the pathogenesis of COPD, whereas the role of pyroptosis in the pathogenesis of COPD has been poorly studied.
A recent study indicated that CS-induced pyroptosis occurs via the ROS/NLRP3 inflammasome pathway in bronchial epithelial cells. Moreover, the caspase-1 inhibitor, VX-765, can inhibit pyroptosis in bronchial epithelial cells, and the ROS inhibitor, N-acetyl-L-cysteine, inhibits the activation of NLRP3 inflammasome and production of IL-1β and IL-18.78 Emerging evidence has revealed that triggering receptor expressed on myeloid cells (TREM-1) is highly expressed in COPD mice and promotes lung injury and inflammation via activation of NLRP3 inflammasome-mediated pyroptosis. Furthermore, inhibition of TREM-1 notably improved the injury in lung tissues of COPD mice and suppressed the activation of NLRP3 inflammasome and pyroptosis, which may provide a novel therapeutic target for COPD treatment.79 Some studies found that inhibition of pyroptosis could ameliorate CS-induced COPD.80,81 For example, (-)-Epicatechin ameliorated CS-induced lung inflammation via inhibiting ROS/NLRP3 inflammasome-mediated pyroptosis in rats with COPD.80 Exosomes derived from adipose-derived stem cells could inhibit alveolar macrophages pyroptosis, thereby attenuated lung injury and inflammation caused by CS.81
Asthma
Asthma is a common chronic inflammatory airway disease that results from the interaction between inflammatory cells (eg mast cells, eosinophils, neutrophils, and T cells) and inflammatory cytokines.82 It is characterized by airway hyper-responsiveness, airway inflammation, airway remodeling, and reversible airflow limitation due to occupational or environmental exposure to microbes, industrial products, and other allergens. In patients with asthma, clumps of epithelial cells in the sputum and increased epithelial cells in bronchoalveolar lavage fluid (BALF) indicate that epithelial cell sloughing is a pathological characteristic of asthma.83,84 Furthermore, recent studies have demonstrated pyroptosis in experimental models of asthma and in cell lines.20,85–87 Therefore, epithelial cell pyroptosis may represent a pathogenic mechanism contributing to inflammatory injury of airway epithelia. Toluene diisocyanate-induced pyroptosis in bronchial epithelial cells is mediated by the NLRP3 inflammasome, resulting in cytomembrane fracture and release of inflammatory mediators. It is observed in asthmatic mice; it enhances airway sensitivity and responsiveness, and causes continuous airway inflammation. MCC950, a specific NLRP3 inhibitor, can inhibit pyroptosis in bronchial epithelial cells and asthmatic mice and slow the progression of asthma.85 Der f1‑induced pyroptosis in bronchial epithelial cells through the NLRP3 inflammasome pathway plays a key role in asthma pathogenesis and airway remodeling, and a caspase-1 inhibitor (Z‑YVAD‑FMK) inhibits the release of IL-1β and subsequently, Der f1-induced pyroptosis.20 Simultaneously, a new study suggests that PARK2, a Parkinson’s disease-associated gene that is involved in house dust mite-induced pyroptosis in BEAS-2B cells negatively regulates NLRP3 protein via ubiquitination; therefore, PARK2 is a negative upstream regulator of the NLRP3 inflammasome.86 In addition, a recent study has shown that pyroptosis plays an important role in promoting airway remodeling in the pathogenesis of asthma.88
GSDMB is highly expressed in differentiated bronchial epithelial cells, and cleavage of GSDMB by caspase-1 induces pyroptosis. Furthermore, multiple coding variants in the GSDMB gene in the 17q21 locus are associated with reduced asthma risk, and the splicing variant, rs11078928, abolishes the pyroptotic activity of GSDMB. This implicates GSDMB-mediated epithelial cell pyroptosis in the pathogenesis of asthma and provides a potential asthma therapy targeting pyroptosis.87 These results confirm that targeting inflammasomes and pyroptosis can provide a new direction for the treatment of asthma.
Acute Lung Injury (ALI)/ARDS
ALI/ARDS as acute diffuse pulmonary inflammation is characterized by hypoxemia, diffuse alveolar injury, and acute respiratory failure, leading to an increase in pulmonary microvascular endothelial permeability, pulmonary edema, and the reduction of lung tissue involved in ventilation.89 Its high mortality rate has a significant impact on public health.90 The inflammasome pathway and its downstream cytokines play critical roles in ARDS development.91 IL-18 and caspase-1 promote the development of ARDS. Moreover, IL-18 is increased in circulation, which correlates with disease severity and mortality in the MICU.91 Notably, pyroptosis in alveolar macrophages plays an essential role in the pathogenesis of ALI/ARDS.92–94 LPS recognized by TLR4 not only activates the NLRP3 inflammasome and subsequently promotes the release of IL-1β, but also upregulates IL-1R1 expression on alveolar macrophage surface through the MyD88/NF-κB dependent pathway. The upregulated expression of IL-1R enables the sensitization of alveolar macrophages to IL-1β and causes pyroptosis. In summary, the LPS-TLR4 pathway triggers alveolar macrophages and augments ALI/ARDS via secondary upregulation of IL-1β-IL-1RI signaling.92 NLRP3 inflammasome-mediated pyroptosis in macrophages may also play a crucial role in the pathogenesis of ALI/ARDS via the p38 MAPK signal pathway and blocking this pathway can ameliorate ALI by inhibiting macrophage pyroptosis and converting pyroptosis to apoptosis.93 Interestingly, extracellular histones promote alveolar macrophages pyroptosis through the NLRP3/caspase-1 pathway, which in turn aggravates lung inflammation in ARDS.94 Phospholipid scramblase 4 (PLSCR4) is a member of single-pass transmembrane proteins that can transfer phospholipids (PS) from the inside to the outside of the cell membrane in a Ca2+-dependent manner.95 PLSCR4 can reduce the binding of the pyroptosis executive protein GSDMD-NT to PS and the formation of pyroptosis pores, thereby reducing the degree of pyroptosis. Furthermore, PLSCR4 is mainly regulated by P62280.95 Recently, Kerr et al have shown that extracellular vesicle-mediated inflammasome and pyroptosis play a role in traumatic brain injury (TBI)-induced ALI model in mice and patients, and ASC can be used as an excellent biomarker for the diagnosis of TBI-induced ALI. In addition, anti-ASC treatment significantly reduces inflammasome activation and TBI-induced ALI.96,97 Therefore, these studies provide new therapeutic strategies for controlling ALI/ARDS. Interestingly, NecroX-5, an inhibitor of necrosis, alleviated lung inflammation in murine models with ALI/ARDS by inhibiting the NF-κB signaling pathway and inhibiting activation of the NLRP3 inflammasome. We speculate that there is crosstalk between necrosis and pyroptosis.98 Therefore, a comprehensive investigation of pyroptosis and its correlation with other cell death pathways would provide a novel therapeutic strategy for the treatment of ARDS.
Pulmonary Fibrosis
Pulmonary fibrosis is characterized by progressive and irreversible destruction of the lungs, leading to progressive decline in lung function. The mechanism underlying pulmonary fibrosis is not yet fully understood. Asbestosis, silica, CS, particulate matter, and bleomycin (BLM) are associated with pulmonary fibrosis.99 Many studies indicate that inflammasomes, such as the NLRP3 and AIM2 inflammasomes, which are tightly associated with pyroptosis, play critical roles in the pathogenesis of pulmonary fibrosis. Several published studies have shown that the NLRP3 inflammasome is involved in lung fibrosis in both experimental models and patients. The NLRP3 inflammasome promotes the release of IL-1β, which can promote fibroblast-to-myofibroblast differentiation and activate the TGF-β1/Smads signal pathway.100–103 NLRP3 inflammasome activation in alveolar epithelial cells promotes myofibroblast differentiation of lung-resident mesenchymal stem cells, and MCC950 attenuates BLM-induced pulmonary fibrosis.104 Moreover, a study has confirmed that NLRP3 inflammasome activation in aged mice increases susceptibility to pulmonary fibrosis.103 AIM2 inflammasome activation has been found in peripheral blood mononuclear cells (PBMCs) from patients with idiopathic pulmonary fibrosis (IPF), which leads to the activation of caspase-4, which induces the release of IL-1α responsible for the release of transforming growth factor beta 1 (TGF-β1) from PBMCs.105 Reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) participates in the destruction of lysosomes and promotes the production of ROS. ROS production and cathepsin B are directly involved in NLRP3 inflammasome activation. Moreover, N-acetyl-L-cysteine can attenuate NLRP3 inflammasome activation and lung fibrosis.106 Treatment targeting NLRP3, AIM2, caspase-1, IL-1β, and ROS can effectively alleviate pulmonary fibrosis, which may also serve to inhibit lung injury and fibrosis. Another study found that the activation of NLRP3 inflammasome was impaired in patients with IPF, and the level of IL-18 in the BALF was lower than that in control patients. This may be because the innate immune response of patients with IPF is impaired, including reduced function of macrophages to kill pathogenic bacteria.107 While the NLRP3 inflammasome was activated, the levels of NLRP3 and caspase-1 were increased in patients with RA-UIP, and IL-1β and IL-18 were significantly elevated in the BALF and BALF macrophages.107 Autophagy and NLRP3 inflammasomes are activated in patients with fibrosis and are positively correlated with oxidation in vivo.108 Angiotensin-II (Ang II) promotes intercellular ROS and NOX4 levels, leading to NLRP3 inflammasome activation and stimulates collagen synthesis through the NLRP3/caspase-1/IL-1β pathway. Furthermore, autophagy suppresses NLRP3 inflammasome activation by eliminating ROS and alleviating mitochondrial dysfunction. Taken together, autophagy alleviates pulmonary fibrosis by regulating NLRP3 inflammasome activation induced by Ang II-mediated via redox balance modulation.108 NLRP3 inflammasome-mediated pyroptosis in macrophages promotes collagen synthesis and the progression of pulmonary fibrosis in BLM-induced pulmonary fibrosis models, and lycorine ameliorates pulmonary fibrosis by inhibiting the NLRP3 inflammasome and pyroptosis.24 Another study reported that bone marrow-derived mesenchymal stem cells can inhibit pyroptosis triggered by NLRP3/caspase-1 in silica-induced pulmonary fibrosis.109 Emerging studies have shown that AIM2 inflammasome activation and pyroptosis in macrophages are involved in the pathogenesis of radiation-induced pulmonary fibrosis. Andrographolide ameliorated radiation-induced pulmonary fibrosis by inhibiting AIM2 inflammasome-mediated pyroptosis in macrophages, thus, identifying andrographolide as a new potential protective agent against for radiation-induced pulmonary fibrosis.110 These studies demonstrated that inflammasomes and induced pyroptosis participate in the pathogenesis of pulmonary fibrosis, and would provide new targets for patients with pulmonary fibrosis.
Pulmonary Infection
Macrophage pyroptosis is a protective mechanism against Klebsiella pneumoniae infection and facilitates the clearance of bacteria.111,112 Pyroptosis mediated by caspase-11 can promote neutrophil recruitment and bacterial clearance against K. pneumoniae infection, and caspase-11 knockout blocks this effect.112 However, different clinical strains of K. pneumoniae have different mechanisms of survival within macrophages. The A28006 strain induced macrophage pyroptosis and high IL-1β production, which is easily cleared; in contrast, the A54970 strain induced high IL-10 production, which inhibited macrophage pyroptosis to evade host defense, resulting in the dissemination of bacteria.111 Studies on Salmonella typhimurium and Legionella pneumophila have revealed that bacteria flagellin is an agonist of NLRC4 inflammasome-mediated pyroptosis in macrophages, which restricts bacterial replication in vivo. However, Salmonella typhimurium avoids innate immune mechanisms by evading NLRC4 inflammasome-mediated pyroptosis.113,114 NLRC4 inflammasome-mediated pyroptosis plays a protective role during Burkholderia pseudomallei infection. Knockdown of NLRC4 in mice causes high susceptibility to bacterial infections due to reduced pyroptosis and an increased bacterial burden. Moreover, IL-18 cleaved by the NLRP3 inflammasome protects against B. pseudomallei lung infection, due to its ability to increase the production of interferon (IFN-γ), whereas IL-1β is deleterious because of excessive recruitment of neutrophils to the lung.115 Wang et al analyzed the protective role of the canonical and noncanonical pyroptosis pathways during B. pseudomallei infection. Caspase-1 triggers pyroptosis, mainly in infected macrophages, and controls the release of IL-18. In contrast, caspase-11-mediated pyroptosis mainly occurs in infected lung epithelial cells.116 In summary, pyroptosis is an efficient effector mechanism that restricts bacterial growth and dissemination during B. pseudomallei infection. NLRC4 inflammasome-mediated pyroptosis plays different roles during Pseudomonas aeruginosa infection, depending on host conditions. Flagella-expressing P. aeruginosa can activate the NLRC4 inflammasome and promote the secretion of IL-1β/IL-18 by macrophages in acute P. aeruginosa pneumonia, and NLRC4 inflammasome signaling not only impairs bacterial clearance but is also involved in increased mortality. Strategies limiting NLRC4 inflammasome activation have limited the pathological consequences of acute P. aeruginosa pulmonary infection.117 However, NLRC4 inflammasome is failed to activate by clinical isolates of P. aeruginosa from chronically infected cystic fibrosis patients during both stable and exacerbation infection, which indicated that P. aeruginosa-activated inflammasomes are not involved in cystic fibrosis pulmonary exacerbations.118 Emerging evidence revealed that neutrophils pyroptosis were essential for host protection during P. aeruginosa lung infection in the absence of NOX2.119 Thus, specifically target neutrophil pyroptosis are likely to be useful for curing highly intractable P. aeruginosa infections. Acinetobacter baumannii-induced pyroptosis contributes to the lung damage and high mortality in a pneumonia mouse model. Interestingly, caspase-1/11 knockout played a protective role in mice infected with A. baumannii. The hLF (1-11) peptide improves the survival of A. baumannii pneumonia in mice by inhibiting caspase-1-mediated pyroptosis.120 Another study claimed that A. baumannii infection induces the activation of multiple cell death pathways, such as pyroptosis and necrosis, and that type I IFN exerts a protective effect against A. baumannii infection by regulating histone modifications.121 GSDME-mediated pyroptosis plays a lethal role in H7N9 virus infection; knockdown of GSDME switched lung epithelial cell pyroptosis to apoptosis upon H7N9 virus infection, and protected mice from an H7N9 virus lethal infection.122 Therefore, specifically targeting GSDME-mediated pyroptosis is a potential therapeutic approach for H7N9 virus infection. Therefore, pyroptosis plays a crucial role as a positive or negative regulator in the pathogenesis of pulmonary infection. It is a potential therapeutic strategy for pulmonary infection, even in multi-drug-resistant bacterial infections.
Lung Cancer
Lung cancer is the most common cancer and leading cause of cancer-related mortality worldwide. GSDMD expression was increased in non-small cell lung cancer (NSCLC) and was associated with tumor metastasis.123 Moreover, high GSDMD expression implies a poor prognosis in lung adenocarcinoma but not in squamous cell carcinoma.123 GSDMD knockdown alleviated tumor proliferation by switching NLRP3/caspase-1-mediated-pyroptosis to apoptosis and suppressing the epidermal growth factor receptor/protein kinase pathway in NSCLC.123 Therefore, GSDMD may be used as an independent prognostic biomarker for lung adenocarcinoma. Furthermore, GSDMD plays an essential role in tumor immunity through GSDMD, contributing to cytotoxic T lymphocyte-mediated killing.124 Emerging evidence has revealed that caspase-3/GSDME-mediated pyroptosis is involved in the treatment of NSCLC with chemotherapy.58,125–127 Paclitaxel and cisplatin, as representative chemotherapeutic drugs for lung cancer, could induce apoptosis in A549 cells, but caspase-3/GSDME-induced pyroptosis was observed in some dying cells. Furthermore, cisplatin induced higher levels of pyroptosis in A549 cells than paclitaxel did, and GSDME knockdown significantly inhibited cisplatin-induced pyroptosis, which revealed that cisplatin may provide additional advantages in the treatment of lung cancers with high levels of GSDME expression.125 Another study explored the effect of GSDME-mediated pyroptosis in the treatment of NSCLC with cisplatin and claimed that GSDME-mediated pyroptosis enhances cisplatin sensitivity to NSCLC, and low GSDME expression in tumor tissues indicates a short survival time and high mortality rate after platinum treatment.126 GSDME-mediated pyroptosis promoted tumor CD3+T cell infiltration via GSDME-NT pores.126 GSDME is silenced in most cancer cells due to methylation of the GSDME gene. The combination of methylation inhibitors and chemotherapy agents can convert apoptosis into caspase-3/GSDME-mediated pyroptosis and significantly improve the efficacy of chemotherapy through immune activation by pyroptosis.127 Therefore, GSDME-mediated pyroptosis may be conducive to improve the chemotherapy sensitivity of lung cancer and may provide a new potential target for the development of lung cancer immunotherapy. miR-556-5p is a small non-coding RNA that is significantly upregulated in patients with cisplatin-resistant NSCLC.128 Silencing of miR-556-5p enhances the sensitivity of cisplatin by inducing NLRP3-mediated pyroptosis in cisplatin-resistance NSCLC cells.128 Long non-coding RNA X-inactive specific transcript (LncRNA-XIST) is an oncogene in various cancers and is aberrantly overexpressed in NSCLC tissues or cell lines, which promotes NSCLC cell proliferation, invasion, and metastasis.129,130 Downregulation of lncRNA-XIST can promote ROS production and NLRP3 inflammasome activation to induce pyroptosis.129,130 Moreover, miR335 is the downstream target of lncRNA-XIST, and lncRNA-XIST regulates superoxide dismutase 2 (SOD2) levels by targeting miR-335. Taken together, knockdown of lncRNA-XIST suppressed NSCLC development by inducing miR-335/SOD2/ROS-mediated pyroptosis.129 Xu et al revealed that lncRNA-XIST was upregulated in NSCLC cells previously treated with cisplatin. Interestingly, knockdown of lncRNA-XIST promoted chemosensitivity to cisplatin by inducing pyroptosis in NSCLC cells.130 Taken together, these findings reveal that pyroptosis may participate in the regulation of chemotherapeutic sensitivity in NSCLC, which provides new therapeutic strategies to overcome chemo-resistance in patients with NSCLC in clinical setting. Additional studies have found that triggering pyroptosis is a novel approach for treating lung cancer. For instance, cucurbitacin B inhibited NSCLC in vivo and in vitro by triggering TLR4/NLRP3/GSDMD-dependent pyroptosis, which supports cucurbitacin B as a potential therapeutic agent for NSCLC.131 Simvastatin inhibited proliferation and metastasis in NSCLC via pyroptosis mediated by the NLRP3/caspase-1 pathway.132 Polyphyllin VI (PPVI) activated the NLRP3 inflammasome and induced the apoptosis-to-pyroptosis switch in NSCLC, which supported that PPVI might be a new candidate for the future treatment of NSCLC.133 L61H10, a heterocyclic ketone derivative exerted anti-tumor effects in NSCLC, mainly through the switch of apoptosis-to-pyroptosis mediated by the NF-κB signaling pathway.134
Inhibitors of Pyroptosis
In recent years, great progress has been made in research on the molecules involved in the process of pyroptosis. Herein, we provide a brief introduction to the therapeutic strategies for the inhibition of pyroptosis (Table 1).
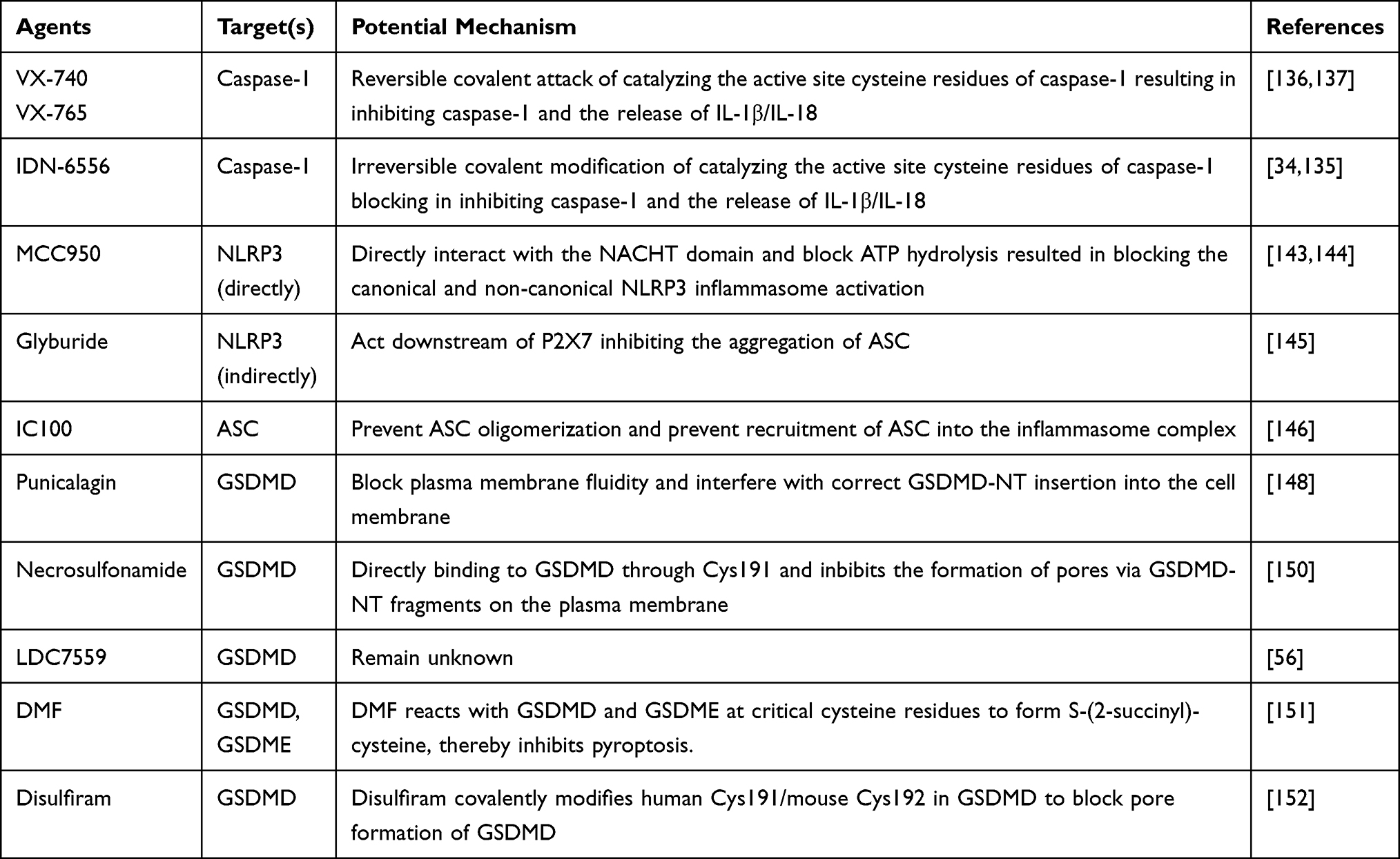 |
Table 1 Inhibitors of Pyroptosis and Their Targets |
Inhibitors of Proinflammatory Caspases
IDN-6556, VX-740 (pralnacasan), and its analog VX-765 are common caspase-1 Inhibitors.135 VX-740 and VX-765 are peptidomimetic drugs, which both catalyze the active site cysteine residues of caspase-1 by reversible covalent attack. IDN-6556 acts through irreversible covalent modification of catalyzing the active site cysteine residues, thus inhibiting caspase-1 and reducing the release of IL-1β/IL-18.34,135 VX-765 and VX-740 were effective in the treatment of RA and osteoarthritis in murine models, and they blocked the release of IL-1β and IL-18.136,137 Furthermore, VX765 reversed cognitive impairment and neuroinflammation in mouse models of AD.138 It reduces the area of myocardial infarction and maintains ventricular function in mice models.139 To date, three caspase-1 inhibitors (VX-740, VX-765 and IDN-6556) have entered clinical trials for RA, epilepsy, psoriasis, and chronic hepatitis C viral infection; however, IDN-6556 has been withdrawn from clinical trials because of undisclosed reasons.140–142 VX-765 was reported to alleviate airway inflammation in the mice models with asthma,21 and rescued the degree of emphysema in CS-exposed mice.78
NLRP3 Inflammasome Inhibitors
MCC950, a diarylsulfonylurea-containing compound, is the best-studied NLRP3 inhibitor; it is highly specific and selective. It inhibits NLRP3 inflammasome activation and IL-1β production by blocking ASC oligomerization, which is specific for the NLRP3 inflammasome, and does not affect the activation of other inflammasomes.143 A recent study revealed that MCC950 directly interacts with the NACHT domain and blocks ATP hydrolysis and can rescue airway remodeling and alleviate neutrophil airway inflammation in asthma murine models.21,144 Another study reported that MCC950 inhibits pyroptosis in human bronchial epithelial cells mediated by the NLRP3 inflammasome.85
Glyburide, a sulfonylurea drug, has recently been found to indirectly inhibit the activation of the NLRP3 inflammasome. Research has shown that glyburide inhibits PAMPs and DAMPs induced by NLRP3 inflammasome activation, while inhibiting the activation of caspase-1 and secretion of IL-1β, which acts downstream of P2X7, inhibiting the aggregation of ASC.145
IC100, a monoclonal antibody targeting ASC, acts extracellularly to prevent ASC oligomerization and recruitment into the inflammasome complex.146 A study found that IC100 inhibits the immune inflammatory response that drives the development and progression of experimental autoimmune encephalomyelitis.146 Anti-ASC treatment significantly suppresses inflammasome activation and TBI-induced ALI.96,97
Gasdermin Inhibitors
Punicalagin was the first compound to block pyroptosis and IL-1β release.147,148 Punicalagin blocks plasma membrane fluidity and interferes with correct GSDMD-NT insertion into the cell membrane.148 Moreover, a recent study suggested that punicalagin affects the activation of NLRC4 and NLRP1 inflammasomes, possibly by affecting membrane fluidity and blocking the delivery of inflammasome activators.149 In contrast, punicalagin has no effect on the generation of GSDMD-NT and activation of NLRP3 and AIM2 inflammasome.148
Necrosulfonamide is a cysteine-reactive drug that inhibits pyroptosis in murine macrophages and human monocytes.150 It directly binds to GSDMD through Cys191 and inhibits the formation of pores via GSDMD-NT fragments, thereby inhibiting the release of IL-1β. Necrosulfonamide is highly specific for GSDMD and has no effect on inflammasome activation and pyroptosis induced by the GSDME-NT terminal.150
LDC7559, a NET formation inhibitor based on the pyrazolo-oxazepane scaffold, binds to GSDMD and blocks neutrophil elastase-dependent pyroptosis in human neutrophils; however, its mechanism is unclear.56
Dimethyl fumarate (DMF) is an intermediate in the citric acid cycle, and is currently used in the treatment of multiple sclerosis. DMF reacts with GSDMD at critical cysteine residues to form S-(2-succinyl)-cysteine. GSDMD succination prevents activation of inflammatory caspases and inhibits pyroptosis. DMF protects against familial Mediterranean fever and experimental autoimmune encephalitis in mice by inhibiting pyroptosis.151 Furthermore, DMF inhibits GSDME-mediated pyroptosis through GSDME succination.151
Disulfiram, an inhibitor of acetaldehyde dehydrogenase, has been approved for the treatment of alcohol addiction. A recent study revealed that disulfiram covalently modified human Cys191/mouse Cys192 in GSDMD to block pore formation of GSDMD. Interestingly, disulfiram had no effect on the processing of GSDMD and IL-1β but abrogates GSDMD pore-formation.152
Potential Medicines for Pyroptosis in Respiratory Diseases
Cucurbitacin B is a natural triterpenoid derived from Cucurbitaceae plants. A previous study found that cucurbitacin B could directly interact with TLR4 to activate the NLRP3 inflammasome and increase mitochondrial ROS production to induce pyroptosis, subsequently inhibiting NSCLC in vivo and in vitro.131 Simvastatin, the most commonly used statin, can suppress proliferation and migration in NSCLC through caspase-1-mediated pyroptosis.132 PPVI, as the main saponin in Trillium tschonoskii Maxim, could activate the NF-κB pathway by increasing the generation of ROS, thereby activating the NLRP3 inflammasome. Moreover, PPVI induced an apoptosis-to-pyroptosis switch. Taken together, PPVI could inhibit the progression of NSCLC via pyroptosis.133 L61H10 is a heterocyclic ketone derivative, that exerts antitumor effects in NSCLC. L61H10 inhibited NF-κB signaling pathway and induced apoptosis-to-pyroptosis switch to exert anti-tumor effect.134 Andrographolide, an active component extracted and purified from Andrographis paniculata, which alleviated radiation-induced lung injury by inhibiting AIM2 inflammasome-mediated pyroptosis in macrophages.110 Resveratrol could suppress autophagic process and NLRP3 inflammasome activation, thereby alleviated particulate matter-induced lung inflammation and fibrosis.153 Lycorine could inhibit NLRP3 inflammasome and pyroptosis by targeting ASC pyrin domain, which exerted anti-fibrotic effect in BLM-induced pulmonary fibrosis.24 Scutellarin and Fluorofenidone attenuated pulmonary fibrosis via inhibiting NLRP3 inflammasome and NF-κB signaling pathway.154,155 NAC also alleviated pulmonary fibrosis by inhibiting the activation of NRLP3 inflammasome, but clinical trials have confirmed that it has no significant effect on IPF.106 Luteolin could regulate the frequency of Tregs and the levels of Treg derived IL-10, which suppressed pyroptosis mediated by caspase-11 in sepsis-induced ALI, thereby alleviated lung injury.156 -Epicatechin (EC), a type of flavonoid, which promoted ubiquitin-mediated Keap1 degradation by upregulating tripartite motif-containing protein 25 expression and enhanced the nuclear localization of Nrf2 protein. Moreover, EC notably inhibited the NLRP3 inflammasome and pyroptosis. Therefore, EC ameliorated cigarette smoke-induced lung inflammation by inhibiting ROS/NLRP3 inflammasome pathway in rats with COPD.80
Conclusions
In summary, pyroptosis is a new type of cell death and the executor is the gasdermin family, which is mainly regulated by the caspase-1-mediated canonical and the caspase-4/5/11-mediated noncanonical pathways. The occurrence and progression of pulmonary diseases are related to pyroptosis, and the NLRP3 inflammasome is the most well-studied mechanism. This review highlighted the significance of pyroptosis in the pathogenesis of COPD, asthma, ALI/ARDS, pulmonary fibrosis, lung cancer, and pulmonary infection. The role of pyroptosis as a key target for the treatment of pulmonary diseases has also been highlighted. In addition, we illustrated potential drugs that may become possible therapeutic targets for the management and treatment of respiratory diseases. These studies have revealed that targeting pyroptosis and inflammasomes also plays a role in the treatment of respiratory diseases, providing new ideas for the treatment of respiratory diseases.
At present, the study of pyroptosis is still in its infancy, and many mechanisms are still unclear. For instance, caspase-3/8 play an important role in apoptosis, but recent studies have shown that it is also involved in the process of pyroptosis. Therefore, when and what conditions does the transition between pyroptosis and apoptosis occur? Is pyroptosis an independent mode of cell death, or is it accompanied by other modes of cell death? In addition, there is crosstalk between pyroptosis and necroptosis, what is the mechanism? Such questions still need to be proven by a great deal of studies.
Several agents regulate the pyroptosis pathway. However, studies have largely focused on cancer treatment and autoimmune diseases. Research and development of medicines for the treatment of pulmonary diseases is ongoing. In the future, medicines targeting pyroptosis could be used to treat lung-related diseases. This will provide a better direction for the development of medications for pulmonary-related diseases.
Abbreviations
AIM2, absent in melanoma; ALI, acute lung injury; Ang II, angiotensin; ARDS, acute respiratory distress syndrome; ASC, apoptosis-associated speck like protein containing a caspase recruitment domain; ATP, adenosine triphosphate; BALF, bronchoalveolar lavage fluid; BLM, bleomycin; CARD, caspase recruitment domain; COPD, chronic obstructive pulmonary disease; CS, cigarette smoke; DAMPs, damage-associated molecular patterns; Der f1, Dermatophagoides farina 1; DMF, dimethyl fumarate; GSDMD, gasdermin D; GSDMD-CT, GSDMD C-terminal; GSDMD-NT, GSDMD N-terminal; HMGB1, high mobility group box 1; IFN, interferon; IL, interleukin; IPF, idiopathic pulmonary fibrosis; LncRNA-XIST, long non-coding RNA X-inactive specific transcript; LPS, lipopolysaccharide; LRR, leucine-rich repeat; MyD88, myeloid differentiation primary response 88; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor kappa B; NLR, nucleotide-binding oligomerization domain-like receptor; NLRC, NLR family CARD containing proteins; NLRP3, NLR family pyrin domain containing proteins; NOD, nucleotide-binding oligomerization domain; NOX, NADPH oxidase; NSCLC, non-small cell lung cancer; PAMPs, pathogen-associated molecular patterns; PBMCs, peripheral blood mononuclear cells; PRRs, pattern recognition receptors; PYD, pyrin domain; RA-UIP, rheumatoid arthritis-associated–usual interstitial pneumonia; ROS, reactive oxygen species; TBI, traumatic brain injury; TGF-β1, transforming growth factor beta 1; TNF-α, tumor necrosis factor alpha; TLR, Toll-like receptor; TREM-1, triggering receptor expressed on myeloid cells; TXNIP, thioredoxin-interacting protein; MAPK, mitogen-activated protein kinase; PLSCR4, Phospholipid scramblase 4; PS, phospholipids.
Author Contributions
TYS and JJL designed the entire subject. JJL, GQF, and NNT: literature collection. TYS and JJL wrote the article. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–114. doi:10.1016/S0966-842X(00)01936-3
2. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18(2):127–136. doi:10.1038/nrm.2016.149
3. Sauler M, Bazan IS, Lee PJ. Cell death in the lung: the apoptosis-necroptosis axis. Annu Rev Physiol. 2019;81:375–402. doi:10.1146/annurev-physiol-020518-114320
4. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26(13):R568–r572. doi:10.1016/j.cub.2016.02.019
5. Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev. 2011;243(1):206–214. doi:10.1111/j.1600-065X.2011.01044.x
6. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165(4):792–800. doi:10.1016/j.cell.2016.03.046
7. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. doi:10.1038/nri3452
8. Diamond CE, Khameneh HJ, Brough D, Mortellaro A. Novel perspectives on non-canonical inflammasome activation. ImmunoTargets Ther. 2015;4:131–141. doi:10.2147/ITT.S57976
9. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514
10. Broz P, Pelegrín P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20(3):143–157. doi:10.1038/s41577-019-0228-2
11. Deng W, Bai Y, Deng F, et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature. 2022;602(7897):496–502. doi:10.1038/s41586-021-04384-4
12. Zhou Z, He H, Wang K, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368(6494). doi:10.1126/science.aaz7548
13. Hou J, Zhao R, Xia W, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22(10):1264–1275. doi:10.1038/s41556-020-0575-z
14. Shen X, Wang H, Weng C, Jiang H, Chen J. Caspase 3/GSDME-dependent pyroptosis contributes to chemotherapy drug-induced nephrotoxicity. Cell Death Dis. 2021;12(2):186. doi:10.1038/s41419-021-03458-5
15. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
16. Han C, Yang Y, Guan Q, et al. New mechanism of nerve injury in Alzheimer’s disease: β-amyloid-induced neuronal pyroptosis. J Cell Mol Med. 2020;24(14):8078–8090. doi:10.1111/jcmm.15439
17. Wu XY, Li KT, Yang HX, et al. Complement C1q synergizes with PTX3 in promoting NLRP3 inflammasome over-activation and pyroptosis in rheumatoid arthritis. J Autoimmun. 2020;106:102336. doi:10.1016/j.jaut.2019.102336
18. Wen S, Deng F, Li L, Xu L, Li X, Fan Q. VX-765 ameliorates renal injury and fibrosis in diabetes by regulating caspase-1-mediated pyroptosis and inflammation. J Diabetes Investig. 2022;13(1):22–33. doi:10.1111/jdi.13660
19. Tan Y, Chen Q, Li X, et al. Pyroptosis: a new paradigm of cell death for fighting against cancer. J Exp Clin Cancer Res. 2021;40(1):153.
20. Tsai YM, Chiang KH, Hung JY, et al. Der f1 induces pyroptosis in human bronchial epithelia via the NLRP3 inflammasome. Int J Mol Med. 2018;41(2):757–764. doi:10.3892/ijmm.2017.3310
21. Kim RY, Pinkerton JW, Essilfie AT, et al. Role for NLRP3 inflammasome-mediated, IL-1β-dependent responses in severe, steroid-resistant asthma. Am J Respir Crit Care Med. 2017;196(3):283–297. doi:10.1164/rccm.201609-1830OC
22. Ghimire L, Paudel S, Jin L, Baral P, Cai S, Jeyaseelan S. NLRP6 negatively regulates pulmonary host defense in Gram-positive bacterial infection through modulating neutrophil recruitment and function. PLoS Pathog. 2018;14(9):e1007308. doi:10.1371/journal.ppat.1007308
23. Perlee D, de Beer R, Florquin S, van der Poll T, van ‘t Veer C, de Vos AF. Caspase-11 contributes to pulmonary host defense against Klebsiella pneumoniae and local activation of coagulation. Am J Physiol Lung Cell Mol Physiol. 2020;319(1):L105–l114. doi:10.1152/ajplung.00422.2019
24. Liang Q, Cai W, Zhao Y, et al. Lycorine ameliorates bleomycin-induced pulmonary fibrosis via inhibiting NLRP3 inflammasome activation and pyroptosis. Pharmacol Res. 2020;158:104884. doi:10.1016/j.phrs.2020.104884
25. Brusselle GG, Provoost S, Bracke KR, Kuchmiy A, Lamkanfi M. Inflammasomes in respiratory disease: from bench to bedside. Chest. 2014;145(5):1121–1133. doi:10.1378/chest.13-1885
26. Barbé F, Douglas T, Saleh M. Advances in Nod-like receptors (NLR) biology. Cytokine Growth Factor Rev. 2014;25(6):681–697.
27. Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11(5):395–402. doi:10.1038/ni.1864
28. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/S1097-2765(02)00599-3
29. Fink SL, Bergsbaken T, Cookson BT. Anthrax lethal toxin and Salmonella elicit the common cell death pathway of caspase-1-dependent pyroptosis via distinct mechanisms. Proc Natl Acad Sci U S A. 2008;105(11):4312–4317. doi:10.1073/pnas.0707370105
30. Vance RE. The NAIP/NLRC4 inflammasomes. Curr Opin Immunol. 2015;32:84–89. doi:10.1016/j.coi.2015.01.010
31. Levy M, Thaiss CA, Zeevi D, et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. 2015;163(6):1428–1443. doi:10.1016/j.cell.2015.10.048
32. Heilig R, Broz P. Function and mechanism of the pyrin inflammasome. Eur J Immunol. 2018;48(2):230–238. doi:10.1002/eji.201746947
33. Franklin BS, Latz E, Schmidt FI. The intra- and extracellular functions of ASC specks. Immunol Rev. 2018;281(1):74–87. doi:10.1111/imr.12611
34. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. 2018;17(9):688. doi:10.1038/nrd.2018.149
35. De Nardo D, De Nardo CM, Latz E. New insights into mechanisms controlling the NLRP3 inflammasome and its role in lung disease. Am J Pathol. 2014;184(1):42–54. doi:10.1016/j.ajpath.2013.09.007
36. Rivers-Auty J, Brough D. Potassium efflux fires the canon: potassium efflux as a common trigger for canonical and noncanonical NLRP3 pathways. Eur J Immunol. 2015;45(10):2758–2761. doi:10.1002/eji.201545958
37. He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–357. doi:10.1038/nature16959
38. Amaral EP, Riteau N, Moayeri M, et al. Lysosomal cathepsin release is required for NLRP3-inflammasome activation by mycobacterium tuberculosis in infected macrophages. Front Immunol. 2018;9:1427. doi:10.3389/fimmu.2018.01427
39. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278–286. doi:10.1038/nature10759
40. Ding J, Shao F. SnapShot: the noncanonical inflammasome. Cell. 2017;168(3):544–544.e541. doi:10.1016/j.cell.2017.01.008
41. Rühl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol. 2015;45(10):2927–2936. doi:10.1002/eji.201545772
42. Yang D, He Y, Muñoz-Planillo R, Liu Q, Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. 2015;43(5):923–932. doi:10.1016/j.immuni.2015.10.009
43. Orning P, Weng D, Starheim K, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362(6418):1064–1069. doi:10.1126/science.aau2818
44. Kayagaki N, Wong MT, Stowe IB, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341(6151):1246–1249. doi:10.1126/science.1240248
45. Boucher D, Monteleone M, Coll RC, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med. 2018;215(3):827–840. doi:10.1084/jem.20172222
46. Liu T, Yamaguchi Y, Shirasaki Y, et al. Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Rep. 2014;8(4):974–982. doi:10.1016/j.celrep.2014.07.012
47. Zeng CY, Li CG, Shu JX, et al. ATP induces caspase-3/gasdermin E-mediated pyroptosis in NLRP3 pathway-blocked murine macrophages. Apoptosis. 2019;24(9–10):703–717. doi:10.1007/s10495-019-01551-x
48. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. doi:10.1038/ncomms14128
49. Schwarzer R, Jiao H, Wachsmuth L, Tresch A, Pasparakis M. FADD and caspase-8 regulate gut homeostasis and inflammation by controlling MLKL- and GSDMD-mediated death of intestinal epithelial cells. Immunity. 2020;52(6):978–993.e976. doi:10.1016/j.immuni.2020.04.002
50. Jiang S, Gu H, Zhao Y, Sun L. Teleost gasdermin E is cleaved by Caspase 1, 3, and 7 and induces pyroptosis. J Immunol. 2019;203(5):1369–1382. doi:10.4049/jimmunol.1900383
51. Fritsch M, Günther SD, Schwarzer R, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575(7784):683–687. doi:10.1038/s41586-019-1770-6
52. Sarhan J, Liu BC, Muendlein HI, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A. 2018;115(46):E10888–e10897. doi:10.1073/pnas.1809548115
53. Demarco B, Grayczyk JP, Bjanes E, et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv. 2020;6(47). doi:10.1126/sciadv.abc3465
54. Ding J, Wang K, Liu W, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535(7610):111–116. doi:10.1038/nature18590
55. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153–158. doi:10.1038/nature18629
56. Sollberger G, Choidas A, Burn GL, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26). doi:10.1126/sciimmunol.aar6689
57. Kambara H, Liu F, Zhang X, et al. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep. 2018;22(11):2924–2936. doi:10.1016/j.celrep.2018.02.067
58. Wang Y, Gao W, Shi X, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103. doi:10.1038/nature22393
59. Zhang JY, Zhou B, Sun RY, et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 2021;31(9):980–997. doi:10.1038/s41422-021-00506-9
60. Vanaja SK, Russo AJ, Behl B, et al. Bacterial outer membrane vesicles mediate cytosolic localization of LPS and Caspase-11 activation. Cell. 2016;165(5):1106–1119. doi:10.1016/j.cell.2016.04.015
61. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–121. doi:10.1038/nature10558
62. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol. 2016;17(10):1176–1186. doi:10.1038/ni.3538
63. Finger JN, Lich JD, Dare LC, et al. Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem. 2012;287(30):25030–25037. doi:10.1074/jbc.M112.378323
64. Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer’s disease. Nat Immunol. 2015;16(3):229–236. doi:10.1038/ni.3102
65. Yan Y, Jiang W, Liu L, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. 2015;160(1–2):62–73. doi:10.1016/j.cell.2014.11.047
66. Sharma A, Tate M, Mathew G, Vince JE, Ritchie RH, de Haan JB. Oxidative stress and NLRP3-inflammasome activity as significant drivers of diabetic cardiovascular complications: therapeutic implications. Front Physiol. 2018;9:114. doi:10.3389/fphys.2018.00114
67. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–241. doi:10.1038/nature04516
68. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: its regulation and involvement in atherosclerosis. J Cell Physiol. 2018;233(3):2116–2132. doi:10.1002/jcp.25930
69. Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell. 2014;5(1):12–20. doi:10.1007/s13238-013-0001-4
70. Karki R, Man SM, Kanneganti TD. Inflammasomes and cancer. Cancer Immunol Res. 2017;5(2):94–99. doi:10.1158/2326-6066.CIR-16-0269
71. Rabe KF, Watz H. Chronic obstructive pulmonary disease. Lancet. 2017;389(10082):1931–1940. doi:10.1016/S0140-6736(17)31222-9
72. Rabe KF, Hurd S, Anzueto A, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2007;176(6):532–555. doi:10.1164/rccm.200703-456SO
73. Beckett EL, Stevens RL, Jarnicki AG, et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol. 2013;131(3):752–762. doi:10.1016/j.jaci.2012.11.053
74. Churg A, Zhou S, Wang X, Wang R, Wright JL. The role of interleukin-1beta in murine cigarette smoke-induced emphysema and small airway remodeling. Am J Respir Cell Mol Biol. 2009;40(4):482–490. doi:10.1165/rcmb.2008-0038OC
75. Franklin BS, Bossaller L, De Nardo D, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15(8):727–737. doi:10.1038/ni.2913
76. Nachmias N, Langier S, Brzezinski RY, et al. NLRP3 inflammasome activity is upregulated in an in-vitro model of COPD exacerbation. PLoS One. 2019;14(5):e0214622. doi:10.1371/journal.pone.0214622
77. Di stefano A, Caramori G, Barczyk A, et al. Innate immunity but not NLRP3 inflammasome activation correlates with severity of stable COPD. Thorax. 2014;69(6):516–524. doi:10.1136/thoraxjnl-2012-203062
78. Zhang MY, Jiang YX, Yang YC, et al. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci. 2021;269:119090. doi:10.1016/j.lfs.2021.119090
79. Wang L, Chen Q, Yu Q, Xiao J, Zhao H. TREM-1 aggravates chronic obstructive pulmonary disease development via activation NLRP3 inflammasome-mediated pyroptosis. Inflamm Res. 2021;70(9):971–980. doi:10.1007/s00011-021-01490-x
80. Tian X, Xue Y, Xie G, et al. (-)-Epicatechin ameliorates cigarette smoke-induced lung inflammation via inhibiting ROS/NLRP3 inflammasome pathway in rats with COPD. Toxicol Appl Pharmacol. 2021;429:115674. doi:10.1016/j.taap.2021.115674
81. Zhu Z, Lian X, Su X, Wu W, Zeng Y, Chen X. Exosomes derived from adipose-derived stem cells alleviate cigarette smoke-induced lung inflammation and injury by inhibiting alveolar macrophages pyroptosis. Respir Res. 2022;23(1):5. doi:10.1186/s12931-022-01926-w
82. GBD 2015 Chronic Respiratory Disease Collaborators. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706. doi:10.1016/S2213-2600(17)30293-X
83. Chanez P. Severe asthma is an epithelial disease. Eur Respir J. 2005;25(6):945–946. doi:10.1183/09031936.05.00038605
84. Fahy JV. Remodeling of the airway epithelium in asthma. Am J Respir Crit Care Med. 2001;164(10 Pt 2):S46–51. doi:10.1164/ajrccm.164.supplement_2.2106066
85. Zhuang J, Cui H, Zhuang L, et al. Bronchial epithelial pyroptosis promotes airway inflammation in a murine model of toluene diisocyanate-induced asthma. Biomed Pharmacother. 2020;125:109925. doi:10.1016/j.biopha.2020.109925
86. Ge X, Cai F, Shang Y, et al. PARK2 attenuates house dust mite-induced inflammatory reaction, pyroptosis and barrier dysfunction in BEAS-2B cells by ubiquitinating NLRP3. Am J Transl Res. 2021;13(1):326–335.
87. Panganiban RA, Sun M, Dahlin A, et al. A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. J Allergy Clin Immunol. 2018;142(5):1469–1478.e1462. doi:10.1016/j.jaci.2017.11.040
88. Chen X, Xiao Z, Jiang Z, Jiang Y, Li W, Wang M. Schisandrin B attenuates airway inflammation and airway remodeling in asthma by inhibiting NLRP3 inflammasome activation and reducing pyroptosis. Inflammation. 2021;44(6):2217–2231. doi:10.1007/s10753-021-01494-z
89. Ferguson ND, Fan E, Camporota L, et al. The Berlin definition of ARDS: an expanded rationale, justification, and supplementary material. Intensive Care Med. 2012;38(10):1573–1582. doi:10.1007/s00134-012-2682-1
90. Villar J, Sulemanji D, Kacmarek RM. The acute respiratory distress syndrome: incidence and mortality, has it changed? Curr Opin Crit Care. 2014;20(1):3–9. doi:10.1097/MCC.0000000000000057
91. Dolinay T, Kim YS, Howrylak J, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185(11):1225–1234. doi:10.1164/rccm.201201-0003OC
92. He X, Qian Y, Li Z, et al. TLR4-upregulated IL-1β and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci Rep. 2016;6:31663. doi:10.1038/srep31663
93. Li D, Ren W, Jiang Z, Zhu L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol Med Rep. 2018;18(5):4399–4409. doi:10.3892/mmr.2018.9427
94. Jiang P, Jin Y, Sun M, et al. Extracellular histones aggravate inflammation in ARDS by promoting alveolar macrophage pyroptosis. Mol Immunol. 2021;135:53–61. doi:10.1016/j.molimm.2021.04.002
95. Liu X, Wang D, Zhang X, et al. Effect and mechanism of phospholipid scramblase 4 (PLSCR4) on lipopolysaccharide (LPS)-induced injury to human pulmonary microvascular endothelial cells. Ann Transl Med. 2021;9(2):159. doi:10.21037/atm-20-7983
96. Kerr NA, de Rivero Vaccari JP, Abbassi S, et al. Traumatic brain injury-induced acute lung injury: evidence for activation and inhibition of a neural-respiratory-inflammasome axis. J Neurotrauma. 2018;35(17):2067–2076. doi:10.1089/neu.2017.5430
97. Kerr NA, de Rivero Vaccari JP, Umland O, et al. Human lung cell pyroptosis following traumatic brain injury. Cells. 2019;8(1):69. doi:10.3390/cells8010069
98. Fang XZ, Ge YL, Chen ZY, et al. NecroX-5 alleviate lipopolysaccharide-induced acute respiratory distress syndrome by inhibiting TXNIP/NLRP3 and NF-κB. Int Immunopharmacol. 2020;81:106257. doi:10.1016/j.intimp.2020.106257
99. Steele MP, Schwartz DA. Molecular mechanisms in progressive idiopathic pulmonary fibrosis. Annu Rev Med. 2013;64:265–276. doi:10.1146/annurev-med-042711-142004
100. Peeters PM, Perkins TN, Wouters EF, Mossman BT, Reynaert NL. Silica induces NLRP3 inflammasome activation in human lung epithelial cells. Part Fibre Toxicol. 2013;10:3. doi:10.1186/1743-8977-10-3
101. Zheng R, Tao L, Jian H, et al. NLRP3 inflammasome activation and lung fibrosis caused by airborne fine particulate matter. Ecotoxicol Environ Saf. 2018;163:612–619. doi:10.1016/j.ecoenv.2018.07.076
102. Hindman B, Ma Q. Carbon nanotubes and crystalline silica stimulate robust ROS production, inflammasome activation, and IL-1β secretion in macrophages to induce myofibroblast transformation. Arch Toxicol. 2019;93(4):887–907. doi:10.1007/s00204-019-02411-y
103. Stout-Delgado HW, Cho SJ, Chu SG, et al. Age-dependent susceptibility to pulmonary fibrosis is associated with NLRP3 inflammasome activation. Am J Respir Cell Mol Biol. 2016;55(2):252–263. doi:10.1165/rcmb.2015-0222OC
104. Ji J, Hou J, Xia Y, Xiang Z, Han X. NLRP3 inflammasome activation in alveolar epithelial cells promotes myofibroblast differentiation of lung-resident mesenchymal stem cells during pulmonary fibrogenesis. Biochim Biophys Acta Mol Basis Dis. 2021;1867(5):166077. doi:10.1016/j.bbadis.2021.166077
105. Terlizzi M, Molino A, Colarusso C, et al. Activation of the absent in melanoma 2 inflammasome in peripheral blood mononuclear cells from idiopathic pulmonary fibrosis patients leads to the release of pro-fibrotic mediators. Front Immunol. 2018;9:670. doi:10.3389/fimmu.2018.00670
106. Sun B, Wang X, Ji Z, et al. NADPH oxidase-dependent NLRP3 inflammasome activation and its important role in lung fibrosis by multiwalled carbon nanotubes. Small (Weinheim an der Bergstrasse, Germany). 2015;11(17):2087–2097. doi:10.1002/smll.201402859
107. Lasithiotaki I, Giannarakis I, Tsitoura E, et al. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur Respir J. 2016;47(3):910–918. doi:10.1183/13993003.00564-2015
108. Meng Y, Pan M, Zheng B, et al. Autophagy attenuates angiotensin II-induced pulmonary fibrosis by inhibiting redox imbalance-mediated NOD-like receptor family pyrin domain containing 3 inflammasome activation. Antioxid Redox Signal. 2019;30(4):520–541. doi:10.1089/ars.2017.7261
109. Zhao Q, Hao C, Wei J, Huang R, Li C, Yao W. Bone marrow-derived mesenchymal stem cells attenuate silica-induced pulmonary fibrosis by inhibiting apoptosis and pyroptosis but not autophagy in rats. Ecotoxicol Environ Saf. 2021;216:112181. doi:10.1016/j.ecoenv.2021.112181
110. Gao J, Peng S, Shan X, et al. Inhibition of AIM2 inflammasome-mediated pyroptosis by Andrographolide contributes to amelioration of radiation-induced lung inflammation and fibrosis. Cell Death Dis. 2019;10(12):957. doi:10.1038/s41419-019-2195-8
111. Codo AC, Saraiva AC, Dos Santos LL, et al. Inhibition of inflammasome activation by a clinical strain of Klebsiella pneumoniae impairs efferocytosis and leads to bacterial dissemination. Cell Death Dis. 2018;9(12):1182. doi:10.1038/s41419-018-1214-5
112. Wang J, Shao Y, Wang W, et al. Caspase-11 deficiency impairs neutrophil recruitment and bacterial clearance in the early stage of pulmonary Klebsiella pneumoniae infection. Int J Med Microbiol. 2017;307(8):490–496. doi:10.1016/j.ijmm.2017.09.012
113. Miao EA, Leaf IA, Treuting PM, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11(12):1136–1142. doi:10.1038/ni.1960
114. Cerqueira DM, Pereira MS, Silva AL, Cunha LD, Zamboni DS. Caspase-1 but not caspase-11 is required for NLRC4-mediated pyroptosis and restriction of infection by flagellated legionella species in mouse macrophages and in vivo. J Immunol. 2015;195(5):2303–2311. doi:10.4049/jimmunol.1501223
115. Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1β is deleterious. PLoS Pathog. 2011;7(12):e1002452. doi:10.1371/journal.ppat.1002452
116. Wang J, Sahoo M, Lantier L, et al. Caspase-11-dependent pyroptosis of lung epithelial cells protects from melioidosis while caspase-1 mediates macrophage pyroptosis and production of IL-18. PLoS Pathog. 2018;14(5):e1007105. doi:10.1371/journal.ppat.1007105
117. Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. 2013;123(4):1630–1637. doi:10.1172/JCI66142
118. Huus KE, Joseph J, Zhang L, et al. Clinical isolates of pseudomonas aeruginosa from chronically infected cystic fibrosis patients fail to activate the inflammasome during both stable infection and pulmonary exacerbation. J Immunol. 2016;196(7):3097–3108. doi:10.4049/jimmunol.1501642
119. Ryu JC, Kim MJ, Kwon Y, et al. Neutrophil pyroptosis mediates pathology of P. aeruginosa lung infection in the absence of the NADPH oxidase NOX2. Mucosal Immunol. 2017;10(3):757–774. doi:10.1038/mi.2016.73
120. Dai M, Pan P, Li H, et al. The antimicrobial cathelicidin peptide hlF(1-11) attenuates alveolar macrophage pyroptosis induced by Acinetobacter baumannii in vivo. Exp Cell Res. 2018;364(1):95–103. doi:10.1016/j.yexcr.2018.01.035
121. Li Y, Guo X, Hu C, et al. Type I IFN operates pyroptosis and necroptosis during multidrug-resistant A. baumannii infection. Cell Death Differ. 2018;25(7):1304–1318. doi:10.1038/s41418-017-0041-z
122. Wan X, Li J, Wang Y, et al. H7N9 virus infection triggers lethal cytokine storm by activating gasdermin E-mediated pyroptosis of lung alveolar epithelial cells. Natl Sci Rev. 2022;9(1):nwab137. doi:10.1093/nsr/nwab137
123. Gao J, Qiu X, Xi G, et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in non‑small cell lung cancer. Oncol Rep. 2018;40(4):1971–1984. doi:10.3892/or.2018.6634
124. Xi G, Gao J, Wan B, et al. GSDMD is required for effector CD8(+) T cell responses to lung cancer cells. Int Immunopharmacol. 2019;74:105713. doi:10.1016/j.intimp.2019.105713
125. Zhang CC, Li CG, Wang YF, et al. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis. 2019;24(3–4):312–325. doi:10.1007/s10495-019-01515-1
126. Peng Z, Wang P, Song W, et al. GSDME enhances Cisplatin sensitivity to regress non-small cell lung carcinoma by mediating pyroptosis to trigger antitumor immunocyte infiltration. Signal Transduct Target Ther. 2020;5(1):159. doi:10.1038/s41392-020-00274-9
127. Xie B, Liu T, Chen S, et al. Combination of DNA demethylation and chemotherapy to trigger cell pyroptosis for inhalation treatment of lung cancer. Nanoscale. 2021;13(44):18608–18615. doi:10.1039/D1NR05001J
128. Shi F, Zhang L, Liu X, Wang Y. Knock-down of microRNA miR-556-5p increases cisplatin-sensitivity in non-small cell lung cancer (NSCLC) via activating NLR family pyrin domain containing 3 (NLRP3)-mediated pyroptotic cell death. Bioengineered. 2021;12(1):6332–6342. doi:10.1080/21655979.2021.1971502
129. Liu J, Yao L, Zhang M, Jiang J, Yang M, Wang Y. Downregulation of LncRNA-XIST inhibited development of non-small cell lung cancer by activating miR-335/SOD2/ROS signal pathway mediated pyroptotic cell death. Aging. 2019;11(18):7830–7846. doi:10.18632/aging.102291
130. Xu X, Zhou X, Chen Z, Gao C, Zhao L, Cui Y. Silencing of lncRNA XIST inhibits non-small cell lung cancer growth and promotes chemosensitivity to cisplatin. Aging. 2020;12(6):4711–4726. doi:10.18632/aging.102673
131. Yuan R, Zhao W, Wang QQ, et al. Cucurbitacin B inhibits non-small cell lung cancer in vivo and in vitro by triggering TLR4/NLRP3/GSDMD-dependent pyroptosis. Pharmacol Res. 2021;170:105748. doi:10.1016/j.phrs.2021.105748
132. Wang F, Liu W, Ning J, et al. Simvastatin suppresses proliferation and migration in non-small cell lung cancer via pyroptosis. Int J Biol Sci. 2018;14(4):406–417. doi:10.7150/ijbs.23542
133. Teng JF, Mei QB, Zhou XG, et al. Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-κB/NLRP3/GSDMD signal axis in non-small cell lung cancer. Cancers. 2020;12(1):193. doi:10.3390/cancers12010193
134. Chen L, Weng B, Li H, et al. A thiopyran derivative with low murine toxicity with therapeutic potential on lung cancer acting through a NF-κB mediated apoptosis-to-pyroptosis switch. Apoptosis. 2019;24(1–2):74–82. doi:10.1007/s10495-018-1499-y
135. Boxer MB, Shen M, Auld DS, Wells JA, Thomas CJ. A small molecule inhibitor of Caspase 1. In: Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): National Center for Biotechnology Information (US); 2010.
136. Rudolphi K, Gerwin N, Verzijl N, van der Kraan P, van den Berg W. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis Cartilage. 2003;11(10):738–746. doi:10.1016/S1063-4584(03)00153-5
137. Wannamaker W, Davies R, Namchuk M, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 2007;321(2):509–516. doi:10.1124/jpet.106.111344
138. Flores J, Noël A, Foveau B, Lynham J, Lecrux C, LeBlanc AC. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat Commun. 2018;9(1):3916. doi:10.1038/s41467-018-06449-x
139. Audia JP, Yang XM, Crockett ES, et al. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y(12) receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res Cardiol. 2018;113(5):32. doi:10.1007/s00395-018-0692-z
140. Siegmund B, Zeitz M. Pralnacasan (vertex pharmaceuticals). IDrugs. 2003;6(2):154–158.
141. Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46(2):324–329. doi:10.1002/hep.21664
142. MacKenzie SH, Schipper JL, Clark AC. The potential for caspases in drug discovery. Curr Opin Drug Discov Devel. 2010;13(5):568–576.
143. Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248–255. doi:10.1038/nm.3806
144. Coll RC, Hill JR, Day CJ, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15(6):556–559. doi:10.1038/s41589-019-0277-7
145. Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187(1):61–70. doi:10.1083/jcb.200903124
146. Desu HL, Plastini M, Illiano P, et al. IC100: a novel anti-ASC monoclonal antibody improves functional outcomes in an animal model of multiple sclerosis. J Neuroinflammation. 2020;17(1):143. doi:10.1186/s12974-020-01826-0
147. Tapia VS, Daniels MJD, Palazón-Riquelme P, et al. The three cytokines IL-1β, IL-18, and IL-1α share related but distinct secretory routes. J Biol Chem. 2019;294(21):8325–8335. doi:10.1074/jbc.RA119.008009
148. Martín-Sánchez F, Diamond C, Zeitler M, et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016;23(7):1219–1231. doi:10.1038/cdd.2015.176
149. de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019;26(1):146–161. doi:10.1038/s41418-018-0106-7
150. Rathkey JK, Zhao J, Liu Z, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3(26). doi:10.1126/sciimmunol.aat2738
151. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369(6511):1633–1637. doi:10.1126/science.abb9818
152. Hu JJ, Liu X, Xia S, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21(7):736–745. doi:10.1038/s41590-020-0669-6
153. Ding S, Wang H, Wang M, Bai L, Yu P, Wu W. Resveratrol alleviates chronic “real-world” ambient particulate matter-induced lung inflammation and fibrosis by inhibiting NLRP3 inflammasome activation in mice. Ecotoxicol Environ Saf. 2019;182:109425. doi:10.1016/j.ecoenv.2019.109425
154. Peng L, Wen L, Shi QF, et al. Scutellarin ameliorates pulmonary fibrosis through inhibiting NF-κB/NLRP3-mediated epithelial-mesenchymal transition and inflammation. Cell Death Dis. 2020;11(11):978. doi:10.1038/s41419-020-03178-2
155. Song C, He L, Zhang J, et al. Fluorofenidone attenuates pulmonary inflammation and fibrosis via inhibiting the activation of NALP3 inflammasome and IL-1β/IL-1R1/MyD88/NF-κB pathway. J Cell Mol Med. 2016;20(11):2064–2077. doi:10.1111/jcmm.12898
156. Zhang ZT, Zhang DY, Xie K, Wang CJ, Xu F. Luteolin activates Tregs to promote IL-10 expression and alleviating caspase-11-dependent pyroptosis in sepsis-induced lung injury. Int Immunopharmacol. 2021;99:107914. doi:10.1016/j.intimp.2021.107914
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.