")
Back to Archived Journals » Hypoxia » Volume 7
Role of pseudohypoxia in the pathogenesis of type 2 diabetes
Authors Song J, Yang X, Yan LJ
Received 24 January 2019
Accepted for publication 23 April 2019
Published 5 June 2019 Volume 2019:7 Pages 33—40
DOI https://doi.org/10.2147/HP.S202775
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Jing Song,1,2 Xiaojuan Yang,1,3 Liang-Jun Yan1
1Department of Pharmaceutical Sciences, UNT System College of Pharmacy, University of North Texas Health Science Center, Fort Worth, TX, USA; 2School of Public Health, Shanxi Medical University, Taiyuan, People’s Republic of China; 3Department of Geriatrics, First Hospital/First Clinical Medical College of Shanxi Medical University, Taiyuan, People’s Republic of China
Abstract: Type 2 diabetes is caused by persistent high blood glucose, which is known as diabetic hyperglycemia. This hyperglycemic situation, when not controlled, can overproduce NADH and lower nicotinamide adenine dinucleotide (NAD), thereby creating NADH/NAD redox imbalance and leading to cellular pseudohypoxia. In this review, we discussed two major enzymatic systems that are activated by diabetic hyperglycemia and are involved in creation of this pseudohypoxic condition. One system is aldose reductase in the polyol pathway, and the other is poly (ADP ribose) polymerase. While aldose reductase drives overproduction of NADH, PARP could in contrast deplete NAD. Therefore, activation of the two pathways underlies the major mechanisms of NADH/NAD redox imbalance and diabetic pseudohypoxia. Consequently, reductive stress occurs, followed by oxidative stress and eventual cell death and tissue dysfunction. Additionally, fructose formed in the polyol pathway can also cause metabolic syndrome such as hypertension and nonalcoholic fatty liver disease. Moreover, pseudohypoxia can also lower sirtuin protein contents and induce protein acetylation which can impair protein function. Finally, we discussed the possibility of using nicotinamide riboside, an NAD precursor, as a promising therapeutic agent for restoring NADH/NAD redox balance and for preventing the occurrence of diabetic pseudohypoxia.
Keywords: diabetes, fructose, nicotinamide riboside, oxidative stress, poly (ADP ribose) polymerase, polyol pathway, pseudohypoxia, redox imbalance, reductive stress
Introduction
The concept of pseudohypoxia in adult-onset diabetes (so called type 2 diabetes) and its complications was first brought up by Williamson et al in 19931 and has since garnered increasing attention in the field of diabetes research.2–5 Pseudohypoxia can be referred to as a compromised cellular capacity of utilizing oxygen due to decreased levels of nicotinamide adenine dinucleotide (NAD),6–8 which can cause accumulation of NADH with occurrence of NADH/NAD redox imbalance.9–11 This redox imbalance initially would cause reductive stress, but would gradually lead to oxidative stress that damages cellular components including proteins, DNA, and lipids.7 It is this widespread oxidative stress in diabetes that wreaks havoc on cellular glucose metabolic pathways and culminates in cell death and tissue dysfunction.12–15 In this review, we discuss the major pathways that can perturb NADH/NAD redox imbalance which leads to pseudohypoxia in diabetes and its complications and the consequences of this pseudohypoxia phenomenon. It should be pointed out that in addition to diabetes, occurrence of pseudohypoxia has also been implicated in the pathogenesis of other diseases including cancers.16–19
While there are numerous enzymes in a cell that use NAD/NADH as their cofactors, there are only two well-recognized enzyme systems that can lead to perturbation of NADH/NAD redox imbalance. These are aldose reductase in the polyol pathway11,20,21 and poly (ADP ribose) polymerases (PARPs).22,23 Both of which use NAD as their substrate. Therefore, when activated by hyperglycemia, aldose reductase (AR) can drive overproduction of NADH while PARP can drive depletion of NAD.24
Aldose reductase
Under euglycemic conditions, AR remains in its inactive state because there is not enough glucose to activate its catalytic function. 25 Therefore, the physiological significance of this enzyme remain enigmatic. Nonetheless, it has been suggested that AR, under normal physiological conditions, is acting as a detoxifying agent that can degrade lipid peroxidation aldehyde byproducts such as hydroxynonenal and its glutathione conjugates.26,27 Chemically, AR catalyzes the first and rate-limiting reaction in the polyol pathway (Figure 1),28 which becomes activated in diabetes due to hyperglycemia and can dispose approximately 30% of the glucose pool in a diabetic patient.29 AR reduces glucose to sorbitol at the consumption of nicotinamide adenine dinucleotide phosphate(NADPH). The second reaction of the polyol pathway is oxidation of sorbitol to fructose with concurrent formation of NADH (Figure 1). Therefore, the products of the polyol pathway are sorbitol as an intermediate, fructose and NADH as final products. All three products have been demonstrated to accumulate in diabetic tissues.30–32 The detrimental role of aldose reductase has been confirmed in AR deletion studies whereby AR deficiency prevents development of diabetes.33 Likewise, AR gene knockdown has also been shown to slow down the development and progression of diabetes complications.34 In fact, many drugs have been designed to inhibit AR for diabetes therapeutic purposes.35–38
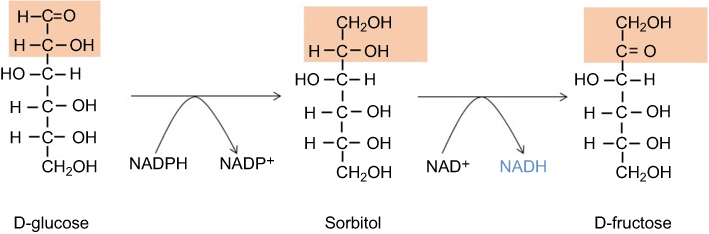 | Figure 1 The polyol pathway catalyzing conversion of glucose to fructose. This pathway has two reactions; the first reaction involves aldose reductase that catalyzes the conversion of glucose to sorbitol; the second reaction involves sorbitol dehydrogenase that catalyzes conversion of sorbitol to fructose with concurrent formation of NADH. Abbreviation: NADH, nicotinamide adenine dinucleotide. |
Poly (ADP ribose) polymerases (PARPs)
PARPs can also be activated in diabetes due to oxidative damage to DNA.39–41 This family of enzymes uses NAD as its substrate by putting multiple ADP molecules onto target proteins with concurrent release of nicotinamide (Figure 2). While the function of activated PARP is to repair damaged DNA,42,43 the enzyme can be over-activated in diabetes, thereby leading to NAD depletion and eventual cell death.22 For example, in our laboratory, we have found that in diabetic lung and pancreas, PARP1 expression is elevated with concurrent decrease in NAD content.44,45 The detrimental role of PARPs in diabetes has also been confirmed by gene knockout studies whereby mouse lacks functional PARP1 does not develop diabetes.46 Similarly, PARP deficiency has also been shown to prevent diabetic development and progression.47 As is the case for AR, PARP has also been explored as a drug target for battling diabetes.48–51 A recent comprehensive review on PARP mechanism and regulation as well as its potential therapeutic applications can be found in an article authored by Alemasova and Lavrik.52
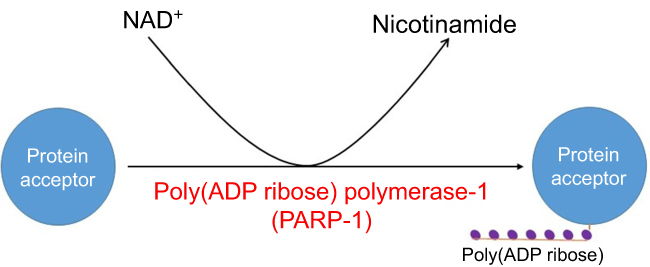 | Figure 2 Poly (ADP ribose) polymerization reaction catalyzed by poly (ADP ribose) polymerase. Shown is PARP1 as an example. Abbreviations: NADH, nicotinamide adenine dinucleotide; PARP, poly (ADP ribose) polymerase. |
Consequences of pseudohypoxia in diabetes
Reductive stress
The immediate consequence of pseudohypoxia due to NADH/NAD redox imbalance is reductive stress.7,53 NADH accumulation can give rise to pseudohypoxia and feedback-inhibit many metabolic enzymes or pathways such as the glycolytic pathway, pyruvate dehydrogenase complex, Krebs cycle, and the electron transport chain. Indeed, it has been reported that redox imbalance can increase aerobic glycolysis54,55 and reductive stress can impair brain blood barrier function and endothelial cell angiogenesis.56,57 Importantly, feedback inhibition of metabolic pathways would further prevent NAD from accepting electrons and accentuate NADH/NAD redox imbalance.7 Moreover, accumulation of NADH could be linked to increase in GSH and NADPH, which could further aggravate reductive stress,58–62 leading to cellular dysfunction and cell death.63,64 It should be noted here that accumulation of NADPH can also contribute to disease development. For example, abrogation of NADH oxidase activity can induce accumulation of NADPH and trigger reductive stress, leading to sensitization of the heart to ischemic/reperfusion injury.58
Oxidative stress
As implicated above, pseudohypoxia is a pathophysiological condition whereby the absolute concentration of cellular NAD is significantly decreased when compared to normal conditions.1 Therefore, the flip side of pseudohypoxia is increased levels of cellular NADH, which would overload mitochondrial electron transport chain. In other words, pseudohypoxia due to NADH/NAD redox imbalance can overload mitochondrial electron transport chain, leading to excess production of reactive oxygen species (ROS).7 In particular, as complex I (NADH-ubiquinone oxidoreductase) is the major site in mitochondria responsible for NAD regeneration, complex I overload of NADH can lead to over-production of ROS because of increased electron leakage from the electron transport chain.65,66 Indeed, our laboratory has found that mitochondrial complex I becomes hyperactive in the diabetic pancreas and lung due to NADH overloading and this hyperactivity is associated with increased ROS production, decreased ATP synthesis, and increased cell death.44,45
Detrimental effects of fructose
Endogenous production of fructose by the polyol pathway can lead to a variety of metabolic abnormalities.67,68 As fructose breakdown bypasses the regulation of the glycolytic pathway (Figure 3),68,69 fructose metabolism can potentially deplete cellular ATP content,70 which can lead to accumulation of uric acid and development of gout and hypertension.71 Moreover, as fructose metabolism leads to overgeneration of acetyl-CoA, more lipids can be made in the liver. This can cause increased protein modification such as glycation and acetylation72 and nonalcohol fatty liver disease that further derange glucose metabolism.73–76 Therefore, detrimental effect of fructose accumulation and metabolism is another consequence of pseudohypoxia that results from the activation of AR in the polyol pathway.
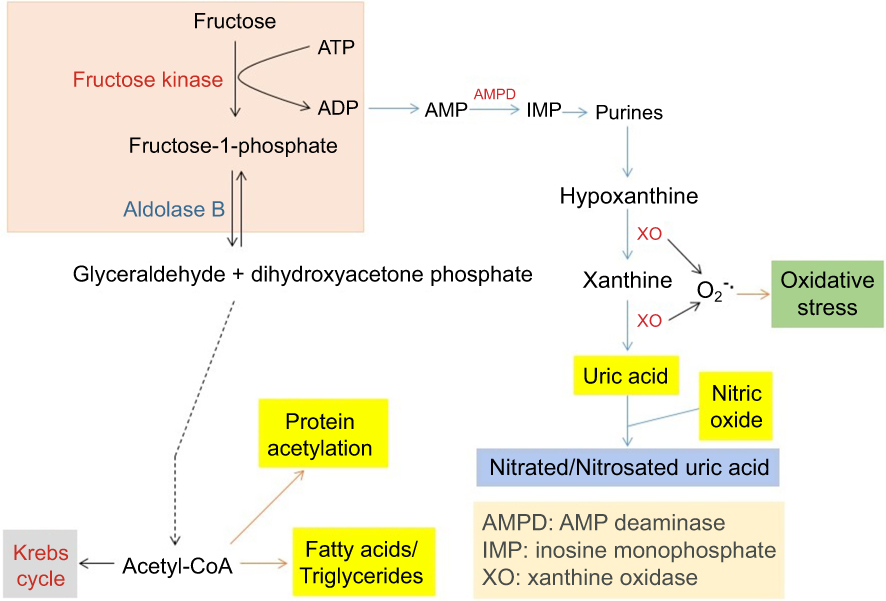 | Figure 3 Pathways of fructose metabolism and detrimental consequences. Major abnormalities resulting from fructose metabolism are shown in the graph, which include potential ATP depletion, accumulation of uric acid that gives rise to gout and hypertension, and nonalcoholic fatty liver disease as well as increased protein acetylation. |
Decreased levels of sirtuins
It is well established in experimental conditions that sirtuin protein expression is positively correlated with cellular NAD content. For example, in obese mouse, Sirt3 expression is decreased because of a decreased NAD content77 while caloric restriction increases Sirt3 expression due to an increased NAD content.78 Therefore, when NAD becomes scarce in diabetes due to NADH overproduction, sirtuin protein contents are decreased.79,80 Several members of the sirtuin family are protein deactylases.81,82 Therefore, dysfunction of sirtuin proteins can cause overcoating of proteins with acetyl groups.83 For example, mitochondrial sirtuin 3 (sirt3) expression was lower in diabetic lung than in healthy controls with concurrent increase in protein acetylation.45 Hence, decreased levels of sirt3 would cause accumulation of acetylated proteins, thereby impairing protein function and derailing metabolic pathways. Conversely, stimulating sirtuin expression or overexpression may serve as approaches to fighting diabetes.84,85
Eliminating pseudohypoxia by restoring NADH/NAD redox balance
While many steps in NAD metabolism can be potentially explored to restore NADH/NAD redox balance in diabetes, supplementation of NAD precursors has been shown to be another promising approach in battling disease or diabetes.86,87 One such precursor that is worth mentioning is nicotinamide riboside (NR).88 This compound is more tolerable89 and has been tested in a variety of experimental systems.88,90,91 For example, NR administration in diabetic mouse decreased fasting and nonfasting glucose levels, decreased weight gain, and lessened hepatic steatosis with concurrent protection against diabetic neuropathy.92 In prediabetic mouse, NR could improve glucose tolerance, decrease body weight gain, prevent liver damage, and retard the development of liver steatosis.92 Moreover, NR supplementation was found to increase NAD levels in tissues and to activate sirt3, thereby improving oxidative metabolism and protecting against metabolic dysfunction induced by high fat diet.89 This study clearly demonstrates that NR supplementation can restore NADH/NAD redox balance by increasing cellular and tissue NAD contents.89 It should also be pointed out that as NAD can be synthesized de novo from either aspartic acid in bacteria or tryptophan in animals,93,94 the supplement of these substrates and enhancement of the pertinent enzymes involved in NAD synthesis can also be explored to fight pseudohypoxia in diabetes by boosting NAD content.95–98
Conclusion
The occurrence of pseudohypoxia in diabetes and its complications is caused by NADH/NAD redox imbalance, which is mainly caused by activation of AR in the polyol pathway and PARPs. Pseudohypoxia can induce reductive stress followed by oxidative stress which eventually leads to cell death and tissue dysfunction (Figure 4). As a means of restoring NADH/NAD redox balance and preventing the occurrence of pseudohypoxia, NR has been shown to be a promising compound as a therapeutic agent for diabetes and its complications. It should be noted that other systems such as mitochondrial complex I may also be explored as a therapeutic target for restoring NADH/NAD redox balance to prevent the occurrence of pseudohypoxia in type 2 diabetes.
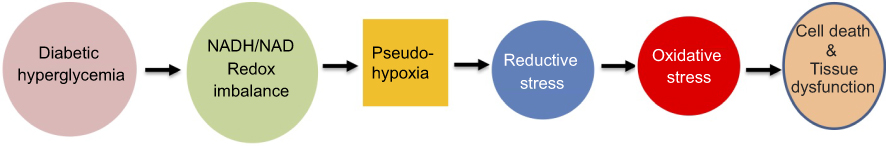 | Figure 4 Scheme showing the summary of hyperglycemia-induced redox imbalance, pseudohypoxia, reductive stress, and oxidative stress. The eventual events are cell death and tissue dysfunction. |
Acknowledgments
LJY was supported in part by UNT HSC grants RI10039 and RI10015 and by the National Institute of Neurological Disorders and Stroke (grant R01NS079792).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Williamson JR, Chang K, Frangos M, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42(6):801–813.
2. Diederen RM, Starnes CA, Berkowitz BA, Winkler BS. Reexamining the hyperglycemic pseudohypoxia hypothesis of diabetic oculopathy. Invest Ophthalmol Vis Sci. 2006;47(6):2726–2731. doi:10.1167/iovs.06-0076
3. Ido Y, Williamson JR. Hyperglycemic cytosolic reductive stress ‘pseudohypoxia‘: implications for diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38(8):1467–1470.
4. Takiyama Y, Haneda M. Hypoxia in diabetic kidneys. Biomed Res Int. 2014;2014:837421. doi:10.1155/2014/837421
5. Obrosova IG, Stevens MJ, Lang HJ. Diabetes-induced changes in retinal NAD-redox status: pharmacological modulation and implications for pathogenesis of diabetic retinopathy. Pharmacology. 2001;62(3):172–180. doi:10.1159/000056091
6. Williamson JR, Ido Y. Understanding retinal cytosolic reductive stress. Invest Ophthalmol Vis Sci. 1998;39(7):1295–1296.
7. Yan LJ. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res. 2014;2014:137919. doi:10.1155/2014/137919
8. Luo X, Wu J, Jing S, Yan LJ. Hyperglycemic stress and carbon stress in diabetic glucotoxicity. Aging Dis. 2016;7(1):90–110. doi:10.14336/AD.2015.0702
9. Hayden MR, Sowers JR. Redox imbalance in diabetes. Antioxid Redox Signal. 2007;9(7):865–867. doi:10.1089/ars.2007.1640
10. Luo X, Li R, Yan LJ. Roles of pyruvate, NADH, and mitochondrial complex I in redox balance and imbalance in β cell function and dysfunction. J Diabetes Res. 2015;2015. doi:10.1155/2015/512618.
11. Yan LJ. Redox imbalance stress in diabetes mellitus: role of the polyol pathway. Animal Model Exp Med. 2018;1(1):7–13. doi:10.1002/ame2.12001
12. Shah S, Iqbal M, Karam J, Salifu M, McFarlane SI. Oxidative stress, glucose metabolism, and the prevention of type 2 diabetes: pathophysiological insights. Antioxid Redox Signal. 2007;9(7):911–929. doi:10.1089/ars.2007.1629
13. Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279(41):42351–42354. doi:10.1074/jbc.R400019200
14. de M Bandeira S, Da Fonseca LJ, Da S Guedes G, Rabelo LA, Goulart MO, Vasconcelos SM. Oxidative stress as an underlying contributor in the development of chronic complications in diabetes mellitus. Int J Mol Sci. 2013;14(2):3265–3284. doi:10.3390/ijms14023265
15. Abdul-Ghani MA, DeFronzo RA. Oxidative stress in type 2 diabetes. In: Miwa S, Beckman KB, Muller FL, editors. Oxidative Stress in Aging. New York: Humana Press; 2008:191–212.
16. Hayashi Y, Yokota A, Harada H, Huang G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1alpha in cancer. Cancer Sci. 2019. doi:10.1111/cas.13990
17. Mohlin S, Wigerup C, Jogi A, Pahlman S. Hypoxia, pseudohypoxia and cellular differentiation. Exp Cell Res. 2017;356(2):192–196. doi:10.1016/j.yexcr.2017.03.007
18. Kantorovich V, Pacak K. New insights on the pathogenesis of paraganglioma and pheochromocytoma. F1000Res. 2018;20(7):pii:F1000 Faculty Rev-1500. doi:10.12688/f1000research.14568.1. eCollection 2018
19. Chappell JC, Payne LB, Rathmell WK. Hypoxia, angiogenesis, and metabolism in the hereditary kidney cancers. J Clin Invest. 2019;129(2):442–451. doi:10.1172/JCI120855
20. Kador PF. The role of aldose reductase in the development of diabetic complications. Med Res Rev. 1988;8(3):325–352.
21. Kador PF, Kinoshita JH. Role of aldose reductase in the development of diabetes-associated complications. Am J Med. 1985;79(5A):8–12.
22. Pacher P, Liaudet L, Soriano FG, Mabley JG, Szabo E, Szabo C. The role of poly(ADP-ribose) polymerase activation in the development of myocardial and endothelial dysfunction in diabetes. Diabetes. 2002;51(2):514–521.
23. Pacher P, Szabo C. Role of poly(ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antioxid Redox Signal. 2005;7(11–12):1568–1580. doi:10.1089/ars.2005.7.1568
24. Wu J, Jin Z, Zheng H, Yan LJ. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes. 2016;9:145–153. doi:10.2147/DMSO.S106087
25. Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625.
26. Maccari R, Ottana R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: new insights and future directions. J Med Chem. 2015;58(5):2047–2067. doi:10.1021/jm500907a
27. Srivastava SK, Yadav UC, Reddy AB, et al. Aldose reductase inhibition suppresses oxidative stress-induced inflammatory disorders. Chem Biol Interact. 2011;191(1–3):330–338. doi:10.1016/j.cbi.2011.02.023
28. Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev. 1998;50(1):21–33.
29. Fantus IG. The pathogenesis of the chronic complications of the diabetes mellitus. Endocrinology Rounds. 2002;2(4):1–8.
30. Li Q, Hwang YC, Ananthakrishnan R, Oates PJ, Guberski D, Ramasamy R. Polyol pathway and modulation of ischemia-reperfusion injury in Type 2 diabetic BBZ rat hearts. Cardiovasc Diabetol. 2008;7:33. doi:10.1186/1475-2840-7-33
31. Obrosova IG, Drel VR, Kumagai AK, Szabo C, Pacher P, Stevens MJ. Early diabetes-induced biochemical changes in the retina: comparison of rat and mouse models. Diabetologia. 2006;49(10):2525–2533. doi:10.1007/s00125-006-0356-7
32. Tilton RG, Baier LD, Harlow JE, Smith SR, Ostrow E, Williamson JR. Diabetes-induced glomerular dysfunction: links to a more reduced cytosolic ratio of NADH/NAD+. Kidney Int. 1992;41(4):778–788.
33. Tang J, Du Y, Petrash JM, Sheibani N, Kern TS. Deletion of aldose reductase from mice inhibits diabetes-induced retinal capillary degeneration and superoxide generation. PLoS One. 2013;8(4):e62081. doi:10.1371/journal.pone.0062081
34. Chang KC, Snow A, LaBarbera DV, Petrash JM. Aldose reductase inhibition alleviates hyperglycemic effects on human retinal pigment epithelial cells. Chem Biol Interact. 2015;234:254–260. doi:10.1016/j.cbi.2014.10.007
35. De Mattia G, Laurenti O, Bravi C, Ghiselli A, Iuliano L, Balsano F. Effect of aldose reductase inhibition on glutathione redox status in erythrocytes of diabetic patients. Metabolism. 1994;43(8):965–968.
36. Demir Y, Isik M, Gulcin I, Beydemir S. Phenolic compounds inhibit the aldose reductase enzyme from the sheep kidney. J Biochem Mol Toxicol. 2017;31:9. doi:10.1002/jbt.21935
37. Drel VR, Pacher P, Stevens MJ, Obrosova IG. Aldose reductase inhibition counteracts nitrosative stress and poly(ADP-ribose) polymerase activation in diabetic rat kidney and high-glucose-exposed human mesangial cells. Free Radic Biol Med. 2006;40(8):1454–1465. doi:10.1016/j.freeradbiomed.2005.12.034
38. Yasunari K, Kohno M, Kano H, Minami M, Yoshikawa J. Aldose reductase inhibitor improves insulin-mediated glucose uptake and prevents migration of human coronary artery smooth muscle cells induced by high glucose. Hypertension. 2000;35(5):1092–1098.
39. Szabo C. Roles of poly(ADP-ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol Res. 2005;52(1):60–71. doi:10.1016/j.phrs.2005.02.015
40. Chiu J, Xu BY, Chen S, Feng B, Chakrabarti S. Oxidative stress-induced, poly(ADP-ribose) polymerase-dependent upregulation of ET-1 expression in chronic diabetic complications. Can J Physiol Pharmacol. 2008;86(6):365–372. doi:10.1139/Y08-033
41. Szabo C, Zanchi A, Komjati K, et al. Poly(ADP-Ribose) polymerase is activated in subjects at risk of developing type 2 diabetes and is associated with impaired vascular reactivity. Circulation. 2002;106(21):2680–2686.
42. Dolle C, Rack JG, Ziegler M. NAD and ADP-ribose metabolism in mitochondria. FEBS J. 2013;280(15):3530–3541. doi:10.1111/febs.12304
43. Francica P, Rottenberg S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018;10(1):101. doi:10.1186/s13073-018-0612-8
44. Wu J, Luo X, Thangthaeng N, et al. Pancreatic mitochondrial complex I exhibits aberrant hyperactivity in diabetes. Biochem Biophys Rep. 2017;11:119–129. doi:10.1016/j.bbrep.2017.07.007
45. Wu J, Jin Z, Yan LJ. Redox imbalance and mitochondrial abnormalities in the diabetic lung. Redox Biol. 2017;11:51–59. doi:10.1016/j.redox.2016.11.003
46. Masutani M, Suzuki H, Kamada N, et al. Poly(ADP-ribose) polymerase gene disruption conferred mice resistant to streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96(5):2301–2304.
47. Pieper AA, Brat DJ, Krug DK, et al. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. 1999;96(6):3059–3064.
48. Long CA, Boulom V, Albadawi H, et al. Poly-ADP-ribose-polymerase inhibition ameliorates hind limb ischemia reperfusion injury in a murine model of type 2 diabetes. Ann Surg. 2013;258(6):1087–1095. doi:10.1097/SLA.0b013e31828cced3
49. Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429.
50. Obrosova IG, Minchenko AG, Frank RN, et al. Poly(ADP-ribose) polymerase inhibitors counteract diabetes- and hypoxia-induced retinal vascular endothelial growth factor overexpression. Int J Mol Med. 2004;14(1):55–64.
51. Byun YS, Kang B, Yoo YS, Joo CK. Poly(ADP-Ribose) polymerase inhibition improves corneal epithelial innervation and wound healing in diabetic rats. Invest Ophthalmol Vis Sci. 2015;56(3):1948–1955. doi:10.1167/iovs.14-16259
52. Alemasova EE, Lavrik OI. Poly(ADP-ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019;47(8):3811–3827. doi:10.1093/nar/gkz120
53. Tilton RG. Diabetic vascular dysfunction: links to glucose-induced reductive stress and VEGF. Microsc Res Tech. 2002;57(5):390–407. doi:10.1002/jemt.10092
54. Oldham WM, Clish CB, Yang Y, Loscalzo J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015;22(2):291–303. doi:10.1016/j.cmet.2015.06.021
55. Van Den Enden MK, Nyengaard JR, Ostrow E, Burgan JH, Williamson JR. Elevated glucose levels increase retinal glycolysis and sorbitol pathway metabolism. Implications for diabetic retinopathy. Invest Ophthalmol Vis Sci. 1995;36(8):1675–1685.
56. Mentor S, Fisher D. Aggressive antioxidant reductive stress impairs brain endothelial cell angiogenesis and blood brain barrier function. Curr Neurovasc Res. 2017;14(1):71–81. doi:10.2174/1567202613666161129113950
57. Fisher D, Mentor S. Antioxidant-induced reductive stress has untoward consequences on the brain microvasculature. Neural Regen Res. 2017;12(5):743–744. doi:10.4103/1673-5374.206640
58. Yu Q, Lee CF, Wang W, et al. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J Am Heart Assoc. 2014;3(1):e000555. doi:10.1161/JAHA.114.000844
59. Banhegyi G, Mandl J, Csala M. Redox-based endoplasmic reticulum dysfunction in neurological diseases. J Neurochem. 2008;107(1):20–34. doi:10.1111/j.1471-4159.2008.05571.x
60. Oka S, Hsu CP, Sadoshima J. Regulation of cell survival and death by pyridine nucleotides. Circ Res. 2012;111(5):611–627. doi:10.1161/CIRCRESAHA.111.247932
61. Farhana A, Guidry L, Srivastava A, Singh AK, Hondalus MK, Steyn JC. Reductive stress in microbes: implications for understanding mycobacterium tuberculosis disease and persistence. In: Poole RK, editor. Advances in Microbial Physiology. Amsterdam: Elsevier; 2010:45–64.
62. Rajasekaran NS, Connell P, Christians ES, et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130(3):427–439. doi:10.1016/j.cell.2007.06.044
63. Lloret A, Fuchsberger T, Giraldo E, Reductive Stress: VJ. A new concept in alzheimer‘s disease. Curr Alzheimer Res. 2016;13(2):206–211.
64. Nishida M, Nishimura A, Matsunaga T, Motohashi H, Kasamatsu S, Akaike T. Redox regulation of electrophilic signaling by reactive persulfides in cardiac cells. Free Radic Biol Med. 2017;109:132–140. doi:10.1016/j.freeradbiomed.2017.01.024
65. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi:10.1042/BJ20081386
66. Vinogradov AD, Grivennikova VG. Oxidation of NADH and ROS production by respiratory complex I. Biochim Biophys Acta. 2016;1857(7):863–871. doi:10.1016/j.bbabio.2015.11.004
67. Lanaspa MA, Ishimoto T, Li N, et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun. 2013;4:2434. doi:10.1038/ncomms3434
68. Jegatheesan P, De Bandt JP. Fructose and NAFLD: the multifaceted aspects of fructose metabolism. Nutrients. 2017;9:3.
69. Diggle CP, Shires M, Leitch D, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem. 2009;57(8):763–774. doi:10.1369/jhc.2009.953190
70. Johnson RJ, Rodriguez-Iturbe B, Roncal-Jimenez C, et al. Hyperosmolarity drives hypertension and CKD–water and salt revisited. Nat Rev Nephrol. 2014;10(7):415–420. doi:10.1038/nrneph.2014.76
71. Bantle JP. Dietary fructose and metabolic syndrome and diabetes. J Nutr. 2009;139(6):1263S–1268S. doi:10.3945/jn.108.098020
72. Gugliucci A. Formation of fructose-mediated advanced glycation end products and their roles in metabolic and inflammatory diseases. Adv Nutr. 2017;8(1):54–62. doi:10.3945/an.116.013912
73. DiNicolantonio JJ, Subramonian AM, O‘Keefe JH. Added fructose as a principal driver of non-alcoholic fatty liver disease: a public health crisis. Open Heart. 2017;4(2):e000631. doi:10.1136/openhrt-2017-000631
74. Choi Y, Abdelmegeed MA, Song BJ. Diet high in fructose promotes liver steatosis and hepatocyte apoptosis in C57BL/6J female mice: role of disturbed lipid homeostasis and increased oxidative stress. Food Chem Toxicol. 2017;103:111–121. doi:10.1016/j.fct.2017.02.039
75. Chiu S, Mulligan K, Schwarz JM. Dietary carbohydrates and fatty liver disease: de novo lipogenesis. Curr Opin Clin Nutr Metab Care. 2018;21(4):277–282. doi:10.1097/MCO.0000000000000469
76. Alwahsh SM, Gebhardt R. Dietary fructose as a risk factor for non-alcoholic fatty liver disease (NAFLD). Arch Toxicol. 2017;91(4):1545–1563. doi:10.1007/s00204-016-1892-7
77. Porter LC, Franczyk MP, Pietka T, et al. NAD(+)-dependent deacetylase SIRT3 in adipocytes is dispensable for maintaining normal adipose tissue mitochondrial function and whole body metabolism. Am J Physiol Endocrinol Metab. 2018;315(4).E520–E530. doi:10.1152/ajpendo.00057.2018
78. Jing E, Emanuelli B, Hirschey MD, et al. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108(35):14608–14613. doi:10.1073/pnas.1111308108
79. Cerutti R, Pirinen E, Lamperti C, et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19(6):1042–1049. doi:10.1016/j.cmet.2014.04.001
80. Vedantham S, Thiagarajan D, Ananthakrishnan R, et al. Aldose reductase drives hyperacetylation of Egr-1 in hyperglycemia and consequent upregulation of proinflammatory and prothrombotic signals. Diabetes. 2014;63(2):761–774. doi:10.2337/db13-0032
81. Morris BJ. Seven sirtuins for seven deadly diseases of aging. Free Radic Biol Med. 2013;56:133–171. doi:10.1016/j.freeradbiomed.2012.10.525
82. Sauve AA. Sirtuin chemical mechanisms. Biochim Biophys Acta. 2010;1804(8):1591–1603. doi:10.1016/j.bbapap.2010.01.021
83. Sol EM, Wagner SA, Weinert BT, et al. Proteomic investigations of lysine acetylation identify diverse substrates of mitochondrial deacetylase sirt3. PLoS One. 2012;7(12):e50545. doi:10.1371/journal.pone.0050545
84. Kitada M, Kume S, Kanasaki K, Takeda-Watanabe A, Koya D. Sirtuins as possible drug targets in type 2 diabetes. Curr Drug Targets. 2013;14(6):622–636.
85. Kim M, Lee JS, Oh JE, et al. SIRT3 overexpression attenuates palmitate-induced pancreatic beta-cell dysfunction. PLoS One. 2015;10(4):e0124744. doi:10.1371/journal.pone.0124744
86. Mendelsohn AR, Larrick JW. Partial reversal of skeletal muscle aging by restoration of normal NAD(+) levels. Rejuvenation Res. 2014;17(1):62–69. doi:10.1089/rej.2014.1546
87. Hou Y, Lautrup S, Cordonnier S, et al. NAD(+) supplementation normalizes key Alzheimer‘s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc Natl Acad Sci U S A. 2018;115(8).E1876–E1885. doi:10.1073/pnas.1718819115
88. Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–130. doi:10.1146/annurev.nutr.28.061807.155443
89. Canto C, Houtkooper RH, Pirinen E, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15(6):838–847. doi:10.1016/j.cmet.2012.04.022
90. Hamity MV, White SR, Walder RY, Schmidt MS, Brenner C, Hammond DL. Nicotinamide riboside, a form of vitamin B3 and NAD+ precursor, relieves the nociceptive and aversive dimensions of paclitaxel-induced peripheral neuropathy in female rats. Pain. 2017;158(5):962–972. doi:10.1097/j.pain.0000000000000862
91. Kourtzidis IA, Stoupas AT, Gioris IS, et al. The NAD(+) precursor nicotinamide riboside decreases exercise performance in rats. J Int Soc Sports Nutr. 2016;13:32. doi:10.1186/s12970-016-0143-x
92. Trammell SA, Weidemann BJ, Chadda A, et al. Nicotinamide riboside opposes type 2 diabetes and neuropathy in mice. Sci Rep. 2016;6:26933. doi:10.1038/srep26933
93. Yang T, Sauve AA. NAD metabolism and sirtuins: metabolic regulation of protein deacetylation in stress and toxicity. Aaps J. 2006;8(4):E632–E643. doi:10.1208/aapsj080476
94. Katoh A, Hashimoto T. Molecular biology of pyridine nucleotide and nicotine biosynthesis. Front Biosci. 2004;9:1577–1586.
95. Matasic DS, Brenner C, London B. Emerging potential benefits of modulating NAD(+) metabolism in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2018;314(4):H839–H852. doi:10.1152/ajpheart.00409.2017
96. Braidy N, Berg J, Clement J, et al. Role of nicotinamide adenine dinucleotide and related precursors as therapeutic targets for age-related degenerative diseases: rationale, biochemistry, pharmacokinetics, and outcomes. Antioxid Redox Signal. 2018;30(2):251–294.
97. Stromland O, Niere M, Nikiforov AA, VanLinden MR, Heiland I, Ziegler M. Keeping the balance in NAD metabolism. Biochem Soc Trans. 2019;47(1):119–130. doi:10.1042/BST20180417
98. Rajman L, Chwalek K, Sinclair DA. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27(3):529–547. doi:10.1016/j.cmet.2018.02.011
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.