")
Back to Journals » Clinical Interventions in Aging » Volume 13
Role of miRNAs in skeletal muscle aging
Authors Zheng Y, Kong J, Li Q, Wang Y, Li J
Received 24 March 2018
Accepted for publication 10 September 2018
Published 22 November 2018 Volume 2018:13 Pages 2407—2419
DOI https://doi.org/10.2147/CIA.S169202
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Zhi-Ying Wu
Yan Zheng,1,* Jian Kong,1,* Qun Li,2 Yan Wang,1 Jie Li1
1Department of Geriatrics, The First Hospital of Jilin University, Changchun 130021, Jilin, China; 2Department of Thyroid Surgery, The First Hospital of Jilin University, Changchun 130021, Jilin, China
*These authors contributed equally to this work
Purpose: The aim of this study was to explore the role of miRNAs in the process of skeletal muscle aging.
Materials and methods: We analyzed the miRNA microarray datasets from 19 young and 17 old skeletal muscle samples by bioinformatic analysis. Differentially expressed miRNAs were identified, followed by function and pathway enrichment analysis. The expression of miRNAs were validated by real-time quantitative PCR (RT-qPCR) analysis.
Results: A total of 23 miRNAs were found to be differentially expressed in old muscle samples based on two platforms. Gene targets of upregulated miRNAs were significantly enriched in the oxytocin signaling pathway, AMP-activated protein kinase (AMPK) signaling pathway, and Notch signaling pathway. The target genes of downregulated miRNAs were significantly related to gap junction, salivary secretion, and estrogen signaling pathway. has-miR-19a and hsa-miR-34a were significant nodes in the miRNA regulatory network. has-miR-19a was closely related to the AMPK signaling pathway. hsa-miR-34a was closely related to cellular senescence and mitogen-activated protein kinase (MAPK) signaling pathway. PCR analysis showed that the expression of has-miR-34a-5p and has-miR-449b-5p was significantly higher in the patient group than in the control group, while no significant difference was observed in the expression of has-miR-19a-3p and has-miR-144-3p between the two groups. Furthermore, the expression of key target genes involved in cellular senescence (sirtuin 1 [SITRI]), MAPK signaling pathway (vascular endothelial growth factor A [VEGFA]), and AMPK signaling pathway (protein kinase AMP-activated catalytic subunit alpha 1 [PRKAA1] and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 [PFKFB3]) were significantly increased in patients with sarcopenia.
Conclusion: has-miR-19a and hsa-miR-34a may play regulatory roles in the aging process of skeletal muscles and may be candidate targets to prevent muscle aging. Further experimental validations are warranted.
Keywords: skeletal muscle aging, differentially expressed miRNA, pathway, miRNA regulatory network
Introduction
Skeletal muscle aging (sarcopenia) is the key feature of age-related degenerative disease, which is characterized by skeletal muscle loss and function degeneration. Patients with sarcopenia present with 0.5%–1% muscle loss per year, which may be related to cachexia and other underlying malignant diseases.1 A previous study conducted in the UK among people with an average age of 67 years suggested that the incidence of sarcopenia was 4.6% in men and 7.9% in women.2 Another study reported that the incidence of sarcopenia was 36.5% in people with an average age of 70.1 years in the USA.3 As the population ages, the incidence of sarcopenia will be highlighted. Therefore, it is necessary to explore the mechanism of age-related sarcopenia.
Contributions have been made to characterize the mechanism associated with age-induced muscle loss. It is reported that age-related muscle loss and function reduction may be due to the abnormal production of aging cells and sensitivity to cytokines.4 Mitochondrial dysfunction is an event in muscle aging and contributes to muscle mass loss and aerobic function decline.5 The superoxide generated by mitochondria is accumulated in aged cells and plays a contributory role in muscle aging.6 Muscle aging is associated with reduced Ca2+ homeostasis, and Ca2+ spark signaling appeared to be suppressed in aged skeletal muscles.7 In addition, Hall et al8 found that myofiber-associated satellite cells showed self-renewal capabilities and the transplantation of these cells was proposed to prevent skeletal muscle aging. However, the mechanism of skeletal muscle aging has not yet been clarified. miRNAs are a class of small noncoding RNAs and have been reported to play a role in the aging process.9,10 Accumulating evidences have confirmed that miRNAs play a crucial role in the age-related changes in skeletal muscle mass, composition, and function.11–13 Moreover, some miRNAs are identified to be regulated with age and may be linked to key regulators of muscle protein synthesis and regeneration in young and old skeletal muscle.14 Despite these, the potential miRNAs involved in skeletal muscle aging have been fully reported.
In the present study, we analyzed the differentially expressed miRNAs between the young and old muscle samples, followed by function and pathway enrichment analysis. We aimed to explore the involvement of miRNAs in the potential mechanisms of muscle aging and provide new perspectives for the prevention of muscle aging.
Materials and methods
miRNA expression data
The miRNA expression profile of human skeletal muscle (GSE23527)15 was downloaded from Gene Expression Omnibus (GEO) database. There were a total of 36 skeletal muscle samples: 19 from young male subjects and 17 from old male subjects. The data from 12 old and 12 young skeletal muscle samples were produced based on the GPL10358 platform, and the remaining data from 5 old and 7 young skeletal muscle samples were produced based on the GPL10415 platform.
Data preprocessing and differentially expressed miRNA analysis
The raw data were processed by limma package (version 3.10.3)16 in R (version 3.3.2), including background correction and expression profile normalization.
After preprocessing, differentially expressed miRNAs between the young and old samples based on the two platforms were analyzed by unpaired t-test through limma package, respectively. P<0.05 and log fold change (log FC) >1 were set as the cutoff values. All the differentially expressed miRNAs were pooled for further analysis.
Prediction of miRNA targets
The target genes of differentially expressed miRNAs were retrieved from DIANA-microT, MicroInspector, miRanda, MirTarget2, miTarget, NBmiRTar, PicTar, PITA, RNA22, RNAhybrid, and TargetScan/TargertScanS databases using the miRecords17 tool. The miRNA–target gene interactions deposited in at least five databases were collected. The miRNA–target gene network was constructed by Cytoscape version 3.2.018 (http://www.cytoscape.org/).
Function and pathway enrichment analysis of miRNA targets
Target genes of upregulated and downregulated miRNAs were subjected to Gene Ontology (GO)19 and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway20 enrichment analysis. The target genes were annotated to GO biological process (BP) terms by DAVID version 6.8 online tool21,22 (https://david.ncifcrf.gov/). The pathways that were closely related to miRNA targets were analyzed by clusterProfiler23 package. P<0.05 was set as the threshold value.
Protein–protein interaction (PPI) network analysis
The miRNA–target gene interactions were predicted by using the STRING database version 10.024 (http://www.string-db.org/). The gene interaction pairs with the PPI score of ≥9 were collected for PPI network construction using Cytoscape version 3.2.0. The significant modules with the score of >10 were mined using the MCODE plugin25 of the Cytoscape software. The module genes were subjected to KEGG pathway analysis with the criteria for significance set as P<0.05.
Transcriptional factor (TF) prediction for target genes
The TF–target gene interactions in the PPI network were predicted by iRegulon plugin version 1.326 (http://apps.cytoscape.org/apps/iRegulon). Minimum identity between orthologous genes was set as 0.05, and the maximum false discovery rate on motif similarity was set as 0.001. The TF–gene interaction pairs with normalized enrichment score (NES) of >4 were used for TF regulatory network construction.
Sample collection
Five patients with sarcopenia and five cases without sarcopenia were enrolled from the First Hospital of the Jilin University. The skeletal muscle samples were collected via orthopedic surgery. All the patients provided written informed consent to participate in this study, and approval was obtained from the ethics committee of the First Hospital of Jilin University.
RNA isolation and real-time quantitative PCR (RT-qPCR) analysis
Total RNA was isolated from 100 mg tissue samples using TRIZOL regent (Thermo Fisher Scientific, Waltham, MA, USA) for further analysis. RNA quality was determined by an Infinite M100 PRO microplate reader (Tecan, Crailsheim, Germany). cDNA was reverse transcribed from a total of 5 μg of RNA by PrimeScript™ RT Master Mix, according to the manufacturer’s instructions. RT-qPCR was performed, with a reaction volume of 20 μL, including 10 μL of the SYBR Premix EX Taq (2×), 1 μL of forward primer, 1 μL of reverse primer, and 8 μL of cDNA. The primer sequences are summarized in Table 1. The PCR reaction conditions used were as follows: 50.0°C for 3 minutes, 95.0°C for 3 minutes, followed by 40 cycles of 95.0°C for 10 seconds and 60.0°C for 30 seconds.
 | Table 1 Primer sequences for PCR analysis |
Statistical analyses
All the data were expressed as mean ± SD and analyzed by the SPSS 22.0 software (IBM Corporation, Armonk, NY, USA). P<0.05 was considered significant.
Results
Differentially expressed miRNAs
Based on the GPL10358 platform, there were 12 differentially expressed miRNAs between the young and old samples, including six downregulated miRNAs and six upregulated miRNAs. In addition, 11 miRNAs were found to be differentially expressed between the young and old samples based on the GPL10415 platform, including five upregulated miRNAs and six downregulated miRNAs (Table 2). All the differentially expressed miRNAs were pooled for further analysis.
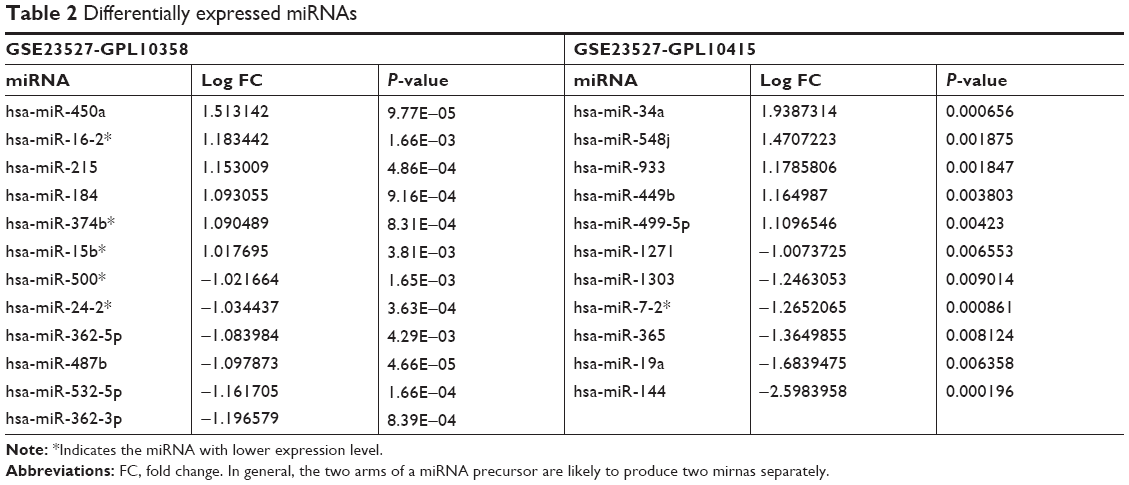 | Table 2 Differentially expressed miRNAs |
miRNA–target gene regulatory network
A total of 1,236 miRNA–target gene interaction pairs were obtained through miRecords, which contained six upregulated miRNAs (hsa-miR-34a, hsa-miR-449b, hsa-miR-215, hsa-miR-499-5p, hsa-miR-184, and hsa-miR-450a) and six downregulated miRNAs (hsa-miR-19a, hsa-miR-144, hsa-miR-532-5p, hsa-miR-365, hsa-miR-487b, and hsa-miR-362-5p). The miRNA–target gene regulatory network is illustrated in Figure 1. The significant nodes in this network included hsa-miR-19a (degree =434), hsa-miR-34a (degree =217), hsa-miR-144 (degree =227), and hsa-miR-532-5p (degree =166). Significant pathways that were closely related to significant miRNAs are shown in Figure 2. Notably, has-miR-19a was significantly enriched in the AMP-activated protein kinase (AMPK) signaling pathway (protein kinase AMP-activated catalytic subunit alpha 1 [PRKAA1] and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 [PFKFB3]), and has-miR-34a was significantly enriched in cellular senescence (sirtuin 1 [SITRI] and cell division cycle 25A [CDC25A]) and mitogen-activated protein kinase (MAPK) signaling pathway (vascular endothelial growth factor A [VEGFA] and microtubule associated protein tau [MAPT]).
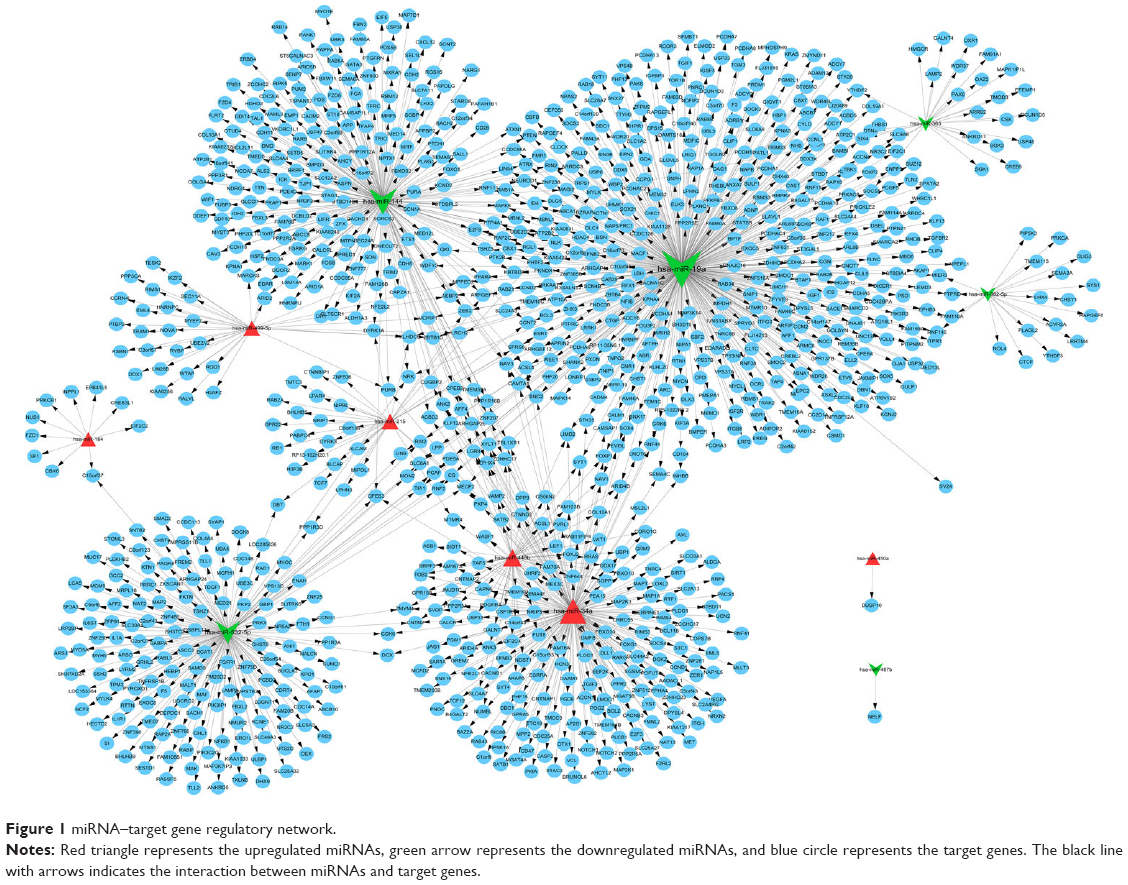 | Figure 1 miRNA–target gene regulatory network. |
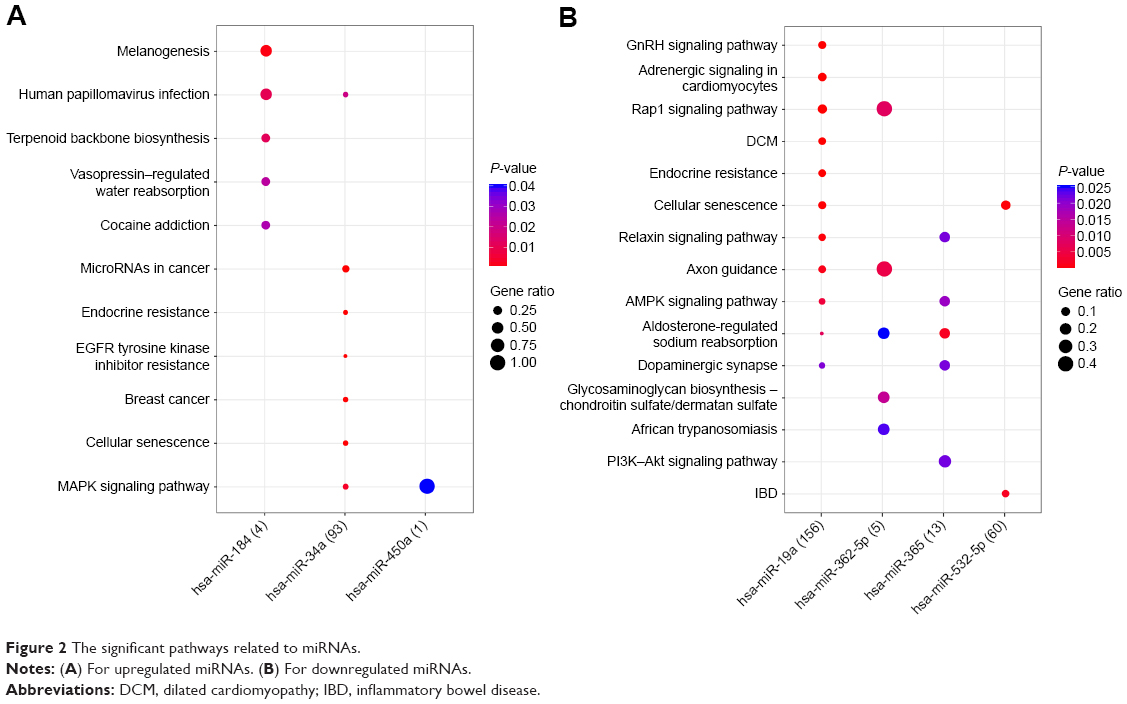 | Figure 2 The significant pathways related to miRNAs. |
Significant function and pathways related to miRNA targets
The target genes of upregulated miRNAs were significantly enriched in 30 pathways, such as the oxytocin signaling pathway, AMPK signaling pathway, and Fc gamma R-mediated phagocytosis. The downregulated miRNAs were significantly enriched in 67 pathways, such as gap junction, estrogen signaling pathway, and prolactin signaling pathway. The top 20 significant pathways for miRNA–target genes are shown in Figure 3.
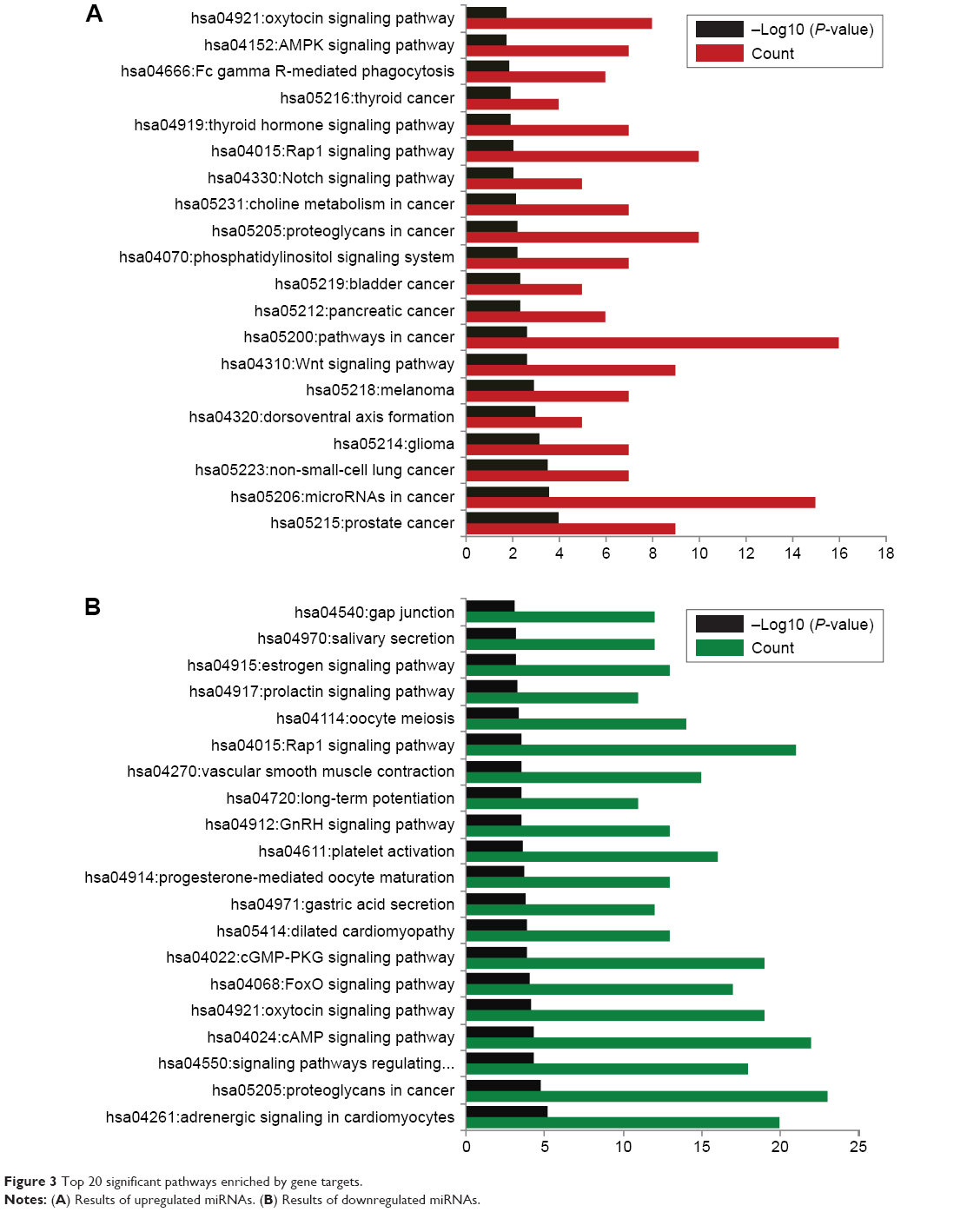 | Figure 3 Top 20 significant pathways enriched by gene targets. |
With the criterion for significance set as P<0.05, 70 GO-BP terms were found to be closely related to gene targets of upregulated miRNAs, while 143 GO functions were related to target genes of downregulated miRNAs. As shown in Figure 4, the top 20 significant BP terms for upregulated miRNAs included the regulation of synaptic vesicle exocytosis, Notch signaling pathway, and histone deacetylation. The top 20 significant BP terms for downregulated miRNAs comprised the regulation of ventricular cardiac muscle cell membrane, response to muscle stretch, and cardiac muscle cell action potential involved in contraction.
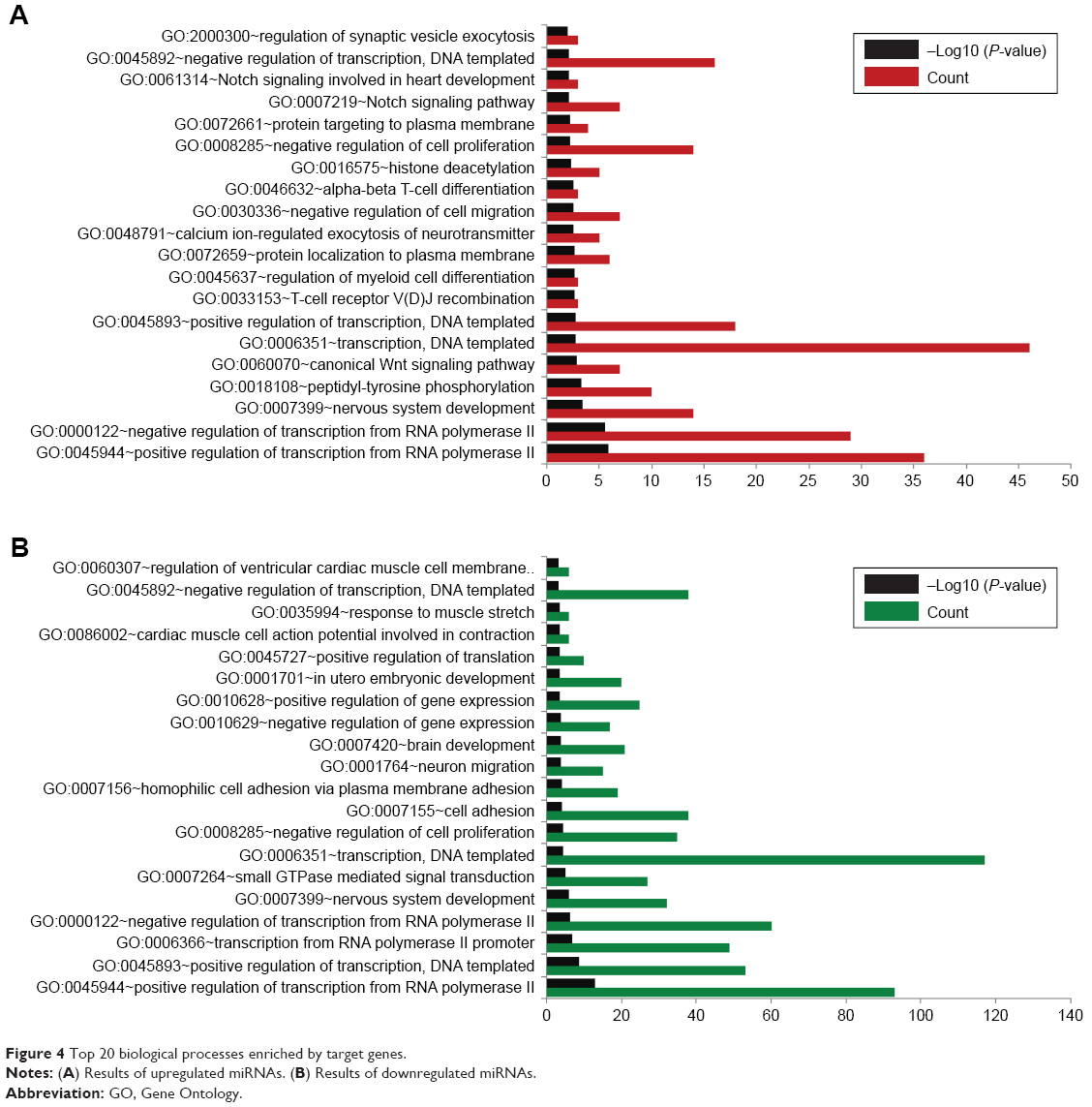 | Figure 4 Top 20 biological processes enriched by target genes. |
PPI network and module analysis
A PPI network of target genes was constructed with 1,291 edges connecting with 414 nodes. On the basis of score of >10, three significant modules were screened, including module-a (score =22, containing 15 nodes and 105 edges), module-b (score =15, containing 15 nodes and 105 edges), and module-c (score =12, containing 12 nodes and 66 interaction pairs; Figure 5).
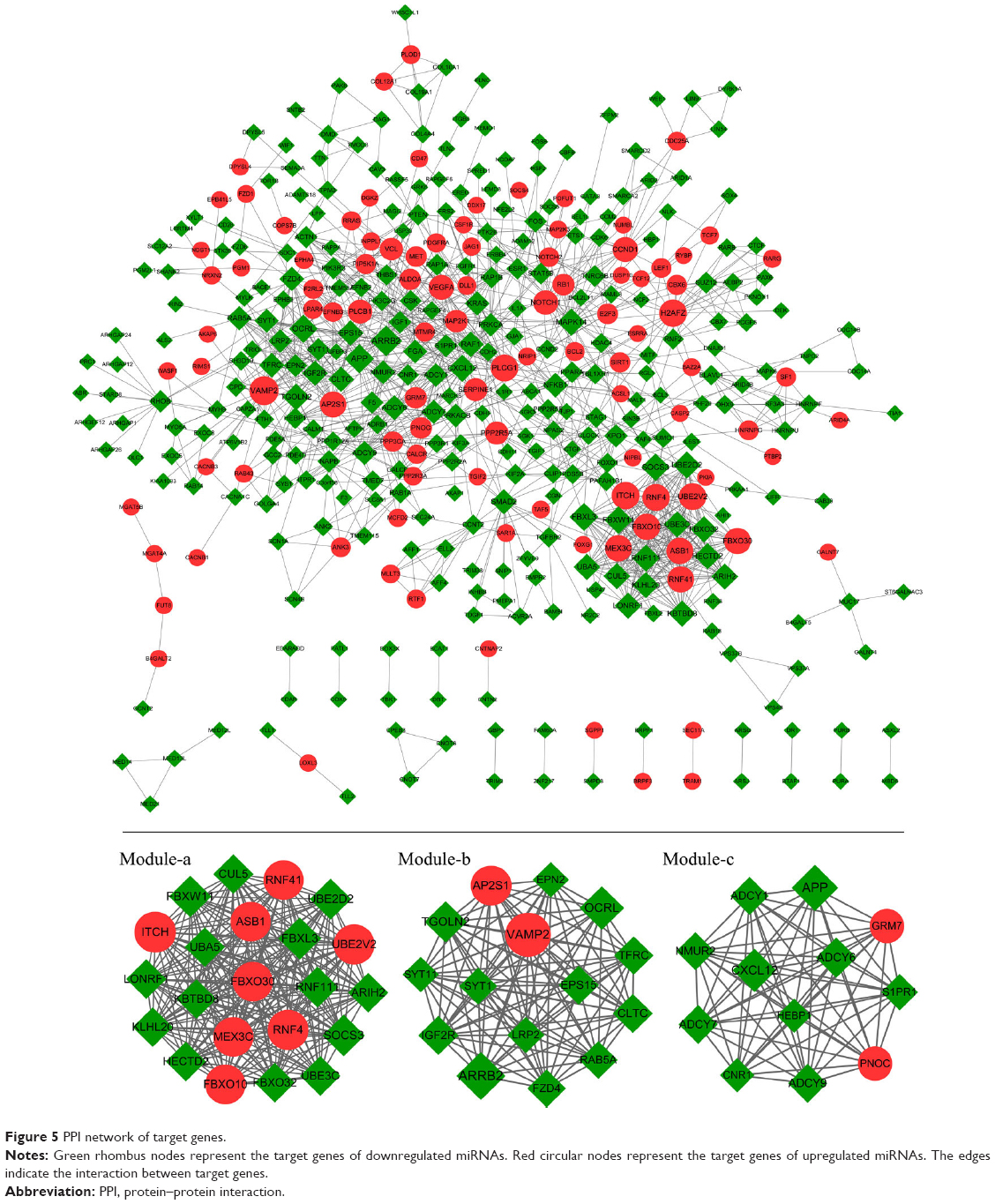 | Figure 5 PPI network of target genes. |
Genes in module-a were significantly enriched in two pathways, including ubiquitin-mediated proteolysis and circadian rhythm. Similarly, two pathways were closely related to module-b genes, including endocytosis and synaptic vesicle cycle. A total of 36 pathways were significantly enriched by module-c genes, such as retrograde endocannabinoid signaling, glutamatergic synapse, ovarian steroidogenesis, and regulation of lipolysis in adipocytes (Table 3).
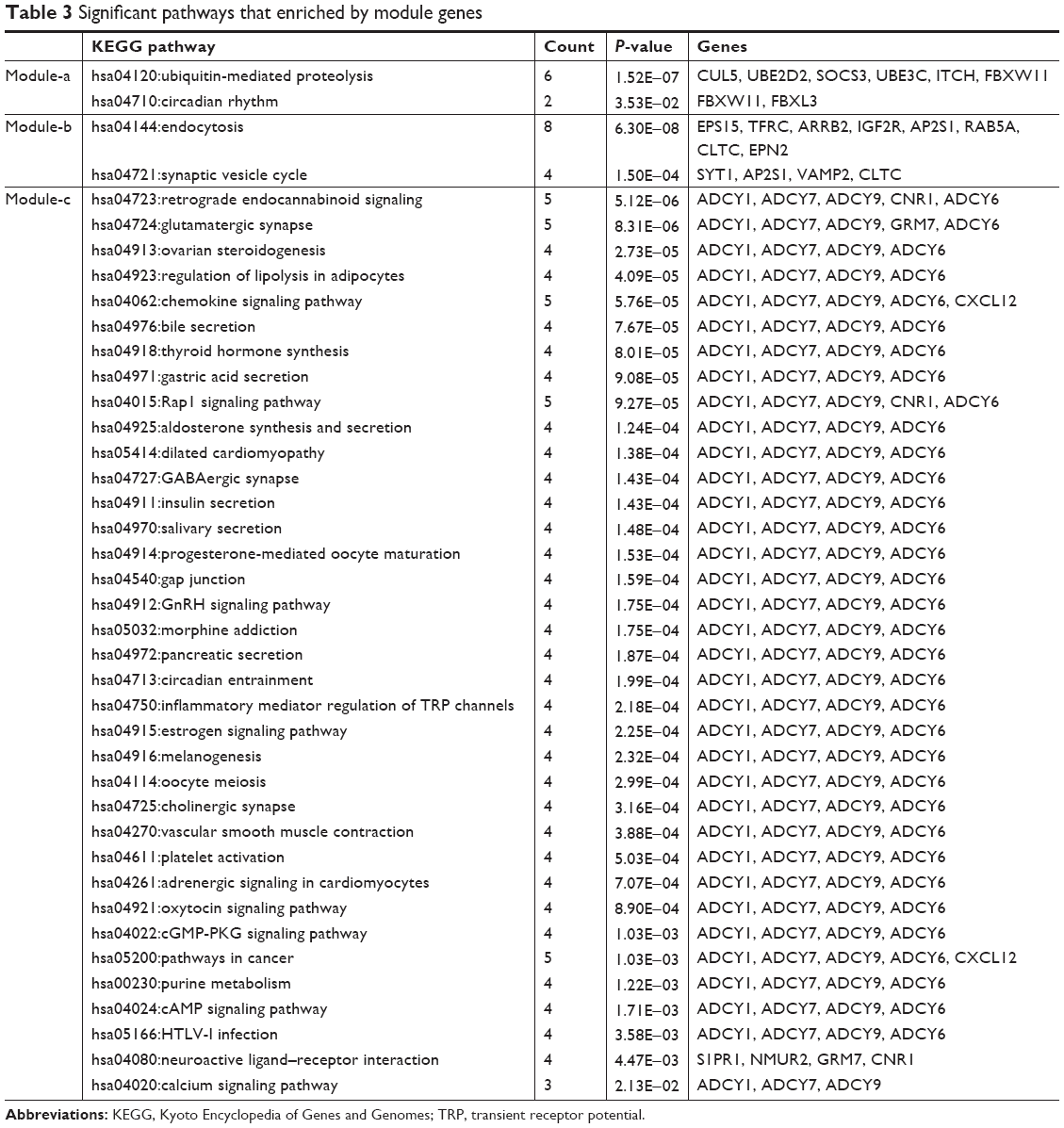 | Table 3 Significant pathways that enriched by module genes |
TF regulatory network
A total of 344 TF–target gene regulatory interactions were predicted, including 58 upregulated miRNA–target genes, 149 downregulated miRNA–target genes, and 2 TFs (early growth response 1 [EGR1] and nanos C2HC-type zinc finger 1 [NANOS1]; Figure 6).
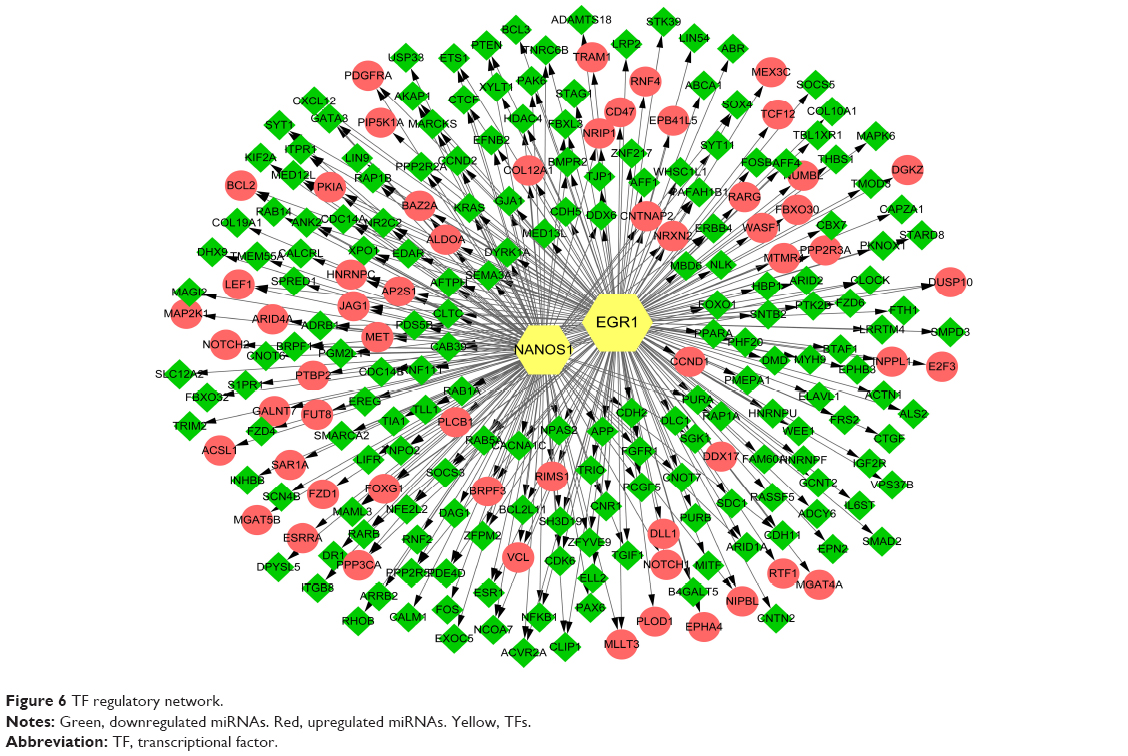 | Figure 6 TF regulatory network. |
Subjects
Subjects (two females and three males) with a mean age of 37.6±14.5 years were included in the control group for fractures. The gender-paired patients with a mean age of 82.4±3.3 years were included in the patient group.
RT-qPCR validation
The expression of has-miR-19a-3p, has-miR-144-3p, has-miR-34a-5p, and has-miR-449b-5p were measured by PCR analysis. Results showed that the expression of has-miR-34a-5p and has-miR-449b-5p was significantly higher in the patient group than in the control group, which was consistent with the analysis described earlier (P<0.01). There was no significant difference in the expression of has-miR-19a-3p and has-miR-144-3p between the two groups, which was not consistent with the bioinformatic prediction (Figure 7).
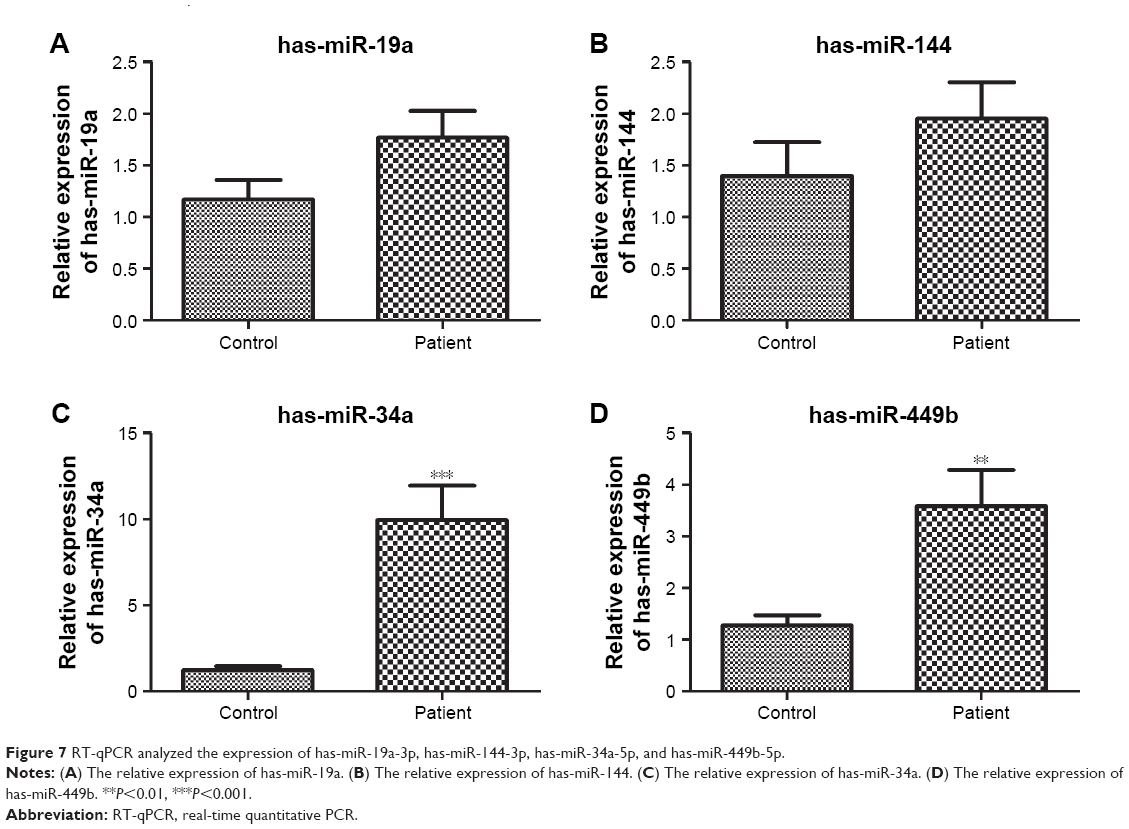 | Figure 7 RT-qPCR analyzed the expression of has-miR-19a-3p, has-miR-144-3p, has-miR-34a-5p, and has-miR-449b-5p. |
Furthermore, the expression of key target genes involved in cellular senescence (SIRT1 and CDC25A), MAPK signaling pathway (VEGFA and MAPT), and AMPK signaling pathway (PRKAA1 and PFKFB3) was also detected. Results showed that the expression levels of SIRT1, VEGFA, PRKAA1, and PFKFB3 were significantly increased in the patient group compared to that in the control group (P<0.05); however, CDC25A and MAPT expression did not exhibit significant difference between the two groups (Figure 8).
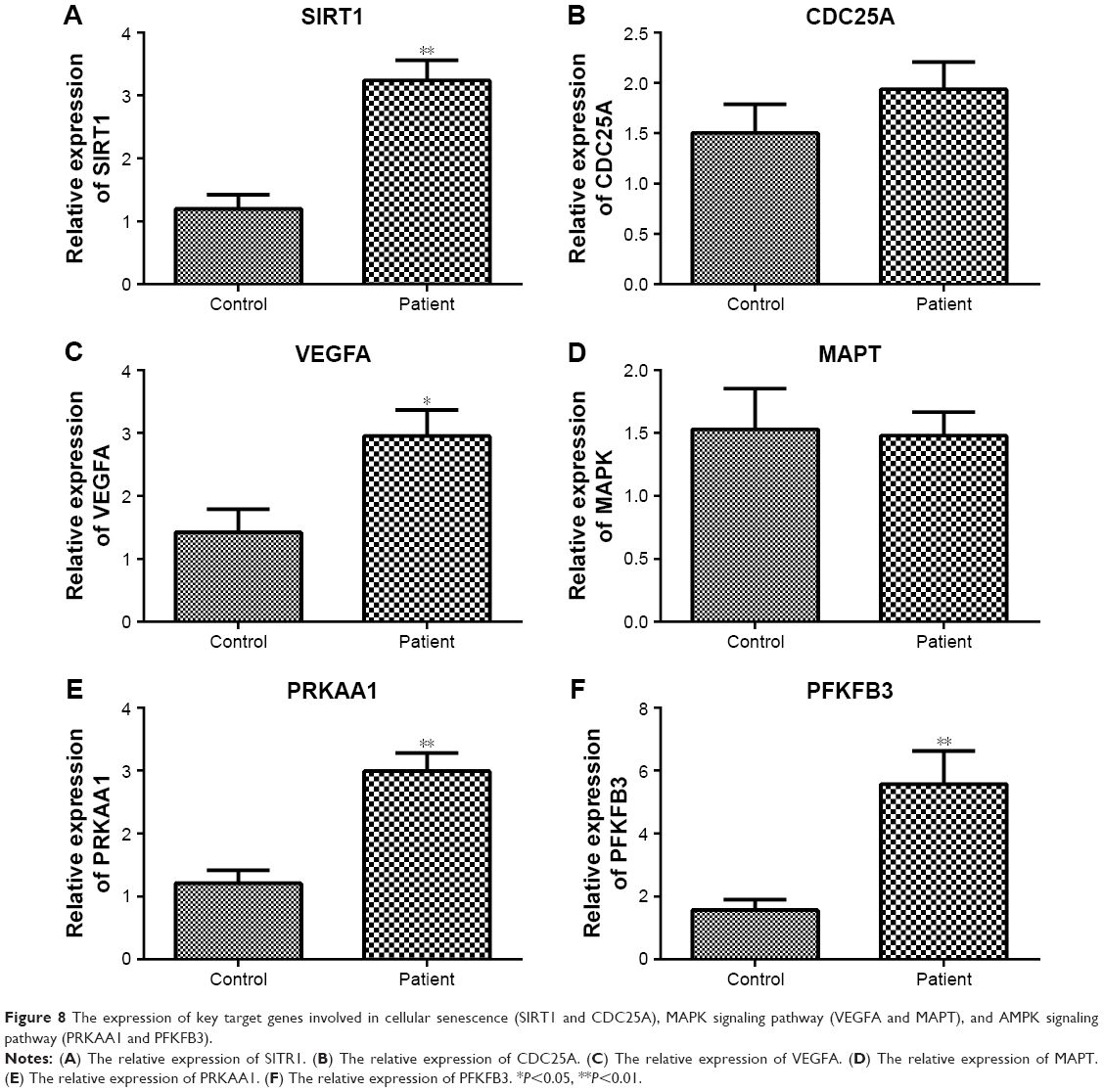 | Figure 8 The expression of key target genes involved in cellular senescence (SIRT1 and CDC25A), MAPK signaling pathway (VEGFA and MAPT), and AMPK signaling pathway (PRKAA1 and PFKFB3). |
Discussion
The role of miRNAs in regulating the aging process has not yet been elucidated. In a previous study, the microarray data (GSE23527) were generated to analyze differentially expressed miRNAs in aging human skeletal muscle, followed by the identification of key cellular functions and canonical pathways related to the progression of muscle loss with aging (sarcopenia) based on ingenuity pathway analysis; the results showed that aging is characterized by an enhanced expression of Let-7 family members that may downregulate genes related to cellular proliferation.15 Compared to this study, we downloaded this microarray data and also identified the differentially expressed miRNAs between young and old skeletal muscles. Furthermore, function and pathway enrichment analyses were also performed. In addition to these, we conducted other analyses, such as PPI network and TF prediction for target genes. These comprehensive analyses will help us identify more significant miRNAs. The results suggested that a total of 23 miRNAs were differentially expressed in old muscle samples, among which 12 were involved in the miRNA–target gene network.
Pathway analysis showed that the gene targets of upregulated miRNAs were significantly enriched in the AMPK signaling pathway. It is reported that AMPK plays a key role in controlling the aging process by regulating energy metabolism, autophagic degradation, and stress resistance.27 AMPK can improve cellular stress resistance and play a critical role in suppressing cell aging. The loss of AMPK activation increased cell stress and suppressed autophagic clearance, which contributed to the cell aging process.27 Another study found that the activation of AMPK signaling was reduced in the skeletal muscles of old rats, suggesting that AMPK activation reduction was a contributory factor in aging process.28 Furthermore, it is reported that deletion of muscle-specific PRKAA1 may lead to delayed development of skeletal muscles and regulate lipid metabolism and mitochondrial oxidation in mice fed a high-fat diet, suggesting that PRKAA1 may be involved in related metabolic diseases.29 In this study, has-miR-19a, with a degree of 434, was the most significant node in the miRNA–target gene network. Pathway analysis also showed that has-miR-19a was closely related to the AMPK signaling pathway. The expression of PRKAA1 and PFKFB3 involved in the AMPK signaling pathway was significantly increased in the patients with sarcopenia. All these indicated that AMPK signaling played a critical role in muscle aging process involved with has-miR-19a. hsa-miR-34a, with a degree of 217, was another significant node in the miRNA–target gene network. Pathway analysis indicated that has-miR-34a was significantly enriched in cellular senescence and MAPK signaling pathway. It is reported that cellular senescence may be involved in the aging process via the accumulation of senescent cells and stem cell loss.30 Cellular senescence may be a marker for the aging process, suggesting that the pathway results were significant. In addition, MAPK plays a critical role in regulating stress responses in skeletal muscles. The activation of MAPK can be induced by exercise, which is involved in preventing skeletal muscle atrophy.31 p38 MAPK signaling pathway affects the autonomous loss and renewal of stem cells.32 The satellite cell homeostatic process was dysregulated by p38 MAPK signaling in the skeletal muscles of aged mice.32 Moreover, we found that the expression levels of SIRT1 involved in cellular senescence and VEGFA involved in MAPK signaling pathway were significantly increased in patients with sarcopenia. Aging gastric mucosa has significantly reduced the expression of VEGF, hinting the role of VEGF in aging-related impairment of angiogenesis.33 Mohamed et al34 revealed that the activation of SIRT1 ameliorated skeletal muscle performance in pathophysiological conditions such as sarcopenia and disuse-induced atrophy in aging mice. Liao et al35 demonstrated that exercise, resveratrol, or their combination played a protective role in sarcopenia in aged rats through regulating AMPK/SIRT1 pathway. These observations suggest that our findings are significant.
Furthermore, in this study, hsa-miR-34a was also found to be closely related to endocrine resistance. It is found that the secondary hormones underlying insulin-like signaling may be involved in the regulation of aging in nematodes and flies.36 In all species, the endocrine system plays a key role in modulating the aging process. Although insulin resistance was found to accelerate muscle protein degradation, which causes muscle atrophy in old people, whether the endocrine system manipulated the aging process by insulin-like signaling remained to be ascertained. Thus, endocrine resistance may be an important event in the aging process regulated by hsa-miR-34a. Further studies are warranted in the future.
However, some limitations merit further consideration. First, whether the identified differentially expressed miRNAs can be used as muscle-specific miRNAs is still needed to be confirmed by more signature analysis. Second, our patient sample size was small, which might influence the stability of statistical power. More than five patient samples are required to be collected for validation of our results. Third, we detected the expression of four miRNAs in skeletal muscle from sarcopenic patients and control subjects. Nevertheless, the connection between aging and sarcopenia in this context was not investigated. In addition, the information on muscle mass or strength in relation to their original analysis was not provided, which hindering us for further patient analysis. Therefore, additional experiments and high throughput data are needed to confirm our findings.
Conclusion
miRNAs play regulatory roles in the aging process of skeletal muscles. AMPK signaling pathway, cellular senescence, and MAPK signaling pathway are significant pathways in the aging process. has-miR-19a and hsa-miR-34a, as significant nodes in miRNA regulatory network, may provide new perspectives to prevent muscle aging.
Disclosure
The authors report no conflicts of interest in this work.
References
Phillips SM. Nutritional supplements in support of resistance exercise to counter age-related sarcopenia. Adv Nutr. 2015;6(4):452–460. | ||
Patel HP, Syddall HE, Jameson K, et al. Prevalence of sarcopenia in community-dwelling older people in the UK using the European Working Group on Sarcopenia in Older People (EWGSOP) definition: findings from the Hertfordshire Cohort Study (HCS). Age Ageing. 2013;42(3):378–384. | ||
Brown JC, Harhay MO, Harhay MN. Sarcopenia and mortality among a population-based sample of community-dwelling older adults. J Cachexia Sarcopenia Muscle. 2016;7(3):290–298. | ||
Cannon JG. Cytokines in aging and muscle homeostasis. J Gerontol A Biol Sci Med Sci. 1995;50 Spec No:120–123. | ||
Hagen JL, Krause DJ, Baker DJ, Fu MH, Tarnopolsky MA, Hepple RT. Skeletal muscle aging in F344BN F1-hybrid rats: I. Mitochondrial dysfunction contributes to the age-associated reduction in VO2max. J Gerontol A Biol Sci Med Sci. 2004;59(11):1099–1110. | ||
Jackson MJ. Skeletal muscle aging: role of reactive oxygen species. Crit Care Med. 2009;37(10 Suppl):S368–S371. | ||
Weisleder N, Brotto M, Komazaki S, et al. Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J Cell Biol. 2006;174(5):639–645. | ||
Hall JK, Banks GB, Chamberlain JS, Olwin BB. Prevention of muscle aging by myofiber-associated satellite cell transplantation. Sci Transl Med. 2010;2(57):57ra83. | ||
Bhaumik D, Scott GK, Schokrpur S, et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging. 2009;1(4):402–411. | ||
Caygill EE, Johnston LA. Temporal regulation of metamorphic processes in Drosophila by the let-7 and miR-125 heterochronic microRNAs. Curr Biol. 2008;18(13):943–950. | ||
Mcgregor RA, Poppitt SD, Cameron-Smith D. Role of microRNAs in the age-related changes in skeletal muscle and diet or exercise interventions to promote healthy aging in humans. Ageing Res Rev. 2014;17(9):25–33. | ||
Murach KA, Mccarthy JJ. MicroRNAs, heart failure, and aging: potential interactions with skeletal muscle. Heart Fail Rev. 2016:1–10. | ||
Mccarthy JJ. microRNA and skeletal muscle function: novel potential roles in exercise, diseases, and aging. Front Physiol. 2014;5(5):290. | ||
Zacharewicz E, della Gatta P, Reynolds J, et al. Identification of microRNAs linked to regulators of muscle protein synthesis and regeneration in young and old skeletal muscle. PLoS One. 2014;9(12):e114009. | ||
Drummond MJ, Mccarthy JJ, Sinha M, et al. Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiol Genomics. 2011;43(10):595–603. | ||
Smyth GK. Limma: Linear models for microarray data. In: Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York: Springer; 2005:397–420. | ||
Xiao F, Zuo Z, Cai G, Kang S, Gao X, Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37(Database issue):D105–D110. | ||
Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. | ||
Ashburner M, Ball CA, Blake JA, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. | ||
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. | ||
Huang Daw, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. | ||
Huang Daw, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. | ||
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. | ||
Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447-52–D452-52. | ||
Bandettini WP, Kellman P, Mancini C, et al. MultiContrast Delayed Enhancement (MCODE) improves detection of subendocardial myocardial infarction by late gadolinium enhancement cardiovascular magnetic resonance: a clinical validation study. J Cardiovasc Magn Reson. 2012;14:83. | ||
Janky R, Verfaillie A, Imrichová H, et al. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput Biol. 2014;10(7):e1003731. | ||
Salminen A, Kaarniranta K, Kai K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11(2):230–241. | ||
Reznick RM, Zong H, Li J, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5(2):151–156. | ||
Wu W, Xu Z, Zhang L, et al. Muscle-specific deletion of Prkaa1 enhances skeletal muscle lipid accumulation in mice fed a high-fat diet. J Physiol Biochem. 2018;74(2):195–205. | ||
Collado M, Blasco MA, Serrano M, Chavent M. Cellular senescence in cancer and aging. Cell. 2007;130(2):223–233. | ||
Kramer HF, Goodyear LJ. Exercise, MAPK, and NF-kappaB signaling in skeletal muscle. J Appl Physiol. 2007;103(1):388–395. | ||
Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, Olwin BB. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med. 2014;20(3):265–271. | ||
Deng X, Ahluwalia A, Xiong X, Szabo S, Sandor Z, Tarnawski A. S1619 MMP9-mediated upregulation of endostatin and downregulation of VEGF in aging gastric mucosa: novel mechanism for impaired angiogenesis. Gastroenterology. 2008;134(4):A-236. | ||
Mohamed JS, Wilson JC, Myers MJ, Sisson KJ, Alway SE. Dysregulation of SIRT-1 in aging mice increases skeletal muscle fatigue by a PARP-1-dependent mechanism. Aging. 2014;6(10):820–834. | ||
Liao ZY, Chen JL, Xiao MH, et al. The effect of exercise, resveratrol or their combination on Sarcopenia in aged rats via regulation of AMPK/Sirt1 pathway. Exp Gerontol. 2017;98:177–183. | ||
Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science. 2003;299(5611):1346–1351. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.