")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Role of p38 Mitogen-Activated Protein Kinase in Asthma and COPD: Pathogenic Aspects and Potential Targeted Therapies
Authors Pelaia C , Vatrella A , Gallelli L , Lombardo N , Sciacqua A, Savino R, Pelaia G
Received 8 January 2021
Accepted for publication 23 February 2021
Published 23 March 2021 Volume 2021:15 Pages 1275—1284
DOI https://doi.org/10.2147/DDDT.S300988
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Corrado Pelaia,1 Alessandro Vatrella,2 Luca Gallelli,1 Nicola Lombardo,3 Angela Sciacqua,3 Rocco Savino,3 Girolamo Pelaia1
1Department of Health Sciences, University “Magna Græcia” of Catanzaro, Catanzaro, Italy; 2Department of Medicine, Surgery and Dentistry, University of Salerno, Salerno, Italy; 3Department of Medical and Surgical Sciences, University “Magna Græcia” of Catanzaro, Catanzaro, Italy
Correspondence: Girolamo Pelaia
Department of Health Sciences, University Magna Græcia of Catanzaro, Viale Europa – Località Germaneto, Catanzaro, 88100, Italy
Tel + 39 0961 3647171
Fax + 39 0961 3647193
Email [email protected]
Abstract: Among the various members of the mitogen-activated protein kinase (MAPK) family, p38 MAPK subgroup is the most involved in airway and lung inflammation underlying asthma and chronic obstructive pulmonary disease (COPD). In particular, several environmental agents including aeroallergens, cigarette smoke, airborne pollutants, viral and bacterial pathogens activate the p38α isoform which in turn up-regulates the expression of multiple proinflammatory cytokines and chemokines, as well as the production of some fibrogenic factors. Therefore, p38 MAPK-induced bronchial inflammation and remodelling significantly contribute to the development, persistence and amplification of airflow limitation, which is the hallmark of asthma and COPD. Such advances in our understanding of p38 role in the pathobiology of the above widespread, chronic obstructive respiratory diseases, have led to consider p38 MAPK as a suitable molecular target for novel treatment strategies. Indeed, many studies have been carried out in both animal and clinical settings, with the aim of evaluating the potential therapeutic effects of p38 MAPK inhibitors in both asthma and COPD.
Keywords: asthma, COPD, airway inflammation, p38-MAPK, p38-MAPK inhibitors
Introduction
Asthma and COPD are widespread, chronic obstructive respiratory diseases, characterized by inflammatory and structural changes causing airflow limitation.1–4 In particular, asthma and COPD are heterogeneous disorders, consisting of different phenotypes originating by the overexpression of proinflammatory and fibrogenic mediators including several cytokines, chemokines, and growth factors.5,6 Within such a pathobiologic framework, the main triggers of asthma and COPD are aeroallergens, cigarette smoke, airborne pollutants, and viral/bacterial infections. Inside the respiratory tract, all these environmental agents are able to activate the p38 subfamily of mitogen-activated protein kinases (MAPK).7,8
The cellular responses promoted by a large variety of extracellular stimuli, capable of inducing airway/lung inflammation and remodelling, are coordinated and amplified by the p38 subgroup of MAPKs, a highly evolutionarily conserved enzymatic system which is also a suitable target for experimental therapies of asthma and COPD.9 The p38 MAPK subfamily includes four isoforms, named p38α, p38β, p38γ, and p38δ, respectively.10 Similarly to all the other members of MAPK superfamily, p38 activation is mediated by a chain of sequential protein phosphorylations initiated by upstream MAPK kinase kinases (MAPKKK), including dual leucine zipper kinase (DLK), transforming growth factor-β-activated kinase 1 (TAK1), and apoptosis signal-regulating kinase 1 (ASK1).10 These MAPKKKs, in turn, phosphorylate and activate the MAPK kinases (MAPKK) MKK3/MKK6, which finally catalyse the phosphorylation-dependent stimulation of p38 MAPK (Figure 1).11 Full activation of p38 MAPK requires dual phosphorylation at the level of its threonine-180 (Thr-180) and tyrosine-182 (Tyr-182) amino acid residues.10 Once activated, p38 MAPK phosphorylates numerous substrates including downstream kinases, transcription factors, and transcriptional regulators. Moreover, p38 MAPK also affects gene expression at the post-transcriptional level. Indeed, p38 stimulates MAP kinase-activated protein kinase 2 (MAPKAPK2/MK2), which in turn inactivates tristetraprolin by phosphorylating its serine residues Ser-52 and Ser-178.12 Tristetraprolin is an important regulatory protein responsible for the destabilisation of various mRNAs transcribed from genes encoding multiple proinflammatory factors.12 Thus, activation of the p38 MAPK signalling machinery stabilises these mRNAs and inhibits their degradation, thereby enhancing the biosynthesis of many cytokines and chemokines involved in the pathophysiology of asthma and COPD.7,8 p38 MAPK activation is abolished by the dual-specificity phosphatase MKP-1 (MAPK phosphatase-1), which dephosphorylates both Thr-180 and Tyr-182.13
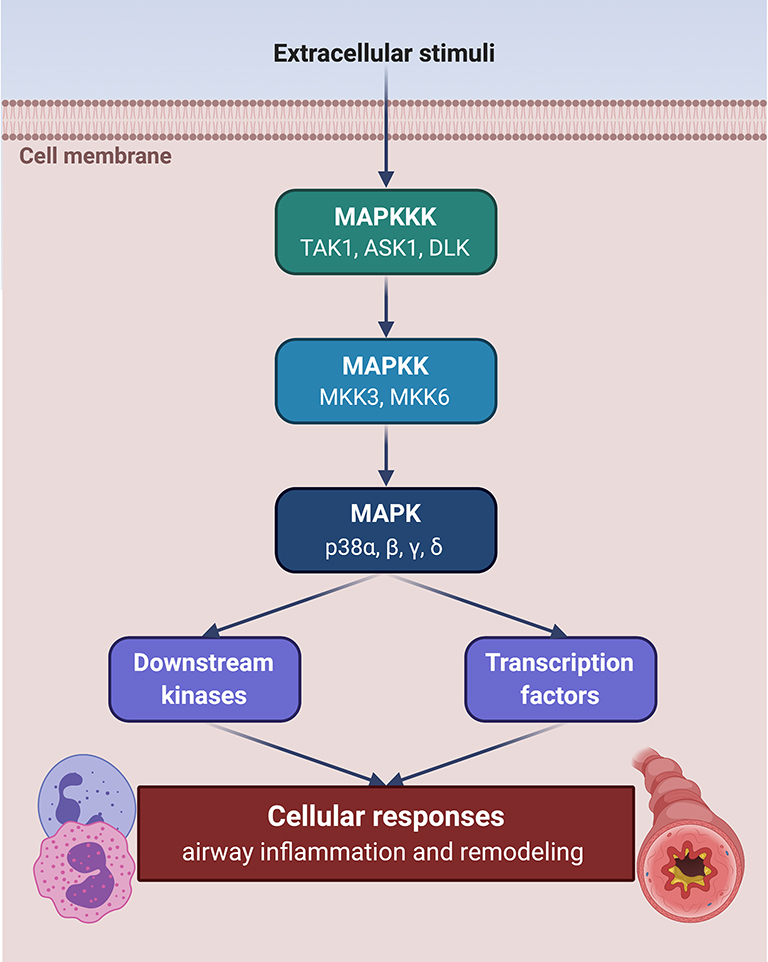 |
Figure 1 Activation of p38 MAPK signalling module. p38 MAPK is activated through a kinase cascade triggered by extracellular inflammatory stimuli. This sequential phosphorylation pathway includes MAPK kinase kinases (MAPKKK) TAK1, ASK1 and DLK, whose targets are MAPK kinases (MAPKK) MKK3 and MKK6, which phosphorylate and activate the α, β, γ, and δ isoforms of p38 MAPK. The latter in turn phosphorylates downstream kinases and transcription factors involved in cellular responses underlying airway inflammation and remodelling. This original figure was created by the authors using BioRender.com. |
With regard to the above considerations, in this concise review article, we will focus our attention on the pathogenic role played by p38 MAPK in asthma and COPD, as well as on potential perspective therapies of such diseases, based on experimental p38 inhibition.
Role of p38 MAPK in Asthma Pathophysiology
Type-2 (T2-high) and non-type-2 (T2-low) asthmatic phenotypes are mainly characterized by airway eosinophilia and neutrophilia, respectively.14 Eosinophilic asthma originates from both allergic and non-allergic mechanisms, driven by T helper 2 (Th2) lymphocytes and group 2 innate lymphoid cells (ILC2).15,16 Th2 and ILC2 cells secrete large amounts of interleukins 4 (IL-4), 5 (IL-5), and 13 (IL-13), which are responsible for immunoglobulin E (IgE) production, eosinophilic inflammation, and bronchial hyperresponsiveness, respectively.5,17 Th2 lymphocytes are activated by IL-4, whereas ILC2 are stimulated by the innate cytokines thymic stromal lymphopoietin (TSLP), interleukin-25 (IL-25), and interleukin-33 (IL-33), also known as alarmins.16,18 p38 MAPK induces the differentiation and activation of Th2 cells, thus promoting the release of Th2 cytokines (IL-4, IL-5, IL-13).7 These effects are due, at least in part, to the bioactivity of the transcription factor GATA-3, which is enabled by p38-catalysed phosphorylation to translocate into the cell nucleus, where GATA-3 activates its target genes coding for Th2 cytokines.19 p38 MAPK-dependent phosphorylation of GATA-3 also stimulates ILC2 to produce IL-5 and IL-13.20 Moreover, it has been recently shown in murine experimental models that p38α/β MAPK plays a pivotal role in mediating ILC2 stimulation induced by IL-33.21 In particular, p38-operated activation of downstream kinases MK2/3 is required for the biosynthesis of IL-6 and IL-13, released by ILC2 upon IL-33-mediated stimulation.21 Furthermore, p38 MAPK exerts many pro-eosinophilic functions, including inhibition of eosinophil apoptosis, as well as induction of eosinophil differentiation, chemotaxis, and secretory activity.7
In addition to being remarkably involved in type-2 eosinophilic asthma, p38 MAPK also significantly contributes to the pathobiology of T2-low neutrophilic inflammation of airways, often associated with the most severe asthmatic phenotypes.22 Indeed, analysis of sputum transcriptomics has recently identified, in subjects with severe asthma, the overexpression of gene networks belonging to the p38 MAPK signalling pathway, associated with neutrophilic bronchial inflammation.23 p38 up-regulates the expression of intercellular adhesion molecule-1 (ICAM-1) on lung vascular endothelial cells, and enhances the release of tumor necrosis factor-α (TNF-α) from neutrophils, thus promoting the accumulation of these cells into the airways.24,25 Activated p38 MAPK is also essential for IL-33-induced potentiation of TNF-α secretion from natural killer (NK) cells stimulated by IL-12.26 In asthmatic patients, inhibition of neutrophil apoptosis is at least in part dependent on bronchial production of survival factors for neutrophils such as IL-6, IL-8, and monocyte chemoattractant protein-1 (MCP-1), whose secretion can be elicited by regulatory proteins S100A8 and S100A9 via activation of p38 MAPK.27 In regard to the role of p38 MAPK in mixed (eosinophilic/neutrophilic) asthmatic phenotypes, it is interesting to mention the findings of a recent study, built on a murine experimental model of airway inflammation associated with high fat diet-induced obesity,28 a frequent comorbidity of severe asthma. In particular, it was shown in obese mice, sensitized to house dust mite, that the expansion of dendritic cell-restricted progenitors occurred concomitantly with both p38 MAPK activation and inflammatory changes consisting of increased counts of eosinophils, neutrophils, and lymphocytes in bronchoalveolar lavage fluid (BALF).28 In another experimental setting, based on the exposure to ozone of mice already challenged with ovalbumin, enhanced BALF numbers of eosinophils and neutrophils were found, associated with p38 MAPK activation and increased levels of interleukin-17A (IL-17A) in lung tissue.29 The biological activities of IL-17A and IL-17F are closely linked to neutrophilic inflammation.14
Besides immune/inflammatory cells, airway resident cells can also be targeted by extracellular stimuli acting through phosphorylation-dependent activation of p38 MAPK. In fact, phospho-p38 expression has been shown to be up-regulated in bronchial epithelial cells taken from atopic asthmatic patients exposed to allergen challenge.30 Via activation of p38 MAPK, transforming growth factor-β (TGF-β) can induce the apoptosis of human airway epithelial cells.31 Indeed, in asthmatic subjects, these cells seem to be highly susceptible to apoptosis.32–35 Moreover, p38 MAPK appears to be implicated in structural changes underlying bronchial remodelling in asthma, such as the thickening of sub-epithelial basement membrane.7 In particular, as a consequence of intercellular contacts with mast cells, lung fibroblasts proliferate and secrete large quantities of collagen through p38 MAPK-mediated release of IL-6.36,37
Role of p38 MAPK in COPD Pathophysiology
Animal models of experimental emphysema have been very useful to demonstrate that cigarette smoke can induce the activation of p38 MAPK and the consequent biosynthesis of pro-inflammatory cytokines and chemokines, leading to neutrophilic lung inflammation.38 Such findings are in agreement with the results of some human studies, which have shown in COPD patients the up-regulation of phospho-p38 MAPK expression.39 For instance, the phosphorylated active form of p38α MAPK resulted to be overexpressed, in comparison to health subjects, in alveolar macrophages obtained from lung surgical samples taken from patients with COPD.40 In addition to alveolar macrophages, increased levels of phospho-p38α MAPK were also detected in CD8+ T cells located within the alveolar walls of COPD patients.40 Therefore, such observations indicate that p38 MAPK contributes to COPD pathophysiology by coordinating intercellular cross-talks between CD8+ T lymphocytes and alveolar macrophages. In COPD patients, the pathogenic role of p38 MAPK activation was found to be extended to structural cellular elements such as small airway epithelial cells.41 Indeed, these cells are involved in COPD pathobiology because they release many proinflammatory cytokines and chemokines when are exposed to cigarette smoke, as well as to other environmental agents that trigger p38 MAPK activation.8 In this regard, it is noteworthy that high sputum levels of phospho-p38 MAPK can be detected in COPD patients.42 Furthermore, such an enhanced activity of p38 MAPK was closely associated with several biological and lung functional parameters, including high concentrations of sputum neutrophils and IL-8, as well as low values of forced expiratory volume in one second (FEV1).42 Taken together, the above findings suggest that in COPD p38 MAPK activation is markedly correlated with both pulmonary inflammation and the progressive deterioration of respiratory function.
COPD exacerbations are characterized by further increments of lung inflammation and p38 MAPK activity. With regard to these aspects, it is interesting to point out that frequent causative agents of COPD exacerbations like bacterial pathogens can cooperate with proinflammatory cytokines such as TNF-α, thereby intensifying the signal transduction function of p38 MAPK.43 Hence, exposure of primary cultures of human bronchial epithelial cells to the joint actions of TNF-α and non-typeable Haemophilus influenzae, significantly enhanced p38 MAPK activation and the consequent IL-8 production.44 Airway inflammation can thus be worsened by the increased neutrophil influx promoted by IL-8.45 Acting together on bronchial epithelial cells, TNF-α and Haemophilus influenzae also triggered cellular apoptosis via p38-MAPK-mediated stimulation of caspase-3 activity.44 The apoptotic death of human bronchial epithelial cells was also caused through p38-MAPK signalling by hydrogen peroxide,46 commonly used as an experimental inducer of oxidative stress. As a consequence of the noxious effects exerted by cytokines, bacterial pathogens, and oxidative stress, the p38 MAPK transduction module is thus remarkably involved in the induction of harmful injuries to airway resident cells. By persistently amplifying deleterious pathogenic circuits, p38 MAPK acts as a converging signalling synapsis for several triggers of COPD exacerbations, which damage the bronchial epithelium and impair its barrier function. The resulting enhanced epithelial permeability makes the airways more susceptible to bacterial infections such as those caused by Haemophilus influenzae and Streptococcus pneumoniae, which synergistically stimulate mucus overproduction via p38α MAPK-dependent activation of MUC5AC mucin gene transcription.47 In addition to bacteria, respiratory viruses can also trigger COPD exacerbations.48 In this regard, it is noteworthy that human rhinovirus (HRV) and IL-17A were able to synergistically induce p38 MAPK activation in bronchial epithelial cells.49 Moreover, in these same cell types, HRV-bacterial coinfections elicited a synergistic stimulation of p38 MAPK activity, thus leading to an increased production of IL-17C and to the consequent enhancement of neutrophil recruitment,50 which represents a common inflammatory feature of COPD exacerbations.51
p38 MAPK also plays a central role in cellular mechanisms underpinning the accelerated rate of lung senescence associated with COPD.52 In particular, at the level of small airways and lung parenchyma of patients with COPD, activated p38 MAPK up-regulates the microRNA miR-570, which inhibits the expression of the anti-aging protein sirtuin-1.52
p38 MAPK Inhibition: A Potential Therapeutic Strategy for Treatment of Asthma and COPD
In experimental murine models of ovalbumin-induced asthma, the pyridinyl imidazole p38 inhibitor SB239063 significantly lowered airway levels of IgE, eosinophils, and Th2 cytokines.53 In asthmatic transgenic mice, pulmonary inflammation nourished by T cells and eosinophils was attenuated by the specific p38α MAPK inhibitor SD-282.54 In other animal models of asthma, p38α MAPK functions can be blocked by inhibition of gene expression; in this regard, a p38α MAPK-targeted antisense oligonucleotide, delivered by inhalation, significantly reduced BALF levels of total cells, eosinophils, and Th2 cytokines.55 Inhibition of p38 MAPK activation also allows to suppress the release of proinflammatory mediators from alveolar macrophages.8 Consistently with these findings, it is noteworthy that the p38 inhibitor doramapimod (BIRB-796) decreased the secretion of TNF-α and IL-6 from alveolar macrophages isolated from COPD patients, and this anti-inflammatory action resulted to be more effective than that one induced by the corticosteroid budesonide.56 Therefore, p38 inhibitors can potentially attenuate chronic airway inflammation.
The majority of p38 MAPK inhibitors under current experimental evaluation, deliverable via oral or inhaled route, have been utilized in COPD patients.9,57
Dilmapimod (SB-681323) is an oral p38 inhibitor which was tested in a Phase 1 trial, showing that at both dosages of 7.5 and 25 mg this drug was able to decrease blood concentrations of TNF-α in patients with COPD.58 Furthermore, when used at the dose of 7.5 mg for 28 days, dilmapimod reduced by 9.4% sputum neutrophils and increased forced vital capacity (FVC) by 210 mL; however, these changes did not reach the threshold of statistical significance.59 Administered to COPD patients at a single dose of 25 mg, dilmapimod was able to inhibit IL-1β gene expression in blood and sputum cells.60 A Phase 2 study evaluated in patients with COPD the oral p38α/β MAPK inhibitor losmapimod (GW856553), which induced a slight increase in pre-bronchodilator FEV1, though not statistically significant when compared to placebo, but did not ameliorate exercise performance investigated through 6 min walking test.61 However, a post hoc analysis of this trial documented that losmapimod lowered the number of COPD exacerbations in subjects with blood eosinophil levels not higher than 2%, only when this drug was used at the dosage of 15 mg.62 Another phase 2 study was designed with the aim of comparatively assessing, in COPD patients, the therapeutic effects of salmeterol/fluticasone propionate (50 µg/500 µg twice a day), losmapimod (7.5 mg twice a day), and placebo.63 None of the above treatments changed the number of blood neutrophils, but losmapimod decreased by 11% plasma fibrinogen levels, and also slightly reduced lung hyperinflation.63 A further phase 2 trial demonstrated that the oral p38α MAPK inhibitor PH-797804 significantly incremented trough FEV1 and improved transition dyspnoea index (TDI), as well as lowered blood levels of C-reactive protein (CRP).64 Acumapimod (BCT197) is another orally active p38 inhibitor, evaluated in a preliminary phase 2 trial which showed that, when used at repeated single doses of 75 mg, after 8 days of treatment this compound elicited a significant FEV1 improvement in comparison to placebo.65 Moreover, acumapimod was capable of reducing the number of hospitalizations caused by COPD exacerbations.66 A very recent pharmacokinetic study, focused on drug–drug interactions, showed that acumapimod can be safely co-administered with azithromycin,67 a therapeutic agent frequently used for the treatment of COPD and COPD exacerbations.68
Some studies have tested the safety of inhaled p38 MAPK inhibitors, as well as their potential efficacy for COPD treatment (Table 1). These drugs are generally safer than oral compounds, which can be characterized by an increased risk of eventual systemic unwanted effects. AZD7624 is a powerful p38α/β MAPK inhibitor, that was able to suppress lipopolysaccharide (LPS)-induced production of IL-6 and TNF-α from alveolar macrophages of patients with COPD.69 Moreover, AZD7624 appeared to be more potent than the inhaled corticosteroid budesonide in inhibiting IL-6 release from bronchial epithelial cells obtained from COPD patients experiencing or not recurrent exacerbations.70 Anyway, AZD7624 did not decrease the number of COPD exacerbations.70 RV-568, another inhaled p38 inhibitor, induced remarkable anti-inflammatory effects in monocytes/macrophages and bronchial epithelial cells, as well as in mice exposed to LPS and cigarette smoke, but elicited only a slight increase of pre-bronchodilator FEV1 in patients with COPD.71 CHF6297 is a potent p38 inhibitor, available as a dry powder preparation, which is undergoing clinical evaluation for inhaled therapy of COPD.8 Another p38 MAPK inhibitor that has been developed as a dry powder inhaler is PF-03715455,72 currently under investigation for treatment of patients with moderate or severe COPD.8
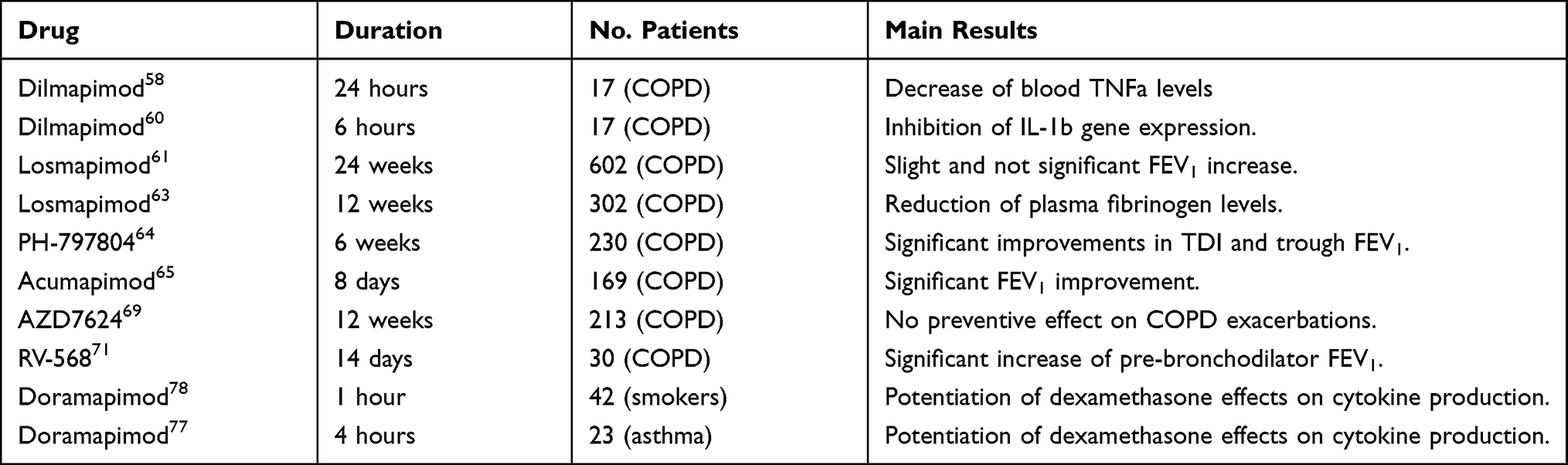 |
Table 1 Summary of the Main Studies Evaluating P38 MAPK Inhibitors for Potential Treatment of COPD and Asthma |
The most common adverse events observed in the above clinical trials were nasopharyngitis, headache, diarrhoea, nausea, and arthralgia.
In both COPD and severe asthma, p38 MAPK inhibitors can be experimentally used to revert corticosteroid resistance.73 Basically, the reciprocal relationships between p38 MAPK and corticosteroids are quite complex. Corticosteroids inhibit p38 activation via dephosphorylation mediated by induction of MKP-1 gene expression (Figure 2).74 However, MKP-1 can be down-regulated in alveolar macrophages collected from patients with severe asthma.75 On the other hand, in subjects with either COPD or severe asthma p38 MAPK can impair the therapeutic activity of corticosteroids through phosphorylation of a serine amino acid residue (Ser-226) of glucocorticoid receptors (GR), which prevents their nuclear translocation and DNA binding (Figure 2), thereby impeding the biological actions of the respective hormone ligands.76 In comparison to healthy subjects, phosphorylation levels of p38 MAPK and GR Ser-226 were found to be significantly increased in bronchial epithelial cells cultured from severe asthmatics.77 When these cells were exposed to both dexamethasone and the p38 inhibitor doramapimod (BIRB-796), an additive inhibitory effect on IL-6 release was observed.77 Furthermore, doramapimod re-established corticosteroid responsiveness in alveolar macrophages taken from COPD patients and infected by non-typeable Haemophilus influenzae, which stimulated p38 MAPK activation and the consequent phosphorylation of GR Ser-226.78 A similar additive inhibitory action, exerted by dexamethasone and doramapimod, was also detected with regard to the release of IL-6 and IL-8 from human lung fibroblasts.79 In peripheral blood monocytes obtained from patients with COPD, losmapimod restored corticosteroid sensitiveness;80,81 this effect was due to abrogation of p38 MAPK-mediated phosphorylation of GR Ser-211.81
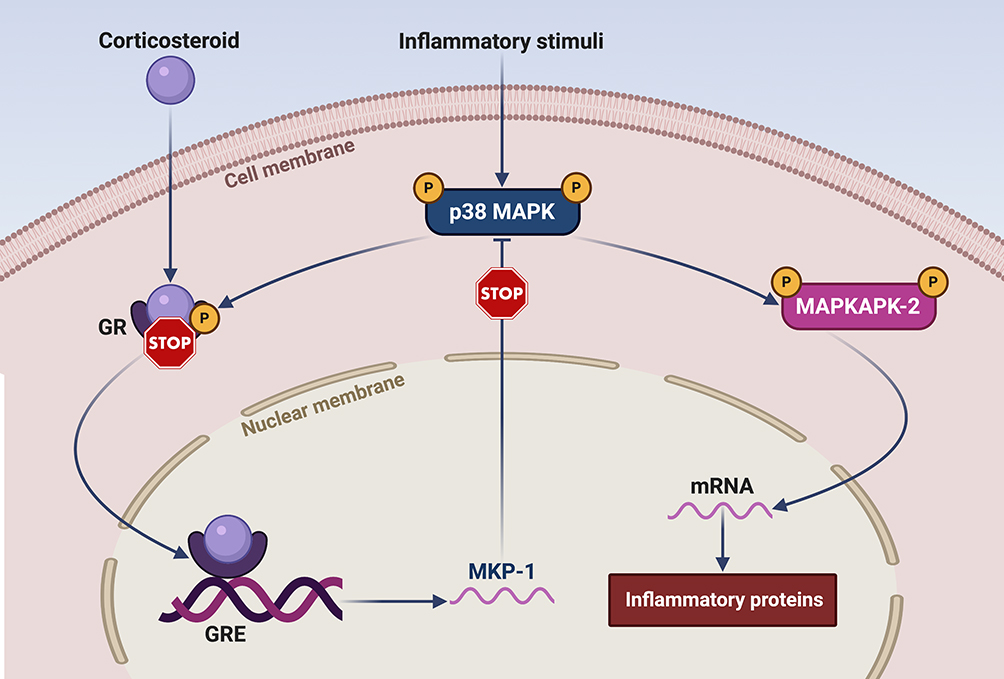 |
Figure 2 Cross-talk between corticosteroids and p38 MAPK. Acting at level of the glucocorticoid response elements (GRE) of target genes, corticosteroids induce the expression of MAP kinase phosphatase-1 (MKP-1), which dephosphorylates and inactivates p38 MAPK. In its active phosphorylated form, p38 in turn phosphorylates the glucocorticoid receptor (GR), thus impeding its nuclear translocation and the consequent biological and pharmacological actions of corticosteroids. In addition to inhibiting the anti-inflammatory effects of corticosteroids, p38 MAPK also plays a key proinflammatory role via phosphorylation-dependent activation of the downstream kinase MAPKAPK-2 (MAP kinase-activated protein kinase 2), which stabilizes several mRNAs encoding multiple cytokines and chemokines. This original figure was created by the authors using BioRender.com. |
Conclusions
The key role played by p38 MAPK activation in the pathobiology of asthma and COPD makes this signalling pathway a suitable potential target for future treatments of such widespread chronic respiratory diseases. However, though preclinical studies have yielded interesting and promising results, so far clinical trials have been quite disappointing. Better outcomes could perhaps be achieved by very selective inhibitors of the p38α isoform, which is the p38 MAPK subtype mostly involved in inflammatory processes, given its predominant expression at level of innate inflammatory cells such as alveolar macrophages.40 Nevertheless, even a very selective p38α inhibition might be counterbalanced by neutralizing mechanisms sustained by a natural redundancy of the MAPK system, possibly leading to a compensative hyperactivation of c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) MAPK subfamilies.82,83
Moreover, within a perspective therapeutic context referring to the treatment of COPD and severe asthma, the overall potential advantages provided by p38 MAPK inhibition might be weakened by objective limitations. Indeed, p38 MAPK is physiologically implicated in the regulation of relevant processes such as innate immunity and anti-bacterial protection, as well as organ differentiation and homeostasis.84 Therefore, the use of p38 MAPK inhibitors could be possibly linked to an increased risk of developing infections and injuries at level of nervous, cardiovascular, and gastrointestinal systems. Obviously, these risks might be remarkably prevented by utilization of the inhaled administration route, even if the latter lacks a significant impact on systemic inflammation, which is especially involved in COPD pathobiology. Hence, though partially clouded by some shadows, pharmacological inhibition of p38 activation might shed a promising light in regard to future therapeutic approaches aimed to improve the management of COPD and severe asthma.
Funding
This paper was not funded.
Disclosure
The authors report no conflicts of interest for this work and no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
References
1. Papi A, Brightling C, Pedersen SE, Reddel HK. Asthma. Lancet. 2018;391(10122):783–800. doi:10.1016/S0140-6736(17)33311-1
2. Khalaf K, Paoletti G, Puggioni F, et al. Asthma from immune pathogenesis to precision medicine. Semin Immunol. 2019;46:101294. doi:10.1016/j.smim.2019.101294
3. Agusti A, Hogg JC. Update on the pathogenesis of chronic obstructive pulmonary disease. N Engl J Med. 2019;381:1248–1256. doi:10.1056/NEJMra1900475
4. Barnes PJ. Inflammatory mechanisms in COPD. J Allergy Clin Immunol. 2016;138:16–27. doi:10.1016/j.jaci.2016.05.011
5. Lambrecht BN, Hammad H, Fahy JV. The cytokines of asthma. Immunity. 2019;50:975–991.
6. Barnes PJ. Inflammatory endotypes in COPD. Allergy. 2019;74:1249–1256. doi:10.1111/all.13760
7. Pelaia C, Vatrella A, Crimi C, Gallelli L, Terracciano R, Pelaia G. Clinical relevance of understanding mitogen-activated protein kinases involved in asthma. Expert Rev Respir Med. 2020;14(5):501–510. doi:10.1080/17476348.2020.1735365
8. Pelaia C, Vatrella A, Sciacqua A, Terracciano R, Pelaia G. Role of p38-mitogen-activated protein kinase in COPD: pathological implications and therapeutic perspectives. Expert Rev Respir Med. 2020;14:485–491. doi:10.1080/17476348.2020.1732821
9. Barnes PJ. Kinases as novel therapeutic targets in asthma and chronic obstructive pulmonary disease. Pharmacol Rev. 2016;68:788–815. doi:10.1124/pr.116.012518
10. Han J, Wu J, Silke J. An overview of mammalian p38 mitogen-activated protein kinases, central regulators of cell stress and receptor signaling. F100Res. 2020;9:653.
11. Khorasasanizadeh M, Eskian M, Gelfand EW, Rezaei N. Mitogen-activated protein kinases as therapeutic targets for asthma. Pharmacol Ther. 2017;174:112–126. doi:10.1016/j.pharmthera.2017.02.024
12. O’Neil JD, Ammit AJ, Clark AR. MAPK p38 regulates inflammatory gene expression via tristetraprolin: doing good by stealth. Int J Biochem Cell Biol. 2018;94:6–9. doi:10.1016/j.biocel.2017.11.003
13. Moosavi SM, Prabhala P, Ammit AJ. Role and regulation of MKP-1 in airway inflammation. Respir Res. 2017;18(1):154. doi:10.1186/s12931-017-0637-3
14. Kuruvilla ME, Lee F, Lee GB. Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol. 2019;56:219–233. doi:10.1007/s12016-018-8712-1
15. Nelson RK, Bush A, Stokes J, Nair P, Akuthota P. Eosinophilic asthma. J Allergy Clin Immunol Pract. 2020;8(2):465–473. doi:10.1016/j.jaip.2019.11.024
16. Cosmi L, Annunziato F. ILC2 are the earliest recruiters of eosinophils in lungs of allergic asthmatic patients. Am J Respir Crit Care Med. 2017;196:666–668. doi:10.1164/rccm.201704-0799ED
17. Pelaia C, Paoletti G, Puggioni F, et al. Interleukin-5 in the pathophysiology of severe asthma. Front Physiol. 2019;10:1514. doi:10.3389/fphys.2019.01514
18. Pelaia C, Crimi C, Vatrella A, Tinello C, Terracciano R, Pelaia G. Molecular targets for biological therapies of severe asthma. Front Immunol. 2020;11:603312. doi:10.3389/fimmu.2020.603312
19. Maneechotesuwan K, Xin Y, Ito K, et al. Regulation of Th2 cytokine genes by p38 MAPK-mediated phosphorylation of GATA-3. J Immunol. 2007;178:2491–2498. doi:10.4049/jimmunol.178.4.2491
20. Kabata H, Moro K, Koyashu S. The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol Rev. 2018;286:37–52. doi:10.1111/imr.12706
21. Petrova T, Pesic J, Pardali K, Gaestel M, Arthur JSC. p38 MAPK signalling regulates cytokine production in IL-33 stimulated type 2 innate lymphoid cells. Sci Rep. 2020;10:3479. doi:10.1038/s41598-020-60089-0
22. Ray A, Kolls JK. Neutrophilic inflammation in asthma and association with disease severity. Trends Immunol. 2017;38(12):942–954. doi:10.1016/j.it.2017.07.003
23. Baines KJ, Fricker M, Mcdonald VM, et al. Sputum transcriptomics implicates increased p38 signalling activity in severe asthma. Respirology. 2020;25:709–718. doi:10.1111/resp.13749
24. Tamura DY, Moore EE, Johnson JL, Zallen G, Aiboshi J, Silliman CC. p38 mitogen-activated protein kinase inhibition attenuates intercellular adhesion molecule-1 up-regulation on human pulmonary microvascular endothelial cells. Surgery. 1998;124:403–407. doi:10.1016/S0039-6060(98)70147-3
25. Nick JA, Young SK, Brown KK, et al. Role of p38 mitogen-activated protein kinase in a murine model of pulmonary inflammation. J Immunol. 2000;164(4):2151–2159. doi:10.4049/jimmunol.164.4.2151
26. Ochayon DE, Ali A, Alarcon PC, et al. IL-33 promotes type 1 cytokine expression via p38 MAPK in human NK cells. J Leukoc Biol. 2020;107(4):663–671. doi:10.1002/JLB.3A0120-379RR
27. Kim DH, Gu A, Lee JS, et al. Suppressive effect of S100A8 and S100A9 on neutrophil apoptosis by cytokine release of human bronchial epithelial cells in asthma. Int J Med Sci. 2020;17:498–509. doi:10.7150/ijms.37833
28. Jaiswal AK, Makhija S, Stahr N, et al. Dendritic cell-restricted progenitors contribute to obesity-associated airway inflammation via Adam17-p38 MAPK-dependent pathway. Front Immunol. 2020;11:363. doi:10.3389/fimmu.2020.00363
29. Zhang Y, Li X, He M, et al. The effects of neutralizing anti-murine interleukin-17A monoclonal antibody on ozone-induced inflammation and glucocorticoids insensitivity in a murine model of asthma. Biomed Pharmacother. 2019;114:108786. doi:10.1016/j.biopha.2019.108786
30. Southworth T, Mason S, Bell A, et al. PI3K, p38 and JAK/STAT signalling in bronchial tissue from patients with asthma following allergen challenge. Biomark Res. 2018;6(1):14. doi:10.1186/s40364-018-0128-9
31. Pelaia G, Cuda G, Vatrella A, et al. Effects of transforming growth factor-β and budesonide on mitogen-activated protein kinase activation and apoptosis in airway epithelial cells. Am J Respir Cell Mol Biol. 2003;29(1):12–18. doi:10.1165/rcmb.2002-0074OC
32. Bucchieri F, Puddicombe SM, Lordan JL, et al. Asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis. Am J Respir Cell Mol Biol. 2002;27(2):179–185. doi:10.1165/ajrcmb.27.2.4699
33. Trautmann A, Schmid-Grendelmeier P, Kruger K, et al. T cells and eosinophils cooperate in the induction of bronchial epithelial cell apoptosis in asthma. J Allergy Clin Immunol. 2002;109:329–337. doi:10.1067/mai.2002.121460
34. O’Sullivan MP, Tyner JW, Holtzman MJ. Apoptosis in the airways: another balancing act in the epithelial program. Am J Respir Cell Mol Biol. 2003;29(1):3–7. doi:10.1165/rcmb.F273
35. Vignola AM, Mirabella F, Costanzo G, et al. Airway remodeling in asthma. Chest. 2003;123(3 Suppl):417S–422S. doi:10.1378/chest.123.3_suppl.417S
36. Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. J Clin Invest. 1999;104(8):1001–1006. doi:10.1172/JCI8124
37. Fitzgerald SM, Lee SA, Hall HK, Chi DS, Krishnaswamy G. Human lung fibroblasts express IL-6 in response to signaling following mast cell contact. Am J Respir Cell Mol Biol. 2004;30:585–593. doi:10.1165/rcmb.2003-0282OC
38. Marumo S, Hoshino Y, Kiyokawa H, et al. p38 mitogen-activated protein kinase determines the susceptibility to cigarette smoke-induced emphysema in mice. BMC Pulm Med. 2014;14:79. doi:10.1186/1471-2466-14-79
39. Wang C, Zhou J, Wang J, et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. 2020;5:248.
40. Renda T, Baraldo S, Pelaia G, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31(1):62–69. doi:10.1183/09031936.00036707
41. Gaffey K, Reynolds S, Plumb J, Kaur M, Singh D. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur Respir J. 2013;42(1):28–41. doi:10.1183/09031936.00170711
42. Huang C, Xie M, He X, Gao H. Activity of sputum p38 MAPK is correlated with airway inflammation and reduced FEV1 in COPD patients. Med Sci Monitor. 2013;19:1229–1235. doi:10.12659/MSM.889880
43. Watanabe T, Jono H, Han J, Lim DJ, Li JD. Synergistic activation of NF-αB by nontypeable Haemophilus influenzae and tumor necrosis factor. Proc Natl Acad Sci USA. 2004;101:3563–3568. doi:10.1073/pnas.0400557101
44. Gallelli L, Pelaia G, Fratto D, et al. Effects of budesonide on p38 MAPK activation, apoptosis and IL-8 secretion, induced by TNF-α and Haemophilus influenzae in human bronchial epithelial cells. Int J Immunopathol Pharmacol. 2010;23:471–479. doi:10.1177/039463201002300209
45. Chung KF. Inflammatory mediators in chronic obstructive pulmonary disease. Curr Drug Targets Inflamm Allergy. 2005;4(6):619–625. doi:10.2174/156801005774912806
46. Pelaia G, Cuda G, Vatrella A, et al. Effects of hydrogen peroxide on MAPK activation, IL-8 production and cell viability in primary cultures of human bronchial epithelial cells. J Cell Biochem. 2004;93(1):142–152. doi:10.1002/jcb.20124
47. Shen H, Yoshida H, Yan F, et al. Synergistic induction of MUC5AC mucin by nontypeable Haemophilus influenzae and Streptococcus pneumoniae. Biochem Biophys Res Commun. 2008;365:795–800. doi:10.1016/j.bbrc.2007.11.060
48. Wedzicha JA, Seemungal TAR. COPD exacerbations: defining their cause and prevention. Lancet. 2007;370(9589):786–796. doi:10.1016/S0140-6736(07)61382-8
49. Wiehler S, Proud D. Interleukin-17A modulates human airway epithelial responses to human rhinovirus infection. Am J Physiol Lung Cell Mol Physiol. 2007;293(2):L505–L515.
50. Jamieson KC, Traves SL, Kooi C, et al. Rhinovirus and bacteria synergistically induce IL-17C release from human airway epithelial cells to promote neutrophil recruitment. J Immunol. 2019;202(1):160–170. doi:10.4049/jimmunol.1800547
51. Bathoorn E, Kerstjens H, Postma D, Timens W, MacNee W. Airways inflammation and treatment during acute exacerbations of COPD. Int J Chron Obstruct Pulmon Dis. 2008;3:217–229. doi:10.2147/COPD.S1210
52. Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med. 2019;200(5):556–564. doi:10.1164/rccm.201810-1975TR
53. Underwood DC, Osborn RR, Kotzer CJ, et al. SB 239063, a potent p38 MAP kinase inhibitor, reduces inflammatory cytokine production, airways eosinophil infiltration, and persistence. J Pharmacol Exp Ther. 2000;293:281–288.
54. Ma JY, Medicherla S, Kerr I, Mangadu R, Protter AA, Higgins LS. Selective p38 mitogen-activated protein kinase inhibitor attenuates lung inflammation and fibrosis in IL-13 transgenic mouse model of asthma. J Asthma Allergy. 2008;1:31–44. doi:10.2147/JAA.S4199
55. Duan W, Chan JH, McKay K. Inhaled p38- mitogen-activated protein kinase antisense oligonucleotide attenuates asthma in mice. Am J Respir Crit Care Med. 2005;171:571–578. doi:10.1164/rccm.200408-1006OC
56. Ratcliffe MJ, Dougall IG. Comparison of the anti-inflammatory effects of cilomilast, budesonide and a p38 mitogen activated protein kinase inhibitor in COPD lung tissue macrophages. BMC Pharmacol Toxicol. 2012;13(1):15. doi:10.1186/2050-6511-13-15
57. Norman P. Investigational p38 inhibitors for the treatment of chronic obstructive pulmonary disease. Expert Opin Investig Drugs. 2015;24(3):383–392. doi:10.1517/13543784.2015.1006358
58. Singh D, Smyth L, Borrill Z, Sweeney L, Tal-Singer R. A randomized, placebo-controlled study of the effects of the p38 MAPK inhibitor SB-681323 on blood biomarkers of inflammation in COPD patients. J Clin Pharmacol. 2010;50(1):94–100. doi:10.1177/0091270009347873
59. Barnes N, Pavord I, Maden C. Evaluation of an oral p38 mitogen activated protein kinase (MAPK) inhibitor SB-681323 in COPD patients. Eur Respir J. 2009;34(Suppl. 53):648s.
60. Betts JC, Mayer RJ, Tal-Singer R, et al. Gene expression changes caused by the p38 MAPK inhibitor dilmapimod in COPD patients: analysis of blood and sputum samples from a randomized, placebo-controlled clinical trial. Pharmacol Res Perspect. 2015;3(1):e00094. doi:10.1002/prp2.94
61. Watz H, Barnacle H, Hartley B, Chan RA. Randomised, double-blind, placebo-controlled trial of the efficacy and safety of losmapimod in patients with chronic obstructive pulmonary disease. Lancet Respir Med. 2014;2:63–72. doi:10.1016/S2213-2600(13)70200-5
62. Marks-Konczalik J, Costa M, Robertson J, McKie E, Yang S, Pascoe S. A post-hoc subgroup analysis of data from a six month clinical trial comparing the efficacy and safety of losmapimod in moderate-severe COPD patients with < 2% and > 2% blood eosinophils. Respir Med. 2015;109:860–869. doi:10.1016/j.rmed.2015.05.003
63. Lamas DA, Lipson DA, Miller BE, et al. Losmapimod Study Investigators. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J Clin Pharmacol. 2012;52:416–424. doi:10.1177/0091270010397050
64. MacNee W, Allan RJ, Jones I, De Salvo MC, Tan LF. Efficacy and safety of the oral p38 inhibitor PH-797804 in chronic obstructive pulmonary disease: a randomized clinical trial. Thorax. 2013;68:738–745.
65. Strambu IR, Kobalava ZD, Magnusson BP, MacKinnon A, Parkin JM. Phase II study of single/repeated doses of acumapimod (BCT197) to treat acute exacerbations of COPD. COPD. 2019;16:344–353. doi:10.1080/15412555.2019.1682535
66. Wedzicha JA, MacKinnon A, Parkin JM. Effectiveness of acumapimod oral p38 inhibitor in the treatment of acute severe exacerbations of COPD: results of the AETHER phase II trial. Am J Respir Crit Care Med. 2018;197:A7710.
67. Agyemang A, Farrell C, Moore W, Parkin J. A physiologically based pharmacokinetic model to predict potential drug-drug interactions and inform dosing of acumopimod, an oral p38 MAPK inhibitor. CPT Pharmacometrics Syst Pharmacol. 2021;10:30–39. doi:10.1002/psp4.12565
68. Ramos FL, Criner GJ. Use of long-term macrolide therapy in chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2014;20(2):153–158. doi:10.1097/MCP.0000000000000028
69. Patel NR, Cunoosamy DM, Fageras M, et al. The development of AZD7624 for prevention of exacerbations in COPD: a randomized controlled trial. Int J COPD. 2018;13:1009–1019. doi:10.2147/COPD.S150576
70. Higham A, Karur P, Jackson N, Cunnoosamy DM, Jansson P, Singh D. Differential anti-inflammatory effects of budesonide and a p38 MAPK inhibitor on COPD pulmonary cells. Int J COPD. 2018;13:1279–1288. doi:10.2147/COPD.S159936
71. Charron CE, Russell P, Ito K, et al. RV568, a narrow-spectrum kinase inhibitor with p38 MAPK-α and -selectivity, suppresses COPD inflammation. Eur Respir J. 2017;50:1700188. doi:10.1183/13993003.00188-2017
72. Millan DS, Bunnage ME, Burrows JL, et al. Design and synthesis of inhaled p38 inhibitors for the treatment of chronic obstructive pulmonary disease. J Med Chem. 2011;54(22):7797–7814. doi:10.1021/jm200677b
73. Mei D, Tan WSD, Wong WSF. Pharmacological strategies to regain steroid sensitivity in severe asthma and COPD. Curr Opin Pharmacol. 2019;46:73–81. doi:10.1016/j.coph.2019.04.010
74. Pelaia G, Vatrella A, Busceti MT, et al. Molecular and cellular mechanisms underlying the therapeutic effects of budesonide in asthma. Pulm Pharmacol Ther. 2016;40:15–21. doi:10.1016/j.pupt.2016.07.001
75. Bhavsar P, Hew M, Khorasani N, et al. Relative corticosteroid insensitivity of alveolar macrophages in severe asthma compared to non-severe asthma. Thorax. 2008;63:784–790. doi:10.1136/thx.2007.090027
76. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2013;131:636–645. doi:10.1016/j.jaci.2012.12.1564
77. Lea S, Li J, Plumb J, et al. p38 MAPK and glucocorticoid receptor crosstalk in bronchial epithelial cells. J Mol Med. 2020;98(3):361–374. doi:10.1007/s00109-020-01873-3
78. Khalaf RM, Lea SR, Metcalfe HJ, Singh D. Mechanisms of corticosteroid insensitivity in COPD alveolar macrophages exposed to NTHi. Respir Res. 2017;18(1):61. doi:10.1186/s12931-017-0539-4
79. Higham A, Singh D. Dexamethasone and p38 MAPK inhibition of cytokine production from human lung fibroblasts. Fundam Clin Pharmacol. 2021.
80. Goldstein DM, Kuglstatter A, Lou Y, Soth MJ. Selective p38α inhibitors evaluated for the treatment of chronic inflammatory disorders. J Med Chem. 2010;53:2345–2353. doi:10.1021/jm9012906
81. Khorasani N, Baker J, Johnson M, Chung KF, Bhavsar PK. Reversal of corticosteroid insensitivity by p38 MAPK inhibition in peripheral blood mononuclear cells from COPD. Int J COPD. 2015;10:283–291.
82. Saba-El-Leil MK, Fremin C, Meloche S. Redundancy in the world MAP kinases: all for one. Front Cell Dev Biol. 2016;4:31. doi:10.3389/fcell.2016.00067
83. Hammaker D, Firestein GS. “Go upstream, young man”: lessons learned from the p38 saga. Annals Rheum Dis. 2010;69:177–182. doi:10.1136/ard.2009.119479
84. Chopra P, Kanoje V, Semwal A, Ray A. Therapeutic potential of inhaled p38 mitogen-activated protein kinase inhibitors for inflammatory pulmonary diseases. Expert Opin Investig Drugs. 2008;17(10):1411–1425. doi:10.1517/13543784.17.10.1411
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.