")
Back to Archived Journals » Research and Reports in Biology » Volume 7
Role of Ocrl1 and Inpp5E in primary cilia assembly and maintenance: a phosphatidylinositol phosphatase relay system?
Authors Madhivanan K, Ramadesika S, Aguilar RC
Received 12 September 2015
Accepted for publication 25 December 2015
Published 19 February 2016 Volume 2016:7 Pages 15—29
DOI https://doi.org/10.2147/RRB.S62510
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Zvi Kelman
Kayalvizhi Madhivanan,* Swetha Ramadesikan,* R Claudio Aguilar
Department of Biological Sciences, Purdue University, West Lafayette, IN, USA
*These authors contributed equally to this work
Abstract: The primary cilium (PC) is a plasma membrane-derived structure of great importance for cell and organismal physiology. Indeed, abnormalities in assembly or function of the PC trigger the onset of a group of genetic diseases collectively known as ciliopathies. In recent years, it has become evident that the integrity and function of the PC depends substantially on signaling elements such as phosphoinositides (PI) and their regulators. Because phospholipids such as PI(4,5)P2 constitute recruitment platforms for cytoskeleton, signaling, and trafficking machinery, control over their levels is critical for PC function. Although information about phosphoinositol phosphate (PIP) kinases in the PC is scarce, a growing body of evidence supports a role for PIP phosphatases in cilia assembly/maintenance. Indeed, deficiencies in two 5′ PIP phosphatases, Inpp5E and Ocrl1, are clearly linked to ciliopathies like Joubert/MORM syndromes, or ciliopathy-associated diseases like Lowe syndrome. Here, we review the unique roles of these proteins and their specific site of action for ensuring ciliary integrity. Further, we discuss the possibility that a phosphatase relay system able to pass PI control from a preciliary to an intraciliary compartment is in place to ensure PC integrity/function.
Keywords: primary cilia, Ocrl1, Inpp5E, Pip2, Pip3
Introduction
The primary cilium (PC) is an antenna-like, microtubule-based structure that senses and transduces extracellular signals to the intracellular space, leading to changes in cellular activity.1,2 Given the crucial function that this plasma membrane specialization plays in the control of developmental signaling pathways, a substantial amount of effort has been invested in understanding how signaling molecules are functionally articulated in the PC. For example, given their signaling relevance, the role of phosphoinositides in integrity and function of the cilia has been the object of intense research in recent times.
In this review, we summarize the importance of phosphoinositides for the general cell physiology to then focus on known details concerning their ciliary distribution, regulation, and proposed functions. The emerging role of the lipid phosphatases Inpp5E and Ocrl1 (as well as its paralog, Inpp5B) in ciliogenesis and cilia maintenance is highlighted. We also describe the impact on protein function and signaling of disease-causing mutations in the INPP5E and OCRL1 genes. Finally, we propose and discuss the existence of a “relay” mechanism by which Inpp5E and Ocrl1 act in a sequential manner to control phosphoinositide function to sustain cilia assembly and maintenance.
Relevance of phosphoinositides for general cell physiology
Phosphatidylinositol (PI) phosphates or PIPs are low- abundance, highly labile phospholipids that play a substantial role in cell physiology. These phospholipids transiently accumulate in discrete areas of different cell membranes, where they constitute platforms for the recruitment of specific PIP-binding proteins. Turnover of PIPs chiefly contributes to the disassembly of PIP-binding protein complexes and their consequent deactivation. Because proteins involved in the regulation of cell signaling, cytoskeleton assembly, and vesicle trafficking are among the PIP-recruited elements, PIP dynamics directly affect these cellular processes, and therefore, impact crucial cell functions such as cell homeostasis, phagocytosis, cytokinesis, and cell migration. Therefore, it is not surprising that mutations affecting PIP regulators give rise to serious genetic diseases.3–5
The seven PIP species [PI3P, PI4P, PI5P, PI(3,4)P2, PI(3,5)P2, PI(4,5)P2, and PI(3,4,5)P3] present in eukaryotic cells are spatiotemporally regulated by several kinases and phosphatases, which add and remove phosphate groups from their substrates, respectively (Figure 1A).6 Concerted action of these regulators results in the accumulation of specific PIPs in distinct cellular compartments that confers identity and triggers characteristic signaling events (Figure 1B).
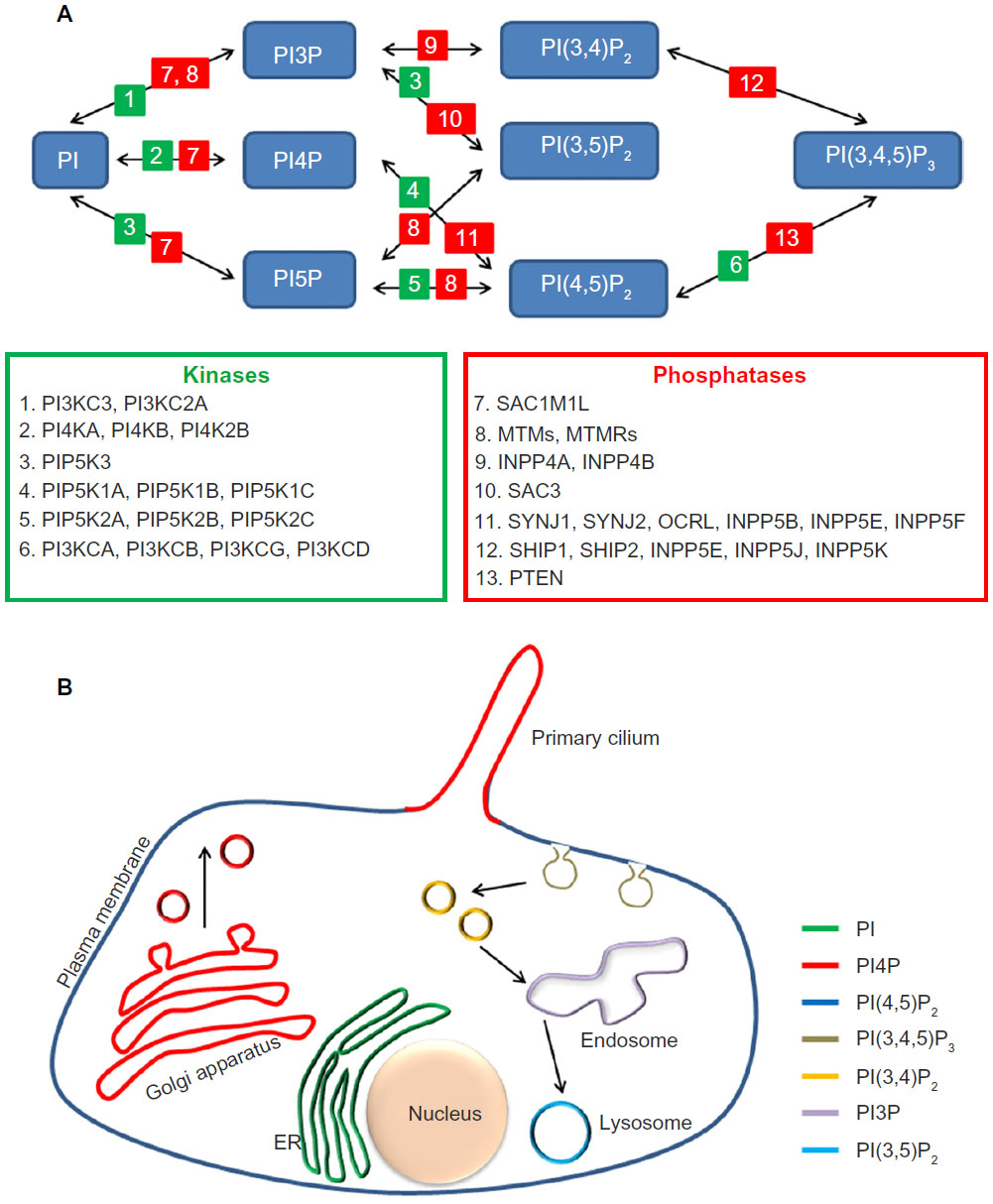 | Figure 1 Phosphoinositides metabolism and distribution. |
One of the best studied PIP species is PI(4,5)P2, which is often produced by action of PI4P-5 kinase (PI5K) on PI4P. PI(4,5)P2 is plasma membrane (PM) enriched and is required for initiation and regulation of several processes, such as Arp2/3-mediated actin nucleation, cell–cell and cell–matrix adhesion, and assembly of endocytic sites. Like many other cell signaling elements, PI(4,5)P2 is further modified to participate in different pathways or to terminate its signaling activity. For example, upon activation by G-protein coupled receptors (GPCR), phospholipase C acts on PI(4,5)P2 to produce two second messengers: diacylglycerol (DAG) and inositol trisphosphate (IP3).7 Among the multiple activities of these agonists, DAG activates members of the protein kinase C family, while IP3 promotes the release of Ca2+ from intracellular stores resulting in the activation of signaling cascades.7 Along the same lines, in response to growth factors, PI3K is activated and phosphorylates PI(4,5)P2 at position 3′ leading to the production of PI(3,4,5)P3 at the PM. The resulting increase in the levels of this phospholipid provides sites for the recruitment of the basic components of the Akt signaling pathway.7 This signaling event can be turned off by the action of the PI3 phosphatase PTEN that removes the phosphate group from position 3′ in the inositol ring of PI(3,4,5)P3.7 Similarly, PI(4,5)P2 is depleted from membranes by the action of 5′ phosphatases such as synaptojanin or Ocrl1 (Figure 1A), leading to disassembly of PI(4,5)P2-binding complexes and termination of their biochemical activity. Therefore, inability to regulate these phosphoinositide surely leads to abnormal signaling and protein trafficking.
The PIP content of different cellular compartments has been well established with PI(4,5)P2 found predominantly in the PM, PI4P in the Golgi, PI3P in early endosomes, and PI(3,5)P2 in late endocytic organelles (Figure 1B). Recent studies have also unveiled the PIP content of the specialized signaling organelle known as the primary cilium,8,9 which has a strikingly different PIP content from the PM, though in principle they are continuous with each other. The following section will highlight the importance of PIP regulation in the maintenance of the PC.
Phosphoinositides in the primary cilium
During the last decade, we have witnessed a surge of studies centered on the PC, an immotile, hair-like membrane structure present in almost every cell type. The PC is a dynamic structure that assembles/disassembles in a cell cycle–dependent manner. At the core of the PC lies a microtubule-based core (9+0 arrangement) comprising the axoneme nucleated from the basal body. The axoneme is ensheathed by the ciliary membrane that is continuous with the PM. However, several lines of evidence indicate the presence of barriers at the ciliary base that avoids free diffusion of molecules, maintaining the composition of the ciliary membrane different from that of the bulk of the PM (Figure 1B).10–15
This PM protrusion houses a high concentration of receptors that can sense and respond to a number of chemical, mechanical, and other extracellular cues. Examples of receptors localized in the PC include rhodopsin, polycystin-1 and 2, transforming growth factor-β, somatostatin receptor-3, melanin-concentrating hormone receptor-1, serotonin receptor, platelet-derived growth factor-α, and members of the Hedgehog and Wnt signaling pathways.16–23 As a result, the PC hosts a plethora of signaling pathways critical for growth, differentiation, development, sensory perception, hormonal regulation, and mechanosensing.2 Consequently, abnormal PC function leads to widespread effects on the physiology of the organism. Indeed, abnormal PC have been linked to a heterogeneous group of multiorgan diseases known as ciliopathies, caused by single-gene mutations in more than 50 loci.24 These illnesses can be lethal and are characterized by overlapping phenotypes that may include retinal degeneration, kidney dysfunction, infertility, cognitive impairment, polydactyly, situs inversus, obesity, diabetes, and other manifestations. The medical relevance of this organelle has resulted in an immense surge of research aimed at understanding the mechanisms of assembly, maintenance, and function of the PC.
What PIPs localize to the PC?
Although PI3P is known to be enriched at the base of the cilia in the PI3K-C2α-containing pericentriolar endosomal compartment (PRE),25 information concerning the PIP composition of the PC has only recently started to emerge. Indeed, two recent studies8,9 specifically addressed PIP composition within the PC. While one study used PI-specific antibodies to examine the neural stem cell cilium,8 the other relied on genetically encoded markers known to bind PI4P (EGFP-2xp4MSidM) and PI(4,5)P2 (EYFP-PHPLCδ1) in different cell lines.9 Both these studies concluded that the major PIP in the cilium is PI4P, while PI(4,5)P2 is restricted to the ciliary base.
What is known in terms of PIP functional relevance?
The PI3P pool at the PRE compartment is required for Rab8 and Rab11 activation,25 and in consequence, for ciliary elongation and cargo (eg, Smoothened and PKD2) translocation.25,26 Although less is known about the role of PI4P, depletion of PI4P-binding protein FAPP2 (involved in the transport of newly synthesized apical proteins) led to a delay in ciliogenesis with accumulation of vesicles at the ciliary base.27 PI(4,5)P2 is only transiently present in the PC where it is involved, for example, in the recruitment of the bridging protein TULP3, that in turn is required for the transport of GPCRs.28 The lack of PI(4,5)P2 regulation inside the cilia is expected to result in aberrant signaling, in part due to abnormal GPCRs levels.8
The concentration of PI(4,5)P2 is controlled by both 5′ kinases and phosphatases.7 However, besides immunofluorescence-based detection of PIP5Kγ at the ciliary base,8 no information is available concerning the localization and function of PIP5 kinases in the PC. Also taking into account the abundance of intraciliary PI4P, proteins such as the PIP5 phosphatases – Inpp5B, Inpp5E, and Ocrl1 – are predicted to be among the major PIP regulators implicated in ciliary function.29 Further, results that will be summarized in this review are consistent with the existence of a relay mechanism by which, during vesicle trafficking to the PC, PI(4,5)P2 control is transferred from Ocrl1 at the PC base to Inpp5E within the cilia.
Phosphatases in ciliogenesis and ciliary maintenance
Three 5′ phosphatases Ocrl1, Inpp5B, and Inpp5E (EC 3.1.3.36) have been localized to the ciliary base and shaft.30–32 In general 5′ phosphatases are classified in four categories (I–IV) based on their substrate specificity.7 While the Ocrl1 and Inpp5B paralogs (~45% identity) belong to type II, Inpp5E is the sole member of the type IV category. Phosphatase activity of all members requires the presence of two signature motifs, (F/Y)WXGDXN(F/Y)R and P(A/S)(W/Y)(C/T)DR(I/V)L(W/Y), separated by ~60–75 residues. Ocrl1, Inpp5B, and Inpp5E are conserved in mice, zebrafish, and frogs. However, only one homologue is present in flies and worms, which are similar to Ocrl1 and Inpp5B. The closest homologue to Inpp5E in worms is the Cil-1 phosphatase.33
Ocrl1 and Inpp5B have similar substrate specificity wherein they can remove the 5-phosphate from PI(4,5)P2, PI(3,4,5)P3, PI(1,4,5)P3, and PI(1,3,4,5)P4.34,35 However, further studies using purified, recombinant phosphatase domains showed that Ocrl1 displays its highest affinity for PI(4,5)P2,36 while Inpp5E does it for PI(3,4,5)P3.37
In the following sections, we will review domain organization, interactions, and function of these 5′ phosphatases as well as the diseases associated with mutations in their corresponding genes.
Oculocerebrorenal syndrome of Lowe protein 1, Ocrl1
Domain organization
Both Ocrl1 and its paralog Inpp5B display four major domains, from N- to C-terminus: PH (pleckstrin homology), 5′ phosphatase, ASH (ASPM-SPD2-Hydin), and a RhoGAP (Rho GTPase-activating protein) domains (Figure 2). The PH domains of Ocrl1 and Inpp5B are structurally very similar and do not have any lipid-binding partners described yet.38 In contrast to Innp5b, the PH domain of Ocrl1 contains a clathrin-binding motif (CBM) and is followed by a linker region containing a clathrin-associated adaptor protein AP2 binding site (ABS). Ocrl1 has a second CBM toward its C-terminal region (Figure 2), with enhanced affinity for clathrin in the nervous system-enriched isoform A.39 Therefore, a major difference between Ocrl1 and Inpp5B is the lack of CBM and ABS in the latter. The 5′ phosphatase domain displays conserved motifs required for its phosphatase function and substrate binding.40 Although found across the gene, majority of mutations in Lowe Syndrome (LS) patients affect the phosphatase domain in the mutated protein.
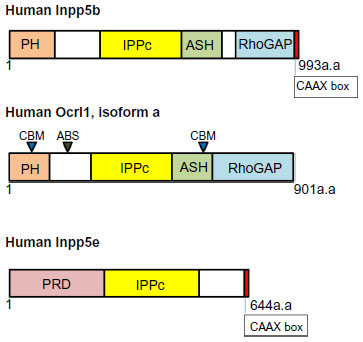 | Figure 2 Domain organization of phosphatidylinositol 5′ phosphatases Inpp5B, Ocrl1, and Inpp5E. |
The ASH and RhoGAP-like domains synergistically interact with the endosomal F&H motif-containing proteins, APPL1 and IPIP27A/B (the latter also known as Ses1/Ses2).41–43 The ASH domain also constitutes a binding site for Rabs, with the highest affinity for Rab8a,44–46 leading to an interaction that is critical for Ocrl1 role in ciliogenesis.30,31,47 The RhoGAP-like domain does not exhibit GTPase-activating protein (GAP) activity but mediates binding to the Rho GTPases Rac1 and Cdc42.48,49
The ability of Ocrl1 to interact with several Rabs ensures its localization in several cellular compartments. Ocrl1 has been observed in early endosomes (Rab5), recycling endosomes (Rab35), and at the Golgi apparatus (Rab1, Rab6),50,51 and it localizes at the ciliary base because of its interaction with Rab8.30 Ocrl1 also has been observed in membrane ruffles following Rac1 activation.49 Notably, Ocrl1 phosphatase activity requires Rab GTPase binding and membrane recruitment.51
Functions of Ocrl1
Most functions of Ocrl1 can be rationalized taking into consideration its PIP phosphatase activity and substrate preference. Specifically, Ocrl1-dependent hydrolysis of PI(4,5)P2 and PI(3,4,5)P3 removes from targeted membranes the recruitment platforms for different protein complexes, such as those involved in actin cytoskeleton remodeling and vesicle trafficking.
Ocrl1 function is also intimately linked with its ability to localize to different intracellular stations, which in turn depends on binding to different Rab GTPases (see “Domain organization” section) and perhaps clathrin and AP2. In addition, some of these interactions can affect Ocrl1’s catalytic activity (eg, by binding to Rabs) or may have a phosphatase-independent signaling relevance (eg, by recognition of the Rho GTPases Rac1 and Cdc42).
Vesicle trafficking and cilium assembly
Several studies have shown that upon depletion or abnormal function of Ocrl1, endosome-to-TGN trafficking and recycling to the plasma membrane is affected, leading to cargo accumulation in enlarged endosomes.52–54 In addition, mannose-6-phosphate receptor (M6PR) missorting to the plasma membrane was also observed upon Ocrl1 deficiency.54,55 Although some authors found no major defects in uptake of transferrin, epidermal growth factor, and low-density lipoprotein upon Ocrl1 deficiency, other studies described aberrant endocytic vesicles and abnormal internalization.53,56,57 Recycling defects in LS patient proximal tubule cells lead to low cell surface levels (and endosomal accumulation) of the multiligand receptor megalin and explains the low molecular weight (LMW) proteinuria characteristic of LS patients.54 This phenotype in kidney cells was also confirmed by knockdown (KD) of Ocrl1 in the HK2 and MDCK cell lines. A recent study showed in vivo evidence in Ocrl1–/– zebrafish of defective megalin recycling and hence reduced uptake of its ligand RAP.58 However, another study reported no defect in megalin recycling or megalin ligand uptake upon Ocrl1 KD.55 Absence of Ocrl1 function also triggers the accumulation of PI(4,5)P2 on endosomes that in turn leads to enhanced, N-WASP-dependent actin polymerization producing the so-called “actin comet tails”.54,55 This phenotype could be rescued by Ocrl1 with a catalytically active phosphatase domain or by use of low concentrations of actin-depolymerizing drugs.54
Ocrl1 also participates in the trafficking of membrane proteins (but not soluble cargoes) to the PC30 (Figure 3). Results suggest two PC routes with Ocrl1 involvement: 1) a Rab8-dependent route from the TGN or from clathrin-independent recycling to the PC; and 2) an indirect route in which cargo is rerouted from the plasma membrane to the PC via the endosomal compartment30 (Figure 3). The latter route, however, does not seem to require Ocrl1 binding to clathrin and AP2, but its interaction with the endosomal proteins APPL1 and IPIP27/Ses instead.30 It has been shown that these proteins recognize the same binding site in Ocrl1.42 Interestingly, experimental evidence indicates that Ocrl1 first binds APPL1 on Rab5-positive endosomes to then transition to IPIP27/Ses-containing complexes on a different endosomal compartment (Figure 3A).42 This Ocrl1-binding relay event suggests a maturation of the Ocrl1-containing endosomal compartment.
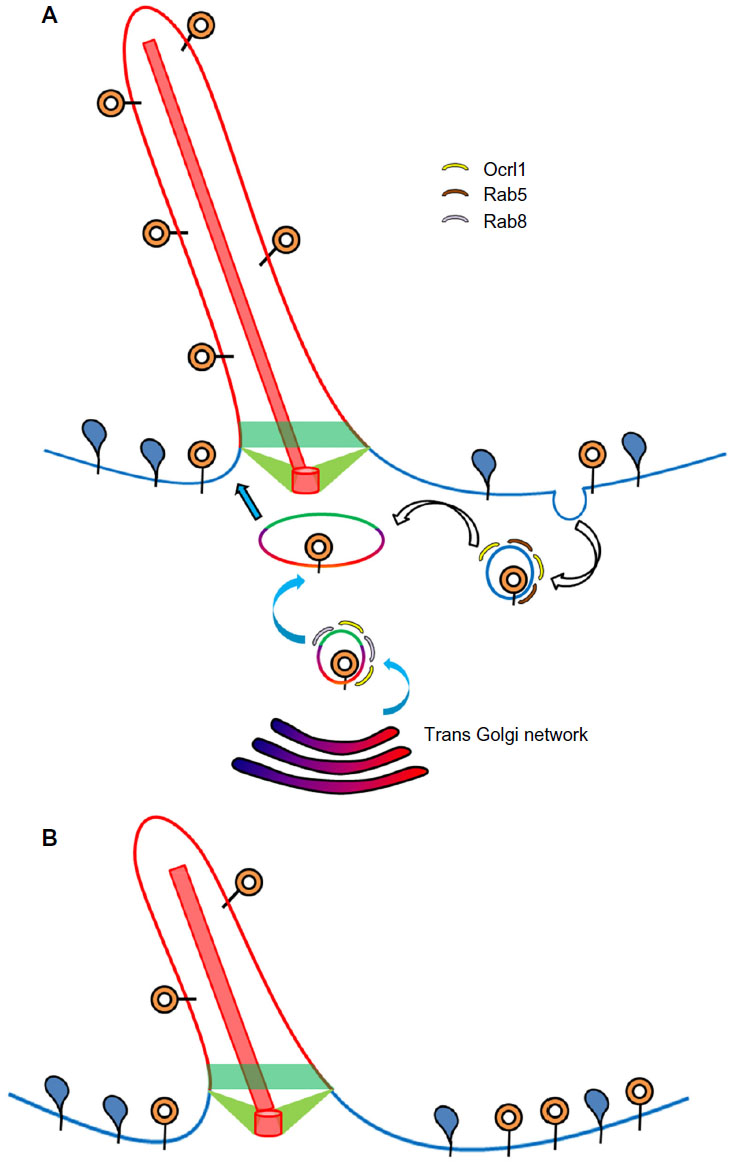 | Figure 3 Ciliary role of Ocrl1. |
During ciliogenesis, Ocrl1 localizes to different intracellular compartments including the basal body of the cilium30 and to the PC itself in some cell types.31 Importantly, Ocrl1 depletion resulted in reduced targeting of ciliary-localized rhodopsin30 (Figure 3B), suggesting a defect in protein sorting to the PC. Further, Ocrl1-deficient patient fibroblasts as well as 3T3 fibroblasts and RPE Ocrl1 KD cells showed a reduction in their ability to assemble PC and a reduction in cilia length.30,31 However, Ocrl1 KD in MDCK cells was reported to cause an increase in ciliary length.47
Ocrl1 has been implicated in the ciliary trafficking and localization of transient receptor potential vanilloid 4 (TRPV4).59 TRPV4 plays a role in the ciliary-dependent pressure-sensing pathways and in the regulation of the transcriptional activity of TNF-α and TGF-β1. In normal ocular trabecular meshwork (TM), an increase in pressure results in a decrease in ciliary length and an increase in transcription of TNF-α and TGF-β1.59 Consequently, loss of Ocrl1 resulted in nonresponsiveness to an increase in intraocular pressure in the ocular TM.59
Ocrl1-deficient zebrafish exhibit underdeveloped eyes and brain, laterality defects, and cystic kidneys, which are characteristics seen in other ciliopathy models.30,31 In addition, defective cilia were also observed in Ocrl1 morphant fish, especially in pronephros and in Kupffer’s vesicle.30,31,47
Membrane dynamics
Other cellular processes requiring PI(4,5)P2/PI(3,4,5)P3 control also showed Ocrl1-dependence, such as phagocytosis, macropinocytosis, cell migration, and cytokinesis. These processes involve Rho GTPase signaling and active actin cytoskeleton reorganization leading to substantial membrane reorganization. Although the RhoGAP domain in Ocrl1 does not have GAP activity, it binds to both activated and inactivated forms of Rac1.48 In fact, although mechanistically unclear, loss of Ocrl1 has been linked to Rac1 inactivation and Rho hyperactivation.53,60 Abnormalities in the actin cytoskeleton organization upon Ocrl1 lack of function have been reported as well.61
Phagocytosis and macropinocytosis
Ocrl1’s phosphatase activity is required for the hydrolysis of PI(4,5)P2 and the consequent termination of actin polymerization at the phagocytic cup during closure.62–64 A similar mechanism was proposed to explain defects in fluid phase uptake observed in LS patient fibroblasts as compared to normal cells.56
Cell migration
Wound healing and transwell migration assays using LS patient fibroblasts and in Ocrl1 KD NIH3T3 cells established a role for Ocrl1 in cell migration.56 Further, the cell migration defects were also confirmed in a zebrafish model of LS as deficiencies in melanophore and neural crest cell (NCC) migration.30 Notably, the phosphatase function of Ocrl1 and its binding to the endocytic machinery (clathrin and AP2) were required for its role in cell migration and for localization to membrane ruffles.56
Cytokinesis
Cytokinesis defects have been observed in fly and mammalian cells lacking Ocrl1, causing the accumulation of multinucleated cells.50,65 In the absence of Ocrl1, high levels of PI(4,5)P2 were detected on endomembranes, leading to the mistargeting of the cytokinesis machinery. In Ocrl1-deficient mammalian cells, the presence of PI(4,5)P2 on intercellular bridges resulted in the accumulation of actin structures, which blocked abscission of the dividing cells at the last stage of cytokinesis.65
Lowe syndrome versus Dent-2 disease
In 1952, Lowe et al66 described a developmental disease mainly affecting the eye, brain, and kidneys, which is currently known as the oculocerebrorenal syndrome of Lowe (OCRL) or Lowe syndrome (LS). This lethal X-linked disease was later found to be caused by mutations in the OCRL1 gene34 and is known to affect approximately 1 in 500,000 births.67 LS is characterized by mental retardation, congenital bilateral cataracts, and renal dysfunction.68 Kidney symptoms include LMW proteinuria, renal proximal tubule acidosis, hypercalciuria, and hypokalemia. Patients eventually develop end-stage renal disease, which often results in death after the second decade of life.69 Nervous system abnormalities lead to different degrees of mental retardation68 and obsessive compulsive disorder, and motor development delays may become apparent during the third year of life.70
OCRL1 mutations were also identified as causative of another disease mostly restricted to renal symptoms, referred to as Dent-2 disease (to differentiate it from the classical Dent disease caused by CLCN5 mutations71). In some cases, Dent-2 disease patients also developed mild peripheral cataracts and presented cognitive abnormalities, although with later onset than LS.72,73 The contribution of the different phenotypes (eg, PC vs membrane remodeling abnormalities60) to the patient symptomatology still needs to be fully investigated. Also, the genotype–phenotype correlation leading to LS versus Dent-2 disease is still unclear. Moreover, mutations affecting the phosphatase domain can result in a variety of patient symptom manifestations and severity,74 and abnormalities in other domains of the protein can affect protein expression and/or phosphatase activity.74 Strikingly, the same missense mutation may result in a severe LS symptoms in one patient, and Dent-2 in another.29 This suggests that genetic modifiers may contribute to symptom severity. Nevertheless, some mutations seem to be prone to lead to Dent-2 versus LS.74,75 Specifically, frameshift and nonsense mutations clustered in exons 1–7 seem to be linked to Dent-2 disease, while the mutations leading to LS are clustered between exons 8–23 (most frequently affecting the phosphatase domain of the mutated protein).76 Interestingly, the presence of an alternative start site in exon 8 explains the existence of Ocrl1 transcripts lacking exons 1–7.71,75 These alternative transcripts would give origin to Ocrl1 variants starting at the phosphatase domain that would be enriched in Dent-2 patients.75
Functions of Inpp5B
As mentioned earlier, Inpp5B is the closer paralog to Ocrl1 and a partially redundant 5′ phosphatase. Indeed, murine models of LS failed to recapitulate the characteristics of the human disease77 because of the differential expression and splicing patterns of Inpp5B versus Ocrl1 in humans and mice.78
It has been clearly established that the LS fibroblast cellular phenotypes of cell spreading and cell migration are independent of Inpp5B.56 Most recently, fibroblasts of LS and Dent-2 patients were compared for phenotypic defect.79 Fibroblasts from both disease groups showed the same category of defects such as lesser F-actin fibers, disorganized α-actinin staining, and ciliogenesis defects. However, Dent-2 disease fibroblasts exhibited milder phenotypes than those from LS patient cells.79 Notably, the levels of PI(4,5)P2 and Inpp5B were very similar in LS and Dent patient fibroblasts.79 These results suggest that Inpp5B does not contribute to the milder phenotypes exhibited by the Dent-2 patient fibroblasts and that, in addition to the phosphatase activity, other functions contribute to the phenotypic manifestations seen in patients.
Although no disease has been identified as a result of INPP5B mutations, Inpp5B has ciliary localization.80 Indeed, KD of Inpp5B in zebrafish resulted in edema, body asymmetry, kinked tail, and microphthalmia, which are classical characteristics observed in ciliary disease zebrafish models. These animals showed defective ciliary formation in the Kupffer’s vesicle, with lesser and shorter cilia in comparison to controls.80 These defects were rescued upon reintroduction of Inpp5B mRNA, thereby establishing a ciliary role for Inpp5B.80 In addition, Ocrl1 and Inpp5B morpholino coinjected fish showed more severe phenotypes suggesting that they play a synergistic role in the ciliary function in zebrafish.80 Studies using LS fibroblast also showed partial rescue of the ciliogenesis phenotype upon overexpression of Inpp5B.30,80
Inositol polyphosphate-5′ phosphatase, E: Inpp5E
Inpp5E has an N-terminus proline-rich domain (PRD) with approximately 13 PxxP consensus sequences that bind SH3 domain-containing proteins, two immunoreceptor tyrosine-associated motif (ITAM), the 5′ phosphatase domain, and a C-terminus CAAX box that undergoes prenylation for membrane anchoring. An alternative splice variant of Inpp5E that lacks the 34 amino acid region that contains the ITAMs has been described (Figure 2).37,81
Inpp5E is known to be tyrosine phosphorylated by activated insulin receptor82 and Ser/Thr-phosphorylated by Aurora kinase A (AurkA).83 In addition to hydrolyzing the product of PI3K enzymatic activity, it has been reported that Inpp5E interacts with p85 subunit of PI3K, negatively regulating the Akt pathway, and also binds Insulin receptor substrate 2(IRS2).82,84 A yeast two-hybrid screen identified Rab20, a Golgi- and phagosome-localized protein as the only Rab GTPase that interacts with Inpp5E.46 Interestingly, Inpp5E preferentially binds to the GDP-bound form of Rab20 over the GTP-bound form, and they colocalize in the Golgi apparatus.46 However, the role of this interaction in protein targeting is not clear. Indeed, it has been demonstrated that the N-terminus PRD of Inpp5E is sufficient to localize it to the Golgi,46,81 suggesting that Rab20 binding is not the main factor for Inpp5E targeting. In proliferating cells, Inpp5E localizes at the trans-Golgi network81 while in quiescent cells, Inpp5E localizes to the primary cilium.32,85
Tandem affinity purification using Inpp5E as bait has revealed that Inpp5E interacts with several proteins in the ciliary network including the centrosomal protein CEP164, RUVBL1, RUVBL2, and 14-3-3µ/γ.86 Although CEP164 is involved in PC formation and the 14-3-3 signaling proteins have been observed to localize in the cilia, little is known about the relevance of RUVBL1 and 2 in ciliary processes; therefore, the role of these latter interactions is still under investigation.86
Although Inpp5E displays a typical ciliary targeting signal located between the phosphatase domain and the CAAX box, its ciliary localization depends on the highly conserved sequence FDRELYL. Biochemical studies revealed that the ciliary recruitment of Inpp5E is mediated by the GTPase Arl13B, which interacts with the phosphatase via its FDERLYL sequence.86 In addition, the phosphodiesterase and prenyl-binding protein PDE6D was found to form a complex with Inpp5E in a CAAX box-dependent manner and to play a role in Inpp5E ciliary localization.86 Indeed, absence of PDE6D was linked to Inpp5E mislocalization and to Joubert syndrome.87 PDE6D binding to prenylated proteins is known to be regulated by the ARF-like proteins Arl2 and Arl3; however, whether Arl2/3 or Arl13, directly or indirectly control the formation of a PDE6D-Inpp5E-targeted complex needs to be further clarified.
Functions of Inpp5E
The cellular functions of Inpp5E directly depend on its interaction partners and its 5′ phosphatase activity [mainly directed against PI(3,4,5)P3]. As indicated earlier, binding to p85 has been proposed to lead to PI3K inhibition.82 Synergistic to this effect, Inpp5E-mediated hydrolysis of PI(3,4,5)P3 is linked to inhibition of the PI3K/AKT signaling pathway. As expected, Inpp5E-deficient cells displayed high levels of PI(3,4,5)P385 and increased phospho-Akt levels;83 further, cells expressing Joubert syndrome Inpp5E mutants also show high levels of phosphorylated Akt.85 Recently, mouse embryonic fibroblasts (MEFs) from Inpp5E–/– mice and NSCs lacking Inpp5E have been show to express high levels of PI(4,5)P2 at the cilia, underscoring the role of Inpp5E as a 5′ phosphatase.8,9
Importantly, Plotnikova et al83 showed that Inpp5E directly interacts with AurkA, resulting in the phosphorylation of Inpp5E by AurkA and the enhancement of its catalytic activity toward PI(3,4,5)P3 (particularly at the base of the PC where the kinase localizes88). Interestingly, AurkA is one of the targets modulated by the Akt signaling pathway89 and has been involved in the regulation of PC stability through complex formation with histone deacetylase 6 (HDAC6).88
As expected, because of its role in the inhibition of the PI3K/AKT pathway, Inpp5E has a multiplicity of effects on cellular processes. For example, Inpp5E diminishes the anti-apoptotic effects mediated by PI3K/AKT signaling.90 Overexpression of Inpp5E led to up to 20% increase in apoptotic cells and cell accumulation in G1 phase of cell cycle. Prolonged exposure to high levels of Inpp5E shifted the cell population to G2/M phase and induced cell growth arrest; these observations led to propose a tumor-suppressive role for this enzyme.90
In addition, and since PI(3,5)P2 is required for phagocytic cup closure, Inpp5E inhibits phagocytosis.91 Also, Inpp5E has been shown to interact with the phagosome-located Rab20.46,92 In Inpp5E knockdown cells, recruitment of Rab20 to the phagosome was unaffected, but Rab20’s retention time was significantly lower92, and so was Rab5’s retention time.92 These results suggest that Inpp5E is involved in tethering these Rabs to the phagosome. Interestingly, Rab5 inactivation and subsequent removal from phagosome is thought to contribute to phagosomal maturation.93 Therefore, Inpp5E seems to impair phagocytosis by depleting PI(3,5)P2 and by delaying the release of Rabs.
Phylogenetic analysis predicts that Inpp5E is the closest mammalian protein to the Caenorhabditis elegans 5′ phosphatase Cil-1.33 This protein is involved in the trafficking of the transient receptor potential-polycystin (TRPP) complex by regulating the levels of PI(3,5)P2 and PI3P; therefore, it is possible that Inpp5E would play a role in protein trafficking in mammalian cells.33
Neural stem cells (NSCs) rely on cilium for differentiation and proliferation via the Sonic Hedgehog (Shh) pathway.94 Inpp5E has been shown to be the key 5′ phosphatase that maintains high levels of localized PI4P in the cilium of these cells while ensuring low PI(4,5)P2 levels (Figure 4). Studies in mice have shown that absence of the enzyme resulted in activation of Akt signaling and proliferation (likely due to excess of PI(3,4,5)P3).32 Interestingly, lack of Inpp5E also led to PI(4,5)P2 accumulation, but this enrichment was found to be more pronounced at the ciliary tips rather than at the base.8 PI(4,5)P2 served as a site for recruitment of the tubby-like protein Tulp3 and its ciliary cargo the GPCRs Gpr161 (Figure 3), which negatively regulates Shh pathways.28,95 NSCs isolated from INPP5E-null mice exhibited stunted cilium with a bulged tip, which stained positive for accumulation of Tulp3 and Gpr161.8 In consequence, inhibition of Shh signaling was observed in these cells, despite stimulation with Shh ligand and normal levels or trafficking of Shh proteins.8,95 It should be noted that mutations in INPP5E lead to Joubert and mental retardation, truncal obesity, retinal degeneration and micropenis (MORM) syndromes, which are characterized by polydactyly and exencephaly causative of aberrant Shh signaling.96 The work of Chavez et al8 further solidifies the model by showing that lack of Inpp5E causes the PI(4,5)P2 accumulation at the PC tip and subsequently leads to Gpr161 enrichment in the ciliary membrane.
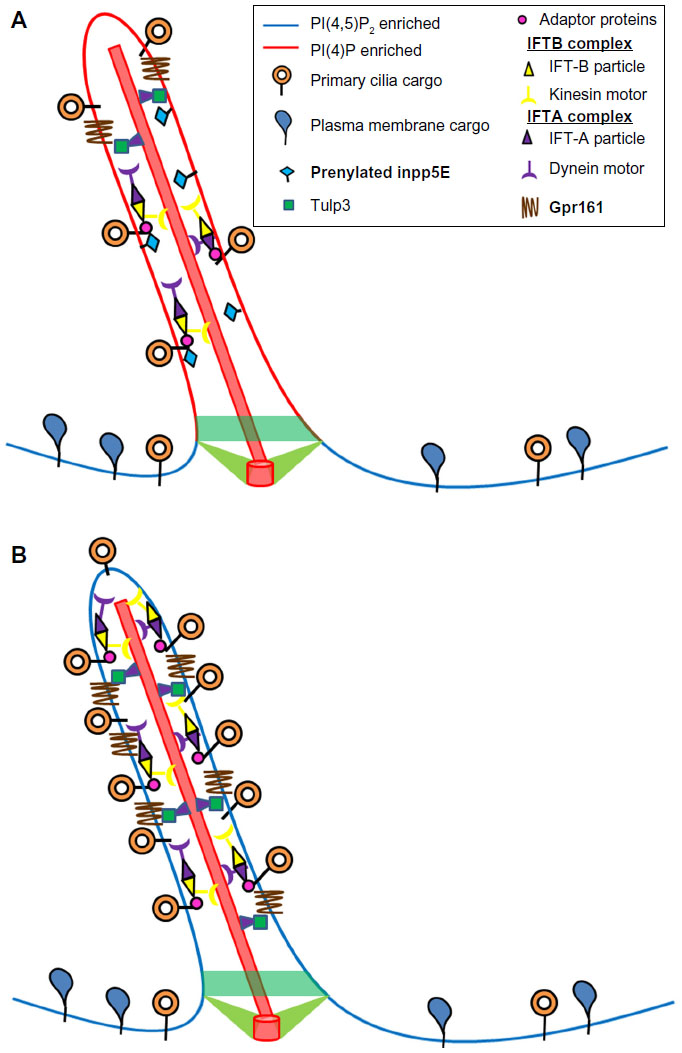 | Figure 4 Ciliary role of Inpp5E. |
A recent study by Garcia-Gonzalo et al9 determined that Inpp5E is critical for limiting the levels of PI(4,5)P2 in the PC. In agreement with the observations of Chavez et al,8 Inpp5E depletion led to accumulation of PI(4,5)P2 in the cilia, followed by enrichment in Tulp3 (which recognizes PI(4,5)P2 and its cargo Gpr161). Further, loss of Inpp5E also led to increase in the levels of negative regulators of Shh signaling such as the IFT-A complex proteins Ift139 (also known as Thm1 or Ttc21b) and Ift140.9 Taken together, these studies provide a mechanistic link between Inpp5E’s phosphatase activity and modulation of Shh signal transduction at the PC for tissue patterning and development.
Insulin signaling and glucose homeostasis
The PI3K/AKT pathway is turned on following receptor tyrosine kinase activation by ligands such as insulin.97 This is the clear in the case of the insulin receptor/insulin system, where the production of PI(3,4,5)P3 is required for initiation of Akt-dependent cell survival and metabolic signaling. Therefore, because of its catalytic activity, Inpp5E is predicted to exert anti-insulin effects, primarily through its phosphatase activity and interaction with the p85 subunit of PI3K.82 Indeed, Inpp5E participates in growth factor signaling, primarily controlling hypothalamic responses to insulin stimulation.82,84,98 It helps regulate basal levels of PI(3,4,5)P3 in the hypothalamus, the main central nervous system (CNS) regulator of insulin signaling.82 Inpp5E is abundantly expressed in the hypothalamus and is tyrosine phosphorylated by activated insulin receptor.82 Physiological consequences of these signaling events are highlighted by a corresponding CNS-mediated reduction in food intake in an insulin-dependent manner and a reinstatement of glucose homeostasis.82,84 Inpp5E overexpression studies resulted in increase in obesity and insulin-resistant diabetes in mice models, suggesting that this protein could constitute a target for diabetes and obesity drug therapies.84
Diseases associated with mutations in INPP5E
Mutations in INPP5E result in two ciliopathies namely, Joubert and MORM syndromes; however, their clinical manifestations are starkly distinct. Most INPP5E missense mutations causative of Joubert syndrome affect the catalytic domain of the protein, impairing its enzymatic activity.85,99 Although the ability of the mutated Inpp5E to localize in the cilium is not compromised, cells bearing these INPP5E mutations exhibit greater PC instability and are more sensitive to serum-stimulated cilium disassembly.85 Further, mice models lacking Inpp5E show embryonic lethality, displaying renal cysts and neurological defects at early stages of development but with very mild or no ocular defects.32 It is interesting to note that cells expressing mutated Inpp5E display normal number and morphology of cilium but have bulged ciliary tips, which is believed to be because of accumulation of cargo.32,85,99 Evidence indicates that Inpp5E is required to regulate phosphoinositide metabolism inside the PC and is particularly critical for the tight regulation of the PDGF–PI3K–Akt pathway, which mediates cell cycle entry.85
The only INPP5E mutation identified to cause MORM syndrome (Q627X) is a nonsense mutation that renders a truncated Inpp5E protein lacking the CAAX box at the C-terminus.32 In contrast to the Inpp5E variants that cause Joubert syndrome, the MORM mutated protein is only found at the base of the cilium but retains full enzymatic activity.32 Further, the major clinical symptoms of MORM include ocular defects not observed in Joubert syndrome. Despite full phosphatase activity, the MORM-related Inpp5E mutation was unable to rescue ciliary instability,32 suggesting that without proper localization, enzymatic activity is necessary but not sufficient for ciliary maintenance. This idea is supported by the observation that mutations in PDE6D produce mice with renal, ocular, and neurological defects reminiscent of defects observed in INPP5E deficient mice.87 As indicated earlier, binding to PDE6D is required for its PC localization,86 and the mutated Inpp5E MORM variant is unable to sustain interaction with PDE6D and therefore, is unable to localize in the PC.87 As a whole, these results indicate that Inpp5E-mediated ciliary stability requires both proper localization to the cilium as well as a fully functional phosphatase.
Inpp5E and cancer
In addition, and similar to the 5′ phosphatases SHIP1 and SHIP2, Inpp5E is upregulated in cancers (non-Hodgkins lymphoma, cervical, and uterine cancers); however, its role in cancer progression is still unclear.100
Conclusions and perspectives: is there a 5′ phosphatase relay mechanism in PC assembly/maintenance?
The PC is a plasma membrane specialization that all eukaryotic cells (except lymphocytes) display at a certain point in their lifespan. The crucial signaling role that this structure plays in cell physiology is highlighted by the fact that assembly or function abnormalities invariably leads to the onset of a group of heterogeneous genetic diseases collectively known as ciliopathies. In fact, the medical relevance of the PC justifies the attention that this subcellular structure has received in recent times. Part of these efforts has been devoted to establish the role of well-known signaling elements such as the phosphoinositides in PC function, assembly, and maintenance.
In addition to the well-known IP3 second messenger, phospholipids such as PI(4,5)P2 and PI(4)P are among the inositol-related species to be found in the PC. These phospholipids are known to serve as recruitment platforms for cytoskeleton signaling and trafficking machinery, and they greatly differ in their PC steady-state levels, with PI(4)P concentration being much higher than the one from PI(4,5)P2. Importantly, abundance of these species is under control of PIP kinases and phosphatases. Although little is known about the presence or activity of PI kinases in the PC (with the exception of PI3K), deficiencies in two 5′ PIP phosphatases, Inpp5E and Ocrl1, have been linked to ciliopathies (Joubert and MORM syndromes) or ciliopathy-associated diseases such as LS. Since 5′ PIP phosphatases are responsible for catalyzing the conversion of PI(4,5)P2 into PI(4)P, these enzymes are predicted to be key for maintaining the characteristic steady-state levels of these PIPs.
Although Ocrl1 plays a role in vesicle trafficking toward the PC, Inpp5E activity is intraciliary and seems to be directed to inhibit the AKT pathway and its PC disassembly effects (Figure 5). The preciliary function of Ocrl1 depends on its interaction with Rab8 (a well-known player in ciliogenesis) and is part of the “supply” route from the Golgi apparatus to the PC. Interestingly, this phosphatase is also involved in an indirect, Rab5-dependent, protein sorting pathway rerouting cargo from the plasma membrane to the PC. Further, this indirect route involves interactions with the endosomal proteins APPL1 and IPIP27/Ses at endosomal compartments. Because both APPL1 and IPIP27/Ses bind the same Ocrl1 region, this competitive reaction implies a relay-type mechanism (Figure 5). Specifically, Ocrl1 is first recruited to Rab5-containing endosomes where it interacts with the adaptor protein APPL1 to then be passed on to another endosomal structure by binding the IPIP27/Ses proteins. Evidence suggests that membrane carriers from both Ocrl1-dependent routes coalesce at the base of the PC. Whether Ocrl1 remains periciliary or also moves inside the PC is still unclear (Figure 5). However, there is no doubt that the C-terminal prenylated Inpp5E is translocated into the PC. In contrast to Ocrl1, Inpp5E localization does not seem to be Rab-dependent (although this phosphatase binds Rab20), but it involves a sequence motif and the immediately downstream prenylation target: the CAAX box. Interestingly, Ocrl1 lacks a CAAX motif that is, nevertheless, present in its functional homologue Inpp5B. Consistent with their localization and function, deficient Ocrl1 phosphatase activity leads to high PI(4,5)P2 cellular levels, whereas Inpp5E abnormalities cause accumulation of this phospholipid at the tip of the PC. This constitutes a second relay event in which the 5′ phosphatase-mediated control over PI(4,5)P2 is transferred from Ocrl1 to Inpp5E. Whether this relay between Ocrl1 and Inpp5E occurs at the PC base (where Ocrl1 and the Inpp5E activator AurkA localize) and/or in the proximal segment of the PC is unclear. Neither is it fully established what role Inpp5B, the prenylation competent, functional homologue of Ocrl1, plays in this process. Although redundancy between these 5′ phosphatases is supported by evidence, patient fibroblasts have been shown to contain substantial levels of Inpp5B and still display Ocrl1-dependent PC defects and other LS cellular phenotypes. In contrast to OCRL1, no disease involving INPP5B mutations has been described yet; however, it is possible that the two different gene products play partially overlapping but unique roles in PC assembly/function.
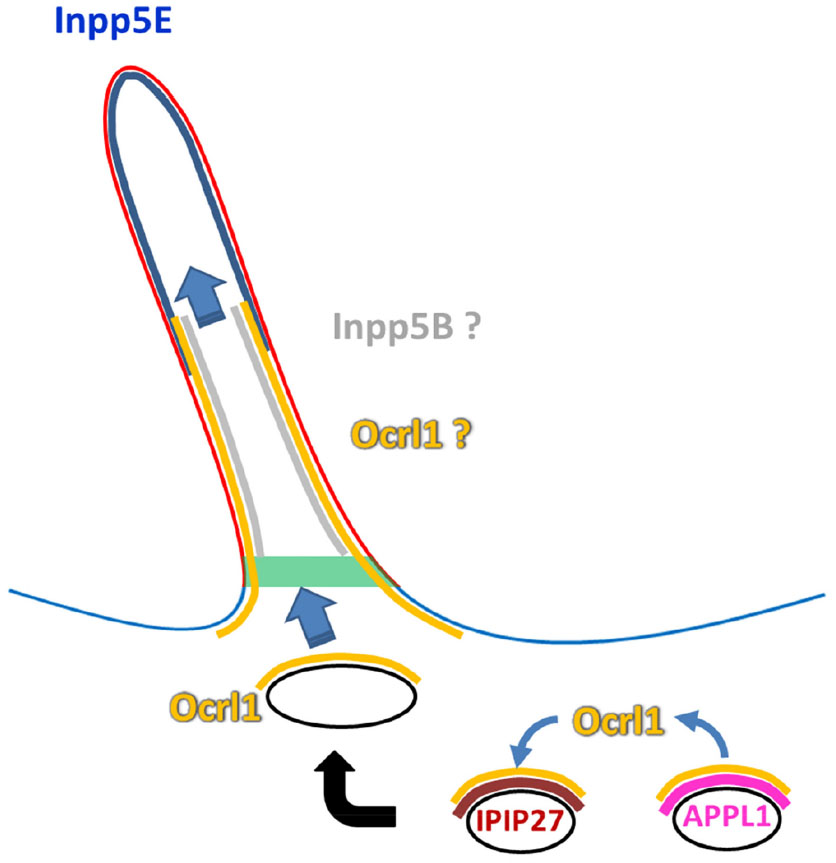 | Figure 5 Putative phosphatase relay model. |
Other questions that remain open in the field are: what are the sources of PI(4,5)P2 at the base and within the PC that requires continuous 5′ phosphatase activity? Are the 5′ phosphatase activities coupled with PI(4, 5)P2 production to generate signaling cycles required for normal assembly, maintenance, and function of the PC?
No doubt, future investigations will be focused on answering these questions, and we will surely witness important developments in the fascinating area of the cell biology of the PC and mechanisms leading to ciliopathies.
Acknowledgments
We thank members of the Aguilar Lab for critical reading of the manuscript. We apologize to all authors whose original contributions we could not cite owing to space limitations. For more complete listings, please refer to the cited reviews and references therein. The Aguilar Lab is supported by grants from the National Science Foundation MCB-1021377 and by the Center for Science of Information (CSoI), an NSF Science and Technology Center, under grant agreement CCF-0939370.
Disclosure
The authors report no conflicts of interest in this work.
References
Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–344. | |
Satir P, Pedersen LB, Christensen ST. The primary cilium at a glance. J Cell Sci. 2010;123(Pt 4):499–503. | |
Hakim S, Bertucci MC, Conduit SE, Vuong DL, Mitchell CA. Inositol polyphosphate phosphatases in human disease. Curr Top Microbiol Immunol. 2012;362:246–314. | |
Billcliff PG, Lowe M. Inositol lipid phosphatases in membrane trafficking and human disease. Biochem J. 2014;461:159–175. | |
Ghigo A, Perino A, Hirsch E. Phosphoinositides and cardiovascular diseases. Curr Top Microbiol Immunol. 2012;362:43–60. | |
Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443(7112):651–657. | |
Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. 2013;93(3):1019–1137. | |
Chavez M, Ena S, Van Sande J, de Kerchove d’Exaerde A, Schurmans S, Schiffmann SN. Modulation of ciliary phosphoinositide content regulates trafficking and Sonic Hedgehog signaling output. Dev Cell. 2015;34(3):338–350. | |
Garcia-Gonzalo FR, Phua SC, Roberson EC, et al. Phosphoinositides regulate ciliary protein trafficking to modulate Hedgehog signaling. Dev Cell. 2015;34(4):400–409. | |
Anderson RG. The three-dimensional structure of basal body from Rhesus-monkey oviduct. J Cell Biol. 1972;54(2):246–265. | |
Gilula NB, Satir P. The ciliary necklace. A ciliary membrane specialization. J Cell Biol. 1972;53(2):494–509. | |
Chih B, Liu P, Chinn Y, et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14(1):61–72. | |
Kee HL, Dishinger JF, Blasius TL, Liu C-J, Margolis B, Verhey KJ. A size-exclusion permeability barrier and nucleoporins characterize a ciliary pore complex that regulates transport into cilia. Nat Cell Biol. 2012;14(4):431–437. | |
Ounjai P, Kim KD, Liu H, et al. Architectural insights into a ciliary partition. Curr Biol. 2013;23(4):339–344. | |
Hu Q, Milenkovic L, Jin H, et al. A septin diffusion barrier at the base of the primary cilium maintains ciliary membrane protein distribution. Science. 2010;329(5990):436–439. | |
Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–1021. | |
Corbit KC, Shyer AE, Dowdle WE, et al. Kif3a constrains β-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol. 2008;10(1):70–76. | |
Clement CA, Ajbro KD, Koefoed K, et al. TGF-β signaling is associated with endocytosis at the pocket region of the primary cilium. Cell Rep. 2013;3(6):1806–1814. | |
Schneider L, Clement CA, Teilmann SC, et al. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15(20):1861–1866. | |
Moritz OL, Tam BM, Papermaster DS, Nakayama T. A functional rhodopsin-green fluorescent protein fusion protein localizes correctly in transgenic Xenopus laevis retinal rods and is expressed in a time-dependent pattern. J Biol Chem. 2001;276(30):28242–28251. | |
Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Vergé D. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res. 2000;872(1–2):271–275. | |
Händel M, Schulz S, Stanarius A, et al. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience. 1999;89(3):909–926. | |
Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Curr Biol. 2002;12(11):R378–R380. | |
Madhivanan K, Aguilar RC. Ciliopathies: the trafficking connection. Traffic. 2014;15(10):1031–1056. | |
Franco I, Gulluni F, Campa CC, et al. PI3K class II α controls spatially restricted endosomal PtdIns3P and Rab11 activation to promote primary cilium function. Dev Cell. 2014;28(6):647–658. | |
Franco I, Margaria JP, De Santis MC, et al. Phosphoinositide 3-kinase-C2α regulates polycystin-2 ciliary entry and protects against kidney cyst formation. J Am Soc Nephrol. Epub August 13, 2015. | |
Vieira OV, Gaus K, Verkade P, Fullekrug J, Vaz WLC, Simons K. FAPP2, cilium formation, and compartmentalization of the apical membrane in polarized Madin-Darby canine kidney (MDCK) cells. Proc Natl Acad Sci U S A. 2006;103(49):18556–18561. | |
Mukhopadhyay S, Wen X, Chih B, et al. TULP3 bridges the IFT-A complex and membrane phosphoinositides to promote trafficking of G protein-coupled receptors into primary cilia. Genes Dev. 2010;24(19):2180–2193. | |
Conduit SE, Dyson JM, Mitchell CA. Inositol polyphosphate 5-phosphatases; new players in the regulation of cilia and ciliopathies. FEBS Lett. 2012;586(18):2846–2857. | |
Coon BG, Hernandez V, Madhivanan K, et al. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly. Hum Mol Genet. 2012;21(8):1835–1847. | |
Luo N, West CC, Murga-Zamalloa CA, et al. OCRL localizes to the primary cilium: a new role for cilia in Lowe syndrome. Hum Mol Genet. 2012;21(15):3333–3344. | |
Jacoby M, Cox JJ, Gayral S, et al. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41(9):1027–1031. | |
Bae Y-K, Kim E, L’Hernault SW, Barr MM. The CIL-1 PI 5-phosphatase localizes TRP Polycystins to cilia and activates sperm in C. elegans. Curr Biol. 2009;19(19):1599–1607. | |
Zhang XL, Jefferson AB, Auethavekiat V, Majerus PW. The protein-deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1995;92(11):4853–4856. | |
Jefferson AB, Majerus PW. Properties of type II inositol polyphosphate 5-phosphatase. J Biol Chem. 1995;270(16):9370–9377. | |
Schmid AC, Wise HM, Mitchell CA, Nussbaum R, Woscholski R. Type II phosphoinositide 5-phosphatases have unique sensitivities towards fatty acid composition and head group phosphorylation. FEBS Lett. 2004;576(1–2):9–13. | |
Kisseleva MV, Wilson MP, Majerus PW. The isolation and characterization of a cDNA encoding phospholipid-specific inositol polyphosphate 5-phosphatase. J Biol Chem. 2000;275(26):20110–20116. | |
Mao Y, Balkin DM, Zoncu R, et al. A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. EMBO J. 2009;28(13):1831–1842. | |
Choudhury R, Noakes CJ, McKenzie E, Kox C, Lowe M. Differential clathrin binding and subcellular localization of OCRL1 splice isoforms. J Biol Chem. 2009;284(15):9965–9973. | |
Tsujishita Y, Guo SL, Stolz LE, York JD, Hurley JH. Specificity determinants in phosphoinositide dephosphorylation: crystal structure of an archetypal inositol polyphosphate 5-phosphatase. Cell. 2001;105(3):379–389. | |
Erdmann KS, Mao Y, McCrea HJ, et al. A role of the Lowe syndrome protein OCRL in early steps of the endocytic pathway. Dev Cell. 2007;13(3):377–390. | |
Swan LE, Tomasini L, Pirruccello M, Lunardi J, De Camilli P. Two closely related endocytic proteins that share a common OCRL-binding motif with APPL1. Proc Natl Acad Sci U S A. 2010;107(8):3511–3516. | |
Noakes CJ, Lee G, Lowe M. The PH domain proteins IPIP27A and B link OCRL1 to receptor recycling in the endocytic pathway. Mol Biol Cell. 2011;22(5):606–623. | |
Hagemann N, Hou X, Goody RS, Itzen A, Erdmann KS. Crystal structure of the Rab binding domain of OCRL1 in complex with Rab8 and functional implications of the OCRL1/Rab8 module for Lowe syndrome. Small GTPases. 2012;3(2):107–110. | |
Hou X, Hagemann N, Schoebel S, et al. A structural basis for Lowe syndrome caused by mutations in the Rab-binding domain of OCRL1. EMBO J. 2011;30(8):1659–1670. | |
Fukuda M, Kanno E, Ishibashi K, Itoh T. Large scale screening for novel Rab effectors reveals unexpected broad Rab binding specificity. Mol Cell Proteomics. 2008;7(6):1031–1042. | |
Rbaibi Y, Cui S, Mo D, et al. OCRL1 modulates cilia length in renal epithelial cells. Traffic. 2012;13(9):1295–1305. | |
Faucherre A, Desbois P, Satre V, Lunardi J, Dorseuil O, Gacon G. Lowe syndrome protein OCRL1 interacts with Rac GTPase in the trans-Golgi network. Hum Mol Genet. 2003;12(19):2449–2456. | |
Faucherre A, Desbois P, Nagano F, et al. Lowe syndrome protein Ocrl1 is translocated to membrane ruffles upon Rac GTPase activation: a new perspective on Lowe syndrome pathophysiology. Hum Mol Genet. 2005;14(11):1441–1448. | |
Dambournet D, Machicoane M, Chesneau L, et al. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol. 2011;13(8):981–988. | |
Hyvola N, Diao A, McKenzie E, Skippen A, Cockcroft S, Lowe M. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. 2006;25(16):3750–3761. | |
Choudhury R, Diao AP, Zhang F, et al. Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol Biol Cell. 2005;16(8):3467–3479. | |
van Rahden VA, Brand K, Najm J, et al. The 5-phosphatase OCRL mediates retrograde transport of the mannose 6-phosphate receptor by regulating a Rac1-cofilin signalling module. Hum Mol Genet. 2012;21(23):5019–5038. | |
Vicinanza M, Di Campli A, Polishchuk E, et al. OCRL controls trafficking through early endosomes via PtdIns4,5P2-dependent regulation of endosomal actin. EMBO J. 2011;30(24):4970–4985. | |
Cui S, Guerriero CJ, Szalinski CM, Kinlough CL, Hughey RP, Weisz OA. OCRL1 function in renal epithelial membrane traffic. Am J Physiol Renal Physiol. 2010;298(2):F335–F345. | |
Coon BG, Mukherjee D, Hanna CB, Riese DJ II, Lowe M, Aguilar RC. Lowe syndrome patient fibroblasts display Ocrl1-specific cell migration defects that cannot be rescued by the homologous Inpp5B phosphatase. Hum Mol Genet. 2009;18(23):4478–4491. | |
Nandez R, Balkin DM, Messa M, et al. A role of OCRL in clathrin-coated pit dynamics and uncoating revealed by studies of Lowe syndrome cells. Elife. 2014;3:e02975. | |
Oltrabella F, Pietka G, Ramirez IB-R, et al. The Lowe syndrome protein OCRL1 is required for endocytosis in the zebrafish pronephric tubule. PLoS Genet. 2015;11(4):e1005058. | |
Luo N, Conwell MD, Chen X, et al. Primary cilia signaling mediates intraocular pressure sensation. Proc Natl Acad Sci U S A. 2014;111(35):12871–12876. | |
Madhivanan K, Mukherjee D, Aguilar RC. Lowe syndrome: between primary cilia assembly and Rac1-mediated membrane remodeling. Commun Integr Biol. 2012;5(6):641–644. | |
Suchy SF, Nussbaum RL. The deficiency of PIP2 5-phosphatase in Lowe syndrome affects actin polymerization. Am J Hum Genet. 2002;71(6):1420–1427. | |
Loovers HM, Kortholt A, de Groote H, Whitty L, Nussbaum RL, van Haastert PJM. Regulation of phagocytosis in Dictyostelium by the inositol 5-phosphatase OCRL homolog Dd5P4. Traffic. 2007;8(5):618–628. | |
Bohdanowicz M, Balkin DM, De Camilli P, Grinstein S. Recruitment of OCRL and Inpp5B to phagosomes by Rab5 and APPL1 depletes phosphoinositides and attenuates Akt signaling. Mol Biol Cell. 2012;23(1):176–187. | |
Marion S, Mazzolini J, Herit F, et al. The NF-κB signaling protein Bcl10 regulates actin dynamics by controlling AP1 and OCRL-bearing vesicles. Dev Cell. 2012;23(5):954–967. | |
El Kadhi KB, Roubinet C, Solinet S, Emery G, Carreno S. The inositol 5-phosphatase dOCRL controls PI(4,5)P2 homeostasis and is necessary for cytokinesis. Curr Biol. 2011;21(12):1074–1079. | |
Lowe CU, Terrey M, Maclachlan EA. Organic-aciduria, decreased renal ammonia production, hydrophthalmos, and mental retardation; a clinical entity. AMA Am J Dis Child. 1952;83(2):164–184. | |
Recker F, Heutter R, Ludwig M. Lowe syndrome/Dent-2 disease: a comprehensive review of known and novel aspects. J Pediatr Genet. 2013;2(2):53–68. | |
Loi M. Lowe syndrome. Orphanet J Rare Dis. 2006;1:16. | |
Mehta ZB, Pietka G, Lowe M. The cellular and physiological functions of the Lowe syndrome protein OCRL1. Traffic. 2014;15(5):471–487. | |
Kenworthy L, Park T, Charnas LR. Cognitive and behavioral profile of the Oculocerebrorenal syndrome of Lowe. Am J Med Genet. 1993;46(3):297–303. | |
Hoopes RR, Shrimpton AE, Knohl SJ, et al. Dent disease with mutations in OCRL1. Am J Hum Genet. 2005;76(2):260–267. | |
Bökenkamp A, Böckenhauer D, Cheong HI, et al. Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr. 2009;155(1):94–99. | |
Bockenhauer D, Bokenkamp A, Nuutinen M, et al. Novel OCRL mutations in patients with Dent-2 disease. J Pediatr Genet. 2012;1(1):15–23. | |
Hichri H, Rendu J, Monnier N, et al. From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat. 2011;32(4):379–388. | |
Shrimpton AE, Hoopes RR Jr, Knohl SJ, et al. OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiol. 2009;112(2):27–36. | |
Recker F, Zaniew M, Boeckenhauer D, et al. Characterization of 28 novel patients expands the mutational and phenotypic spectrum of Lowe syndrome. Pediatr Nephrol. 2015;30(6):931–943. | |
Janne PA, Suchy SF, Bernard D, et al. Functional overlap between murine Inpp5B and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J Clin Invest. 1998;101(10):2042–2053. | |
Bothwell SP, Farber LW, Hoagland A, Nussbaum RL. Species-specific difference in expression and splice-site choice in Inpp5B, an inositol polyphosphate 5-phosphatase paralogous to the enzyme deficient in Lowe Syndrome. Mamm Genome. 2010;21(9–10):458–466. | |
Montjean R, Aoidi R, Desbois P, et al. OCRL-mutated fibroblasts from patients with Dent-2 disease exhibit INPP5B-independent phenotypic variability relatively to Lowe syndrome cells. Hum Mol Genet. 2015;24(4):994–1006. | |
Luo N, Kumar A, Conwell M, Weinreb RN, Anderson R, Sun Y. Compensatory role of inositol 5-phosphatase INPP5B to OCRL in primary cilia formation in oculocerebrorenal syndrome of Lowe. PLoS One. 2013;8(6):e66727. | |
Kong AM, Speed CJ, O’Malley CJ, et al. Cloning, and characterization of a 72-kDa inositol-polyphosphate 5-phosphatase localized to the Golgi network. J Biol Chem. 2000;275(31):24052–24064. | |
Bertelli DF, Araujo EP, Cesquini M, et al. Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits inositide trisphosphate accumulation in hypothalamus and regulates food intake and body weight. Endocrinology. 2006;147(11):5385–5399. | |
Plotnikova OV, Seo S, Cottle DL, et al. INPP5E interacts with AURKA, linking phosphoinositide signaling to primary cilium stability. J Cell Sci. 2015;128(2):364–372. | |
Bertelli DF, Coope A, Caricilli AM, et al. Inhibition of 72 kDa inositol polyphosphate 5-phosphatase E improves insulin signal transduction in diet-induced obesity. J Endocrinol. 2013;217(2):131–140. | |
Bielas SL, Silhavy JL, Brancati F, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41(9):1032–1036. | |
Humbert MC, Weihbrecht K, Searby CC, et al. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A. 2012;109(48):19691–19696. | |
Thomas S, Wright KJ, Le Corre S, et al. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. 2014;35(1):137–146. | |
Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129(7):1351–1363. | |
Liu X, Shi Y, Woods KW, et al. Akt inhibitor A-443654 interferes with mitotic progression by regulating Aurora A kinase expression. Neoplasia. 2008;10(8):828–837. | |
Kisseleva MV, Cao L, Majerus PW. Phosphoinositide-specific inositol polyphosphate 5-phosphatase IV inhibits Akt/protein kinase B phosphorylation and leads to apoptotic cell death. J Biol Chem. 2002;277(8):6266–6272. | |
Horan KA, Watanabe K, Kong AM, et al. Regulation of Fcγ-stimulated phagocytosis by the 72-kDa inositol polyphosphate 5-phosphatase: SHIP1, but not the 72-kDa 5-phosphatase, regulates complement receptor 3-mediated phagocytosis by differential recruitment of these 5-phosphatases to the phagocytic cup. Blood. 2007;110(13):4480–4491. | |
Segawa T, Hazekil K, Nigorikawa K, et al. Inpp5e increases the Rab5 association and phosphatidylinositol 3-phosphate accumulation at the phagosome through an interaction with Rab20. Biochem J. 2014;464:365–375. | |
Fairn GD, Grinstein S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol. 2012;33(8):397–405. | |
Han YG, Spassky N, Romaguera-Ros M, et al. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci. 2008;11(3):277–284. | |
Mukhopadhyay S, Wen X, Ratti N, et al. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic Hedgehog pathway via cAMP signaling. Cell. 2013;152(1–2):210–223. | |
Lopez-Rios J, Speziale D, Robay D, et al. GLI3 constrains digit number by controlling both progenitor proliferation and BMP-dependent exit to chondrogenesis. Dev Cell. 2012;22(4):837–848. | |
Folli F, Saad MJ, Backer JM, Kahn CR. Insulin stimulation of phosphatidylinositol 3-kinase activity and association with insulin receptor substrate 1 in liver and muscle of the intact rat. J Biol Chem. 1992;267(31):22171–22177. | |
Wang F, Ijuin T, Itoh T, Takenawa T. Regulation of IGF-1/PI3K/Akt signalling by the phosphoinositide phosphatase pharbin. J Biochem. 2011;150(1):83–93. | |
Travaglini L, Brancati F, Silhavy J, et al. Phenotypic spectrum and prevalence of INPP5E mutations in Joubert Syndrome and related disorders. Eur J Hum Genet. 2013;21(10):1074–1078. | |
Miyazawa K. Phosphoinositide 5-phosphatases: how do they affect tumourigenesis? J Biochem. 2013;153(1):1–3. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.