")
Back to Journals » Cancer Management and Research » Volume 6
Role of microRNA in epithelial to mesenchymal transition and metastasis and clinical perspectives
Authors Díaz-López A, Moreno-Bueno G, Cano A
Received 14 December 2013
Accepted for publication 9 February 2014
Published 25 April 2014 Volume 2014:6 Pages 205—216
DOI https://doi.org/10.2147/CMAR.S38156
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Antonio Díaz-López,1 Gema Moreno-Bueno,1,2 Amparo Cano1
1Departamento de Bioquímica, Facultad de Medicina, Universidad Autónoma de Madrid, Instituto de Investigaciones Biomédicas “Alberto Sols” (CSIC-UAM), IdiPAZ, Madrid, Spain; 2Fundación MDAnderson Internacional, Madrid, Spain
Abstract: The microRNAs (miRNAs) are a class of small, 20–22 nucleotides in length, endogenously expressed noncoding RNAs that regulate multiple targets posttranscriptionally. Interestingly, miRNAs have emerged as regulators of most physiological and pathological processes, including metastatic tumor progression, in part by controlling a reversible process called epithelial-to-mesenchymal transition (EMT). The activation of EMT increases the migratory and invasive properties fundamental for tumor cell spread while activation of the reverse mesenchymal-to-epithelial transition is required for metastasis outgrowth. The EMT triggering leads to the activation of a core of transcription factors (EMT-TFs) – SNAIL1/SNAIL2, bHLH (E47, E2-2, and TWIST1/TWIST2), and ZEB1/ZEB2 – that act as E-cadherin repressors and, ultimately, coordinate EMT. Recent evidence indicates that several miRNAs regulate the expression of EMT-TFs or EMT-activating signaling pathways. Interestingly, some miRNAs and EMT-TFs form tightly interconnected negative feedback loops that control epithelial cell plasticity, providing self-reinforcing signals and robustness to maintain the epithelial or mesenchymal cell status. Among the most significant feedback loops, we focus on the ZEB/miR-200 and the SNAIL1/miR-34 networks that hold a clear impact in the regulation of the epithelial-mesenchymal state. Recent insights into the p53 modulation of the EMT-TF/miRNA loops and epigenetic regulatory mechanisms in the context of metastasis dissemination will also be discussed. Understanding the regulation of EMT by miRNAs opens new avenues for the diagnosis and prognosis of tumors and identifies potential therapeutic targets that might help to negatively impact on metastasis dissemination and increasing patient survival.
Keywords: EMT, MET, microRNAs, cancer, metastasis
miRNAs: an overview in cancer
The microRNAs (miRNAs) are small noncoding RNA molecules of 20–22 nucleotides (nt) which modulate gene expression posttranscriptionally, binding specific sequences present mostly in the 3′ untranslated region (3′ UTR) of target messenger RNAs (mRNAs). To date, approximately 700 human miRNAs have been identified, and it is estimated that more than 30% of the global mRNA expression is regulated by the miRNAs.1 The miRNAs are either localized within introns, sharing promoter regulation with the host gene, or endowed with their own independent promoters and transcriptional regulation. Remarkably, miRNAs are frequently organized in clusters, containing several miRNAs in tandem and found to map to all human chromosomes, with the exception of the Y chromosome.2
From miRNA synthesis to the generation of an active RNA-induced silencing complex (RISC), miRNAs are subjected to various maturation processes. Briefly, RNA polymerase II transcribes a long miRNA proprecursor in the nucleus, termed primary miRNA (pri-miRNA), of several hundred nt and containing a 5′ prime cap (5′ cap) and a 3′ poly (A) tail end. This primary precursor is then processed by the ribonuclease III Drosha that releases a miRNA precursor (pre-miRNA) of approximately 70–80 nt that presents a RNA hairpin loop structure. Later, the pre-miRNA is actively exported by exportin-5 to the cytoplasm where it is cleaved by the Dicer complex, removing the loop region, and delivering the usually 22 nt mature double-stranded miRNA. One of the RNA strands is then bound by the argonaute protein, resulting in the miRNA-induced silencing complex (miRISC) which specifically binds to the 3′ UTR of mRNA targets.3,4
The specificity of the miRNA-RISC complex is provided by the seed – a sequence of 2–8 nt in the miRNA – that recognizes a complementary sequence in the 3′ UTR of target mRNAs. Interestingly, miRNAs use different mechanisms to control the mRNA expression. A near-perfect match of the seed and the 3′ UTR mRNA sequence leads to mRNA degradation; whereas, partial complementation results in reduced expression by either mRNA removal or translation impairment.3,5 Thus, a single miRNA might affect the expression of several mRNA targets, even within the same signaling pathway, and simultaneously control diverse biological processes, including differentiation, proliferation, migration, or cell survival.3–5
One of the first insights into the potential deregulation of miRNAs in cancer was the finding by Calin et al6 in 2004, which showed that approximately one-half of total miRNAs are located at fragile sites or in the cancer susceptibility loci. Indeed, genomic amplification, chromosomal deletion, point mutation, and aberrant promoter methylation are the main strategies used by cancer cells to gain or lose miRNA function.4,7 The deregulation of miR-15 and miR-16 expression found in chronic lymphocytic leukemia was the original evidence for the miRNA implication in cancer biogenesis.8 From that first report, an increasing number of miRNAs associated with cancer (also called oncomiRs) have been reported with pro- or antitumoral activities. Intriguingly, several miRNAs have either pro- or antitumoral properties, depending on the tumor context. In this sense, miR-29 operates as a tumor suppressor in lung tumors; whereas, in breast cancer, it bears an oncogenic role.9,10 Another example is miR-26, which has been found amplified in gliomas,11 while its overexpression induces either apoptosis in hepatocellular carcinoma12 or metastasis in lung cancer cells.13
Epithelial-to-mesenchymal transition in cancer
Cancer constitutes a conglomerate of diseases characterized by the loss of growth control and cell dissemination. To spread, epithelial tumor cells take advantage of a reversible developmental process called epithelial-to-mesenchymal transition (EMT) to favor cell migration and invasiveness. Once they reach their new niche, they activate the reverse program – mesenchymal-to-epithelial transition (MET) – to form macrometastasis.14–17 Intermediate states between epithelial and mesenchymal phenotypes are presently considered to exist during the metastatic cascade,18,19 implying that epithelial tumor cells are endowed with a high degree of epithelial plasticity that needs to be dynamically regulated and finely tuned during tumor progression and metastasis.15 Epithelial cells are tightly adjoined cells through specialized membrane structures: tight junctions; desmosomes; and adherens junctions that constrain cell movement and detachment from the epithelial layer. Up to 90% of human tumors are carcinomas, which originate from the differentiated epithelium and are susceptible to suffer an EMT process within certain tumor subpopulations in response to external cues.19–21 The hallmarks of the EMT program are the loss of E-cadherin and apical–basal cell polarity, accompanied by the gain of mesenchymal characteristics, including the acquisition of cell migration and invasion abilities, as well as an increased expression of mesenchymal markers, such as vimentin, fibronectin, and N-cadherin.17 The molecular and cellular mechanisms underlying an EMT are complex and can be initiated by multiple extracellular signals that finally activate transcription factors, miRNAs, and/or different signaling pathways, depending on the physiological or pathological contexts. The reverse MET process might be, therefore, promoted by blocking the action of the factors and signaling pathways that activate EMT.22,23
EMT can be induced by several signaling pathways, including transforming growth factor beta (TGF-β), Notch and Wnt, among others that converge in the expression of a plethora of E-cadherin transcriptional repressors that also act as EMT inducers (EMT-TFs).
The EMT-TFs’ core is integrated by members of the SNAIL (SNAIL1/SNAIL2), basic helix-loop-helix (bHLH) (E47, E2-2, and TWIST1/TWIST2) and ZEB (ZEB1/ZEB2) families.16 Several of these EMT-TFs are thereby considered EMT drivers, and their expression is directly correlated with poor outcome.16,17,21 EMT-TFs from the SNAIL, ZEB, and TWIST families govern EMT through a complex EMT-TF network, which entails a strong transcriptional control of EMT. However, there is growing evidence that at least three additional regulatory levels solidly support the EMT/MET programs in parallel – miRNA expression, differential splicing, and translational and posttranslational control – that affect localization and protein stability.14,17,18 In this review, we will discuss the most relevant recent findings concerning epithelial plasticity regulation by miRNA expression.
It is well-known that the miRNAs regulate a plethora of cancer-related pathways, such as cell cycle and proliferation, senescence, DNA damage response, apoptosis, and autophagy, among others. An increasing number of reports indicate that several miRNAs are deregulated in primary tumors compared to normal tissue, with some of them being associated to metastasis.24 Noticeably, during the last years, the implication of a growing number of miRNAs in the regulation of EMT-TFs or EMT-related pathways has been observed in several human tumor contexts (Table 1). Interestingly, poorly differentiated carcinomas present an overall decrease of total miRNA expression, suggesting a role for the miRNAs in the regulation of different mechanisms governing epithelial plasticity.25 Targeting the EMT-TFs’ modulators or the EMT-TFs themselves, pivotal proteins implicated in invasion and migration, and/or regulatory enzymes involved in miRNA biosynthesis constitutes the most common mechanism used by the miRNAs to regulate epithelial plasticity and, hence, promote or inhibit metastatic tumor progression.
 | Table 1 Selected miRNAs implicated in metastatic progression and their EMT-related known targets |
miRNA regulation of epithelial plasticity
As mentioned, a hallmark of EMT is E-cadherin loss, mainly due to its transcriptional repression by EMT-TFs. A few miRNAs have been involved in the direct regulation of E-cadherin expression. An example is miR-9, which directly targets the E-cadherin gene in breast cancer cells.26 Interestingly, miR-9 is upregulated by MYC. Its expression is correlated with MYCN amplification, tumor grade, and the metastasis status in breast cancer patients.26 Conversely, miR-9 acts as a tumor suppressor in melanoma cells by targeting the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-SNAIL1 pathway, thus upregulating the E-cadherin expression.27 Furthermore, additional activities of the miR-9 have been described, including its secretion by the tumor cells within microvesicles to promote endothelial cell migration and angiogenesis,28 revealing the complexity of miRNA activities depending on cell context and tumor type.
Another mechanism to control epithelial plasticity is regulating those pathways controlling EMT. Recently, several miRNAs that directly impinge on the regulation of EMT-TFs have been described; some of them operate in double negative feedback loops that contribute to maintain the epithelial or mesenchymal states29–31 (Figure 1). In addition, these EMT-regulatory miRNA networks can be controlled by EMT-inducing signals, like TGF-β signaling, and/or by p53, implementing additional levels of regulation and providing robustness to the system. In this review, we will discuss the most relevant miRNA-EMT-TF networks discovered to date.
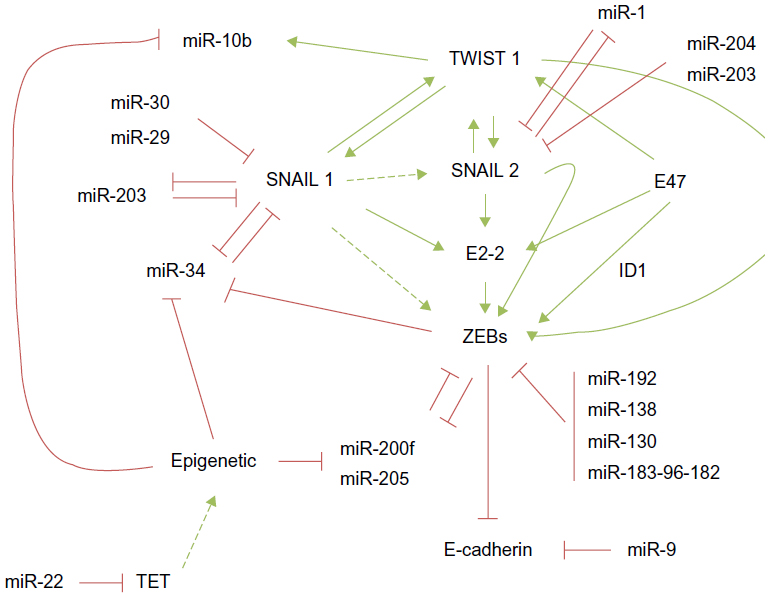 | Figure 1 miRNA and EMT-TF regulatory networks controlling epithelial plasticity. |
ZEB/miR-200 feedback loop
The two members of the zinc finger E-box-binding homeobox (ZEB family), ZEB1/δEF1, and ZEB2/SIP1, are among the first described EMT-TFs32,33 and have emerged as the better-studied EMT-TFs subject to miRNA regulation because their expression is associated with poor prognosis in several tumor types.16,34–36 ZEB1 has been found upregulated in metaplastic breast carcinomas37 as well as in the sarcomatous element of endometrial carcinosarcomas, an aggressive neoplasia with a biphasic histology of carcinomatous and sarcomatous components considered an example of complete EMT in vivo.38,39 Moreover, ZEB1 silencing in a colorectal cancer xenograft model blocks metastasis.40 One of the first pieces of evidence confirming a functional role for the miRNA control of epithelial plasticity is the regulation of ZEB proteins by the miR-200 family (miR-200f) and miR-20541 (Figure 1). The miR-200f is composed of five members located in two polycistronic pri-miRNA loci on chromosome 1 (miR-200b, miR-200a, and miR-429) and chromosome 12 (miR-200c and miR-141).41
In 2007, Christoffersen et al42 reported for the first time the ZEB regulation by miR-200f, identifying nine target sites for miR200f and two for miR-205 in the 3′ UTR of ZEB2. Shortly after that finding, two reports identified several functional targeting sites in both ZEB1/ZEB2.43,44 Park et al43 described a significant positive correlation between high E-cadherin/low vimentin expression and miR-200f levels after miRNA expression profile analyses of a cell panel including 60 epithelial and mesenchymal cell lines from the National Cancer Institute. Ectopic expression of miR-200f in colorectal HCT-116 cells or their silencing in breast carcinoma MDA-MB-231 significantly affected ZEB1 expression, sustaining a nontissue specific regulation.43
Similar results were found by Gregory et al44 in Madin-Darby canine kidney cells after TGF-β-mediated EMT. Furthermore, both Burk et al45 and Bracken et al46 simultaneously demonstrated that ZEB1 silencing in mesenchymal cancer cells induced miR-200f expression, resulting in a negative feedback loop. Remarkably, the ZEB1-mediated repression was leaded by its direct binding to the E-box sites present upstream of miR-200c/miR-14145 or miR-200b/miR-200a/miR-429 promoters.46
In addition to targeting ZEB1/ZEB2, miR-200f regulates the expression of diverse stemness-related genes, such as CD44, BMI1, and c-MYC. Conversely, ZEB1 represses miR-183 and miR-203 which, in turn, target BMI1 and other stemness factors, such as SOX2 and KLF4,47 which support previous studies that showed that EMT confers cell stemness properties.48,49 Importantly, the EMT-stemness association is also found in vivo in basal-like breast tumors, a subtype of breast cancer associated with poor differentiation grade, enriched in numerous EMT, and stemness markers.50 Interestingly, in some cellular contexts, the miR-200f/ZEB1 feedback loop can be maintained and reinforced by autocrine secretion of TGF-β, thus contributing to the maintenance of the mesenchymal phenotype.51 Additionally, in cancer stem cells, E-cadherin is also an indirect target of miR200f through SUZ12, a component of the PRC2 complex, thus resulting in the epigenetic repression of E-cadherin and the upregulation of ZEB1/ZEB2.52
Aside from the established double feedback loop, miR-200f also acts in a ZEB1/ZEB2 independent manner, being associated with the regulation of Sec23a, a modulator of the secretome.53 Targeting Sec23a, a coat protein complex II (COPII) vesicle component, resulted in decreased secretion of the metastasis suppressive factors tubulointerstitial nephritis antigen-like 1 (TINAGL1) and insulin-like growth factor binding protein 4 (IGFBP4) in 4TO7 cells.53 Moreover, miR-200a suppresses β-cat/Wnt signaling by targeting β-catenin. Consequently, miR200a downregulation in some tumors, like meningiomas, activates the β-cat/Wnt pathway, represses E-cadherin, and promotes tumor growth.54
All these findings support a dual role of miR-200f in the tumor progression and the regulation of epithelial plasticity. Thus, miR-200f-silencing facilitates early steps of metastasis impacting on EMT control by ZEB1/ZEB2, while its upregulation at distal metastasis might promote MET and metastatic colonization through Sec23 targeting.53 Interestingly, the tumor suppressor p21 controls miR-200 levels but also the miR-183-96-182 cluster, which represses ZEB1, SNAIL2, ITGB1, and KLF4 expression in HCT-116 colorectal cells.55 However, the biological implication of miR-183-96-182 in EMT and metastasis remains still unknown, since knockdown of individual members of the cluster did not affect cell invasion.55
ZEB factors can be regulated by additional miRNAs, like miR-19256 or miR-13857 targeting ZEB2, and the recently described miR-130 acting on ZEB158 (Figure 1). On the other hand, ZEB2 is indirectly regulated by miR-221/miR-222, which is overexpressed in basal-like breast tumors. The miR-221/miR-222 targets the ZEB2-repressor TRPS1, a GATA family transcription factor, promoting E-cadherin downregulation and increased migration and invasion properties.59
SNAIL1 regulation: the SNAIL1/miR-34 axis
Similar to the ZEB/miR-200 negative feedback loop, SNAIL1, and the miRNAs, the miR-34f (miR-34a, miR-34b, and miR-34c) and miR-203, constitute additional epithelial plasticity regulatory loops60,61 (Figure 1). Interestingly, the seed sequence of miR-34 targets a highly conserved 3′ UTR sequence present in SNAIL1, SNAIL2, ZEB1, and also the stemness factors BMI1, CD44, CD133, and c-MYC. Conversely, SNAIL1 and ZEB1 directly repress the miR-34f60 and miR-200f promoters,45,46 respectively, providing a tight crosstalk between the ZEB/miR-200 and SNAIL1/miR-34 pathways and adding robustness to the network.
SNAIL1 expression is also controlled by miR-29b, miR-30, and by a second negative feedback loop involving miR-20361–63 (Figure 1). Accordingly, the expression of miR-29 induces a complete MET and decreases the invasive phenotype in the prostate cancer cell line PC3.62 Another elegant strategy to silence SNAIL1 expression involves targeting SNAIL1-activation pathways. The lethal-7 (lET-7) family is formed by 12 miRNA members located on eight different chromosomes in humans. Remarkably similar to miR-200, the lET-7 family has no tissue-dependent activity, but rather its role is related to the differentiation states of the tissue, suggesting that both miRNAs families are central differentiation regulators.64 The LET-7 family member levels are downregulated in various cancers, including lung, colon, ovarian, and gastric carcinomas – as well as melanoma – during tumor progression, supporting their central role in epithelial plasticity.
Mechanistically, LET-7 acts as a tumor suppressor in humans, mainly because it directly binds to the transcripts of multiple genes with oncogenic activity, such as RAS, the high mobility group (HMG)A2, or BACH1.65 HMGA2, a chromatin-binding protein that integrates the TGF-β response66 is downregulated by the LET-7 miRNA family in pancreatic human cells67 or by miR-365 in lung adenocarcinoma,68 thus indirectly affecting SNAIL1 expression. Importantly, the LET-7 family coordinates other antimetastatic events by repressing H-RAS and the transcription factor BACH1. Both HMGA2 and BACH1 promote the transcription of proinvasive genes, cell invasion, and the metastasis to the bone of breast cancer cells.69,70
SNAIL2 regulation
SNAIL2 is regulated posttranscriptionally by different miRNAs, including miR-1, miR-203, and miR-20471–73 (Figure 1). In a recent report using a PTEN- and TP53-null prostate adenocarcinoma mice model that progresses via TGF-β-induced EMT, SNAIL2 was the main driver of EMT activation.74 In this model, miR-1 was downregulated by a double-negative feedback loop including SNAIL2 and the miRNAs – miR-1 and miR-200b – in a self-reinforcing regulatory loop, favoring EMT.71 SNAIL2 is also a direct target of miR-20472 and miR-20373 in retinal pigment epithelium and breast cancer cells, respectively.
Remarkably, miR-204 acts using a dual mechanism, targeting TGFBR2, a TGF-β receptor, but also by the direct targeting of SNAIL2.72 On the other hand, the ectopic expression of miR-203 induces apoptosis and blocks cell cycle progression and invasion in breast cancer cell lines mediated, at least partially, by SNAIL2 repression.73
All the present findings indicate a tight regulation of epithelial plasticity by direct and indirect modulation of crucial EMT-TFs. This, together with the interconnection between several core EMT-TFs75–78 (Figure 1), provides robustness to the system as well as the potential to modulate epithelial plasticity in response to different environmental cues.
Other miRNAs implicated in metastasis
Some miRNAs are not directly implicated in EMT-TF regulation but are relevant for metastatic dissemination, either suppressing or promoting metastasis. A significant suppressive miRNA is miR-335, which targets SOX4 and Tenascin-C, thus, promoting cancer cell migration, invasion, and metastasis.79 In the same study, downregulation of miR-206 and miR-126 was found to be correlated with metastatic relapse in breast cancer patients. Restoration of miR-335 or miR-206 expression in a lung metastatic MD-MB231-derived cell line reduces its metastatic dissemination.79 Interestingly, the role of miR-126 is linked to tumor growth, endothelial recruitment, and metastatic initiation by inhibiting the proangiogenic factors: IGFBP2; PITPNC1; and MERTK.80 Another example is miR-31, a metastasis relapse marker in breast tumors. The miR-31 targets the prometastatic proteins ITGA5, RdX, and RhoA – thus blocking invasion, anoikis resistance, and metastasis.81 However, miR-31 silencing is observed in other tumor types, suggesting that miR-31 pro- or antitumorigenic function depends on tumor context.82
On the other hand, overexpression of some miRNAs confers prometastatic ability to the tumor cells, like miR-10b, which is frequently overexpressed in primary invasive breast carcinomas and their metastasis.83,84 However, the mechanism of action of this miRNA is not based on targeting the core EMT-TFs. The miR-10b is, in fact, induced by the EMT-TF TWIST1 (Figure 1), and it is associated with mesenchymal features and invasion properties by inhibiting HOXD10, an inhibitor of the prometastatic protein ROCK.84 Interestingly, miR-106b modulates TWIST1 expression and invasiveness in endometrial cancer cells.85 Besides, the overexpression of miR-373 and miR-520c confers metastatic capacity to breast86 and prostate cancer cells87 by suppressing CD44 expression.
In melanoma, a highly metastatic tumor, miR-199a-3p, miR-199a-5p, and miR-1908 promote metastatic invasion, angiogenesis, and colonization by targeting the apolipoprotein E and the heat shock factor DNAJA4.88 Expression of these three miRNAs directly correlates with melanoma metastatic outcome. Overexpression of miR-30b/miR-30d also correlates with stage, metastatic potential, and reduced overall survival in melanoma patients.89 Accordingly, ectopic expression of miR-30b/miR-30d in melanoma cells increased their metastatic behavior by a dual mechanism – promoting invasion and immunosuppression by directly targeting the GalNAc transferase GALNT7.89
miRNA regulation in epithelial plasticity and metastasis
The dynamic nature of the EMT and MET processes along the metastatic cascade implies a finely tuned, regulated control of tumor cell plasticity that can be exerted, at least in part, through posttranscriptional modulation of the EMT-TFs, such as that mediated by miRNAs. The regulation of the miRNA expression linked to EMT and metastasis is still poorly known. Nevertheless, several mechanisms that have started to be exposed during the last years will be discussed in this section.
The p53-EMT connection
Numerous recent reports highlight a connection between p53 and EMT through the modulation of some miRNAs that regulate EMT-TFs in different tumor models. p53 has been reported to maintain the epithelial phenotype by enhancing miR-34 expression and subsequently repressing SNAIL1. Importantly, a mutated form of p53 is unable to increase miR-34 expression, shifting the equilibrium toward a mesenchymal phenotype in colorectal carcinoma cells.90 A similar situation occurs in the endometrial carcinomas, where a p53 gain-of-function mutant exerts oncogenic properties by transcriptional inhibition of miR-130b, a negative regulator of ZEB1. Interestingly, patients with higher levels of miR-130b survived longer.58
Additionally, p53 indirectly controls EMT by the transcriptional regulation of other miRNAs. AP4, a transcription factor recently described as a new EMT inducer,91 is indirectly regulated by p53 through the expression of miR-15a/16-1 in colorectal cancer cells.92 Ectopic expression of miR-15/miR-16-1 suppresses lung metastatic colonization in a xenograft model. Conversely, AP4 inhibits miR-15/miR-16-1 expression defining a novel negative regulatory loop92 (Figure 2). Wild type p53 also controls EMT and stemness properties in the prostate cancer cell line PC-3, at least in part, by enhancing the expression of miR-145. Accordingly, anti-miR-145 partially reverts this phenotype.93 Collectively, all these findings define a new link between chromosome stability, p53, miRNA expression, cell differentiation, and epithelial plasticity that can be potentially used by cancer cells for the initial stages of metastatic tumor progression, such as invasion or intravasation.
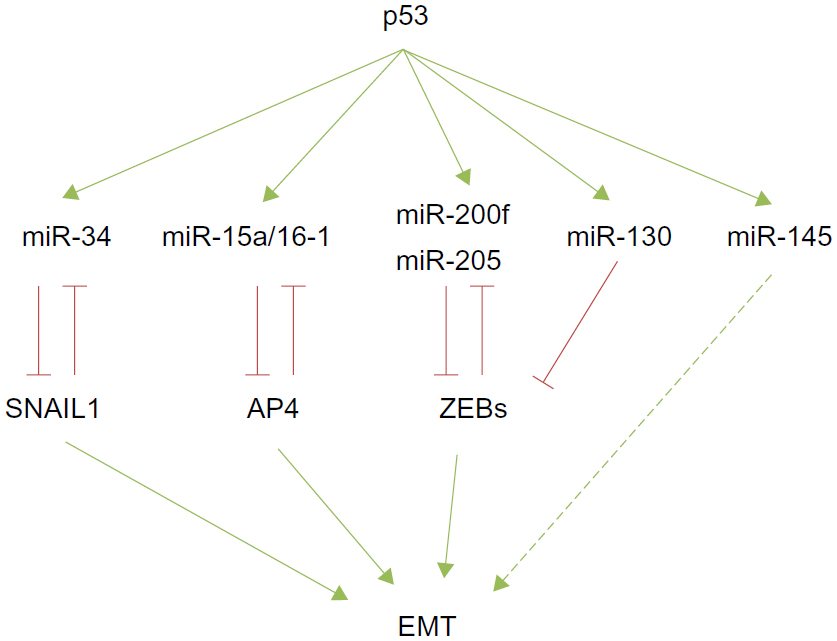 | Figure 2 The p53 modulation of miRNA-EMT-TF networks. |
Epigenetic regulation of miRNAs involved in epithelial plasticity
Since the EMT-TF expression has to be regulated by different miRNAs in a dynamic fashion during the metastatic cascade, CpG miRNA promoter hypermethylation has emerged as a novel mechanism to increase the mesenchymal characteristics of tumor cells due to specific miRNA repression. For example, CpG oligonucleotide hypermethylation of the miR-34 promoter is found in prostate tumors (80%) and in melanoma (63%); it is also present in many cell lines derived from breast, colon, lung, and other types of tumors.94,95 The miR-203, which targets SNAIL2, is also hypermethylated in several tumors, including hepatocellular, prostate, and breast carcinoma.73
Epigenetic regulation of the miR-200 family and its relationship with aggressiveness and chemoresistant behavior have been established in different cancer cell types, including nonsmall cell lung carcinomas, colon, breast carcinomas, as well as endometrial carcinosarcomas.39,95,96 Interestingly, the hypermethylation rate of the miR-200 loci is related to the breast cancer subtype and metastasis outcome. Promoter analysis in 93 breast cancers has identified the presence of a novel CpG promoter region in the miR-200b cluster, mapped 2 kb upstream of the 5′ stem loop region, whose hypermethylation was associated with the loss of either estrogen or progesterone receptors, while hypermethylation of the upstream promoter is associated with lymph node metastasis.96 In addition, a direct correlation between miR-200c-141 loci expression and hypermethylation has been found in endometrial carcinosarcomas.39 In fact, the hypermethylation of this locus seems to be a surrogate marker to differentiate endometrial carcinosarcomas versus carcinomas.39 Hypermethylation of the miR-200f has also been observed in colorectal tumors.95 Microdissection of human primary colorectal samples showed that normal epithelial colon mucosa crypts and stroma (mesenchyme) bear unmethylated and methylated miR-200f promoters, respectively. Colorectal tumors undergo selective miR-200 hypermethylation, sustaining the association between methylation and differentiation status.95 Interestingly, the expression of miR-22 is correlated with miR-200 downregulation and poor outcome in breast cancer patients.96 Mechanistically, miR-22 triggers EMT, enhancing invasiveness, and promoting metastasis in mice xenografts, through the targeting of the ten-eleven translocation (TET) family of methylcytosine dioxygenases, and is implicated in the inhibition of miR-200 promoter demethylation, thereby inducing dedifferentiation90 (Figure 1).
Additional miRNA regulatory mechanisms
The components of the miRNA processing machinery have also been involved in cancer progression, resulting in an overall decrease of mature miRNAs. Interestingly, high levels of miR-103/miR-107 are associated with reduced expression of Dicer and metastasis as well as with poor outcome in breast cancer patients.97 Mechanistically, the overexpression of miR-107 in MB-MDA-231 or SUM149 breast carcinoma cells confers migratory capacities in vitro and empowers metastatic dissemination in vivo by fostering EMT through the downregulation of miR-200 levels.97 Reduced Dicer expression was also found in nonsmall cell lung carcinomas and in ovarian cancer, sustaining a more complex mechanism in tumor progression.98,99 Moreover, the tumor suppressor TP53 also affects the miRNA biogenesis machinery facilitating the processing of pri-miRNAs to pre-miRNAs through its interaction with the Drosha processing complex,100 revealing a wide control regulation by p53 apart from that relying on each specific miRNA’s regulation already discussed (Figure 2).
Potential clinical applications
One of the deleterious effects of tumor progression is the formation of distant tumors or metastasis. In fact, metastases are the main cause of cancer death. Besides understanding the functional miRNAs implicated in cancer, their analyses in cancer patients open new avenues for diagnosis, prognosis, and treatment in oncology. Although we are now beginning to understand how the miRNA regulates epithelial plasticity and metastasis, more efforts have to be made to transfer these findings into the clinic. Here, we will describe the different approaches found from miRNA analysis that might be applied for EMT-related miRNAs into the clinics.
miRNAs as predictive markers
The miRNAs can be found in biological fluids, including: blood; urine; tears; breast milk; bronchial lavage; colostrum; seminal; amniotic; pleural; peritoneal; and cerebrospinal fluids. Moreover, endogenous serum miRNAs are very stable, even after boiling, freeze–thaw cycles, or low or high pH conditions, thus providing an easy substrate for detection and analysis. The quantitative polymerase chain reaction detection is the most feasible and cost-effective method to detect miRNA expression; however, next generation sequencing might be used as an alternative method, overtaking the use of normalizing miRNA expression levels.
Expression levels of circulating miRNA in plasma can be used to discriminate patients with different outcomes. Accordingly, serum levels of miR-141 can distinguish between healthy subjects and patients with prostate cancer.101 The sensitivity of the approach can be highly improved using more than a single miRNA.102 In colorectal cancer patients, elevated mir-92a and mir-29a levels in plasma are useful biomarkers of the disease.103 Importantly, the quantitative polymerase chain reaction analyses in the blood of miR-10b, miR-34, and miR-155 – some of them involved in EMT regulation and/or metastasis – allows the discrimination between breast cancer metastasis patients from the healthy controls,104 and miRNA expression profiles can be a classifier of responder versus nonresponder patients for chemotherapy in colon cancers.105
Many miRNAs have been correlated with tumor patient outcome.3,4,24 Tumors with low expression levels of miR-335 and miR-126 have more probability to develop metastasis than tumors with higher expression.79 The miR-210, a hypoxia-induced miRNA, is an independent prognostic marker in breast cancer and shows inverse correlation with disease-free and overall survival outcomes.106 The well-studied oncomiR miR-21 has been considered a poor prognosis biomarker in breast, squamous cell carcinoma, astrocytoma, and gastric cancer.107 Related to epithelial plasticity, DNA methylation of the miR-200c-141 locus might be a useful surrogate marker of endometrial carcinosarcomas.39 The expression of miR-200f and CpG methylation status of the miR-200c-141 locus is able to discriminate breast cancer subtypes.37 Indeed, metaplastic breast tumors with sarcomatoid differentiation present the higher downregulation of miR-200f and hypermethylation of the miR-200c-141 locus compared with other cancer types. Moreover, basal-like and HER2 positive breast tumors exhibited lower miR-200f expression than estrogen receptor positive tumors.37
miRNAs: a promising therapeutic tool
The miRNAs could potentially be used as therapeutic tools in cancer. Depending on the protumoral or antitumoral properties, different strategies based on blocking miRNA function or specific miRNA delivery to the tumor cells can be used. At present, different preclinical approaches directed to block miRNAs are being developed, which include anti-miRNA oligonucleotides, miRNA sponges, miRNA masks (target protectors), and small molecule inhibitors.108 The miRNA sponges are synthetic miRNAs that contain multiple tandem-binding sites of the endogenous miRNA. They harbor the seed sequence, allowing the inhibition of the complete family of related miRNAs. The miRNA masks are single-stranded 2-O-methyl modified antisense oligonucleotides that lose their repression activity, but they maintain full complementarity to the predicted miRNA-binding sites. Treatment of melanoma cells with locked nucleic acids targeting miR-1908, miR-199a-5p, and miR-199a-3p inhibits metastasis to multiple organs, and the therapeutic delivery of these locked nucleic acids suppresses melanoma metastasis.88 Similar studies have been developed by inhibiting miR-182 to treat metastasis in melanoma mouse models.109
However, the delivery of tumor suppressor miRNAs is a potential therapeutic application, in particular to promote the differentiation of cancer stem cells to increase their sensitivity to traditional therapies. Nevertheless, the delivery of miRNA to solid tumors is technically a challenging issue. Different strategies to deliver miRNAs to tumor cells are being developed based on synthetic miRNA precursors, lentiviral and adenovirus vectors, or nanoparticles.110 For example, miR-26 adenovirus delivery arrests cell cycle progression and induces tumor specific apoptosis in liver cells.12 However, actual strategies are limited by high toxicity and the lack of cell specificity of the delivery.
The miRNAs might also be useful as coadjuvant therapies in cancer treatment. For example, the miR-21 inhibition sensitizes breast cancer cells to the chemotherapeutic drugs, Topotecan and Taxol.111 The expression of miR-205 in SkBR3 breast cancer cells targets HER3, leading to an increased response to the tyrosine kinase inhibitors gefitinib and lapatinib.112 Although these data were obtained in vitro and need to be reevaluated in vivo, miRNA targeting might represent a promising coadjuvant strategy for metastatic cancer patients.
Conclusion
To acquire a migratory/invasive phenotype for metastatic progression, tumor cells activate the developmental EMT program. Remarkably, the activation of an EMT program must be temporarily limited and coupled to the reversible program, MET, for macrometastasis formation. Because epithelial plasticity must be finely regulated along the metastatic cascade, miRNAs have emerged as critical regulators necessary for metastatic progression. Indeed, these small RNA sequences use different mechanisms to regulate epithelial plasticity, such as targeting different signaling pathways, EMT-TFs, and/or their targets, or the proper miRNA maturation machinery. The presence of double negative feedback loops defines flexible and efficient control of EMT and MET processes providing self-reinforcing signals to maintain the epithelial or the mesenchymal status of the cell. The redundancy observed in the targeting of different EMT related genes would contribute to the stability of the regulation during physiological or pathological events. Consequently, modifying a single miRNA might activate or inactivate different pathways required for tumor cells to survive and/or invade during the metastatic cascade. Understanding the different strategies employed by tumor cells to switch on/off EMT will challenge oncologists’ efforts for the next years, providing new avenues for early diagnosis, prognosis, and the treatment of human tumors.
Acknowledgments
AC is funded by grants of the Spanish Ministry of Economy and Competitiveness, formerly Innovation and Sciences, (SAF2010-21143; Consolider-Ingenio CSD2007-00017) and Association for International Cancer Research (grant 12-1057). GMB is funded by SAF2010-20175, AECC-2011 and Instituto de Salud Carlos III (ISCIII) PI13/00132. GMB and AC are funded by the Community of Madrid (S2010/BMD-2303) and the Instituto de Salud Carlos III (RETIC-RD12/0036/0007). ADL is a postdoctoral researcher funded by the Sara Borrell program (ISCIII).
Disclosure
The authors report no conflicts of interest in this work.
References
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. | |
Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011; 39(Database issue):D152–D157. | |
Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13(4):271–282. | |
Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–269. | |
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. | |
Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101(9):2999–3004. | |
Sayed D, Abdellatif M. MicroRNAs in development and disease. Physiol Rev. 2011;91(3):827–887. | |
Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002;99(24):15524–15529. | |
Fabbri M, Garzon R, Cimmino A, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A. 2007;104(40):15805–15810. | |
Gebeshuber CA, Zatloukal K, Martinez J. miR-29a suppresses tristetraprolin, which is a regulator of epithelial polarity and metastasis. EMBO Rep. 2009;10(4):400–405. | |
Huse JT, Brennan C, Hambardzumyan D, et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009;23(11):1327–1337. | |
Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137(6):1005–1017. | |
Liu B, Wu X, Liu B, et al. MiR-26a enhances metastasis potential of lung cancer cells via AKT pathway by targeting PTEN. Biochim Biophys Acta. 2012;1822(11):1692–1704. | |
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. | |
Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6159):1234850. | |
Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–428. | |
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | |
Nieto MA, Cano A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Semin Cancer Biol. 2012;22(5–6):361–368. | |
Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438–1449. | |
Scheel C, Onder T, Karnoub A, Weinberg RA. Adaptation versus selection: the origins of metastatic behavior. Cancer Res. 2007;67(24):11476–11479. | |
Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. | |
Ocaña OH, Córcoles R, Fabra A, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22(6):709–724. | |
Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–736. | |
Pencheva N, Tavazoie SF. Control of metastatic progression by microRNA regulatory networks. Nat Cell Biol. 2013;15(6):546–554. | |
Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. | |
Ma L, Young J, Prabhala H, et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat Cell Biol. 2010;12(3):247–256. | |
Liu S, Kumar SM, Lu H, et al. MicroRNA-9 up-regulates E-cadherin through inhibition of NF-kappaB1-Snail1 pathway in melanoma. J Pathol. 2012;226(1):61–72. | |
Zhuang G, Wu X, Jiang Z, et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012;31(17):3513–3523. | |
Brabletz S, Brabletz T. The ZEB/miR-200 feedback loop – a motor of cellular plasticity in development and cancer? EMBO Rep. 2010;11(9):670–677. | |
Hill L, Browne G, Tulchinsky E. ZEB/miR-200 feedback loop: at the crossroads of signal transduction in cancer. Int J Cancer. 2013;132(4):745–754. | |
Brabletz T. MiR-34 and SNAIL: another double-negative feedback loop controlling cellular plasticity/EMT governed by p53. Cell Cycle. 2012;11(2):215–216. | |
Comijn J, Berx G, Vermassen P, et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7(6):1267–1278. | |
Eger A, Aigner K, Sonderegger S, et al. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24(14):2375–2385. | |
Sánchez-Tilló E, Liu Y, de Barrios O, et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69(20):3429–3456. | |
Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28(1–2):151–166. | |
Siebzehnrubl FA, Silver DJ, Tugertimur B, et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5(8):1196–1212. | |
Castilla MÁ, Díaz-Martín J, Sarrió D, et al. MicroRNA-200 family modulation in distinct breast cancer phenotypes. PLoS One. 2012;7(10):e47709. | |
Castilla MÁ, Moreno-Bueno G, Romero-Pérez L, et al. Micro-RNA signature of the epithelial-mesenchymal transition in endometrial carcinosarcoma. J Pathol. 2011;223(1):72–80. | |
Díaz-Martín J, Díaz-López A, Moreno-Bueno G, et al. A core microRNA signature associated with inducers of the epithelial-to-mesenchymal transition. J Pathol. 2014;232(3):319–329. | |
Spaderna S, Schmalhofer O, Wahlbuhl M, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008;68(2):537–544. | |
Cano A, Nieto MA. Non-coding RNAs take centre stage in epithelial-to-mesenchymal transition. Trends Cell Biol. 2008;18(8):357–359. | |
Christoffersen NR, Silahtaroglu A, Orom UA, Kauppinen S, Lund AH. miR-200b mediates post-transcriptional repression of ZFHX1B. RNA. 2007;13(8):1172–1178. | |
Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907. | |
Gregory PA, Bert AG, Paterson EL, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601. | |
Burk U, Schubert J, Wellner U, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9(6):582–589. | |
Bracken CP, Gregory PA, Kolesnikoff N, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68(19):7846–7854. | |
Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–1495. | |
Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. | |
Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008;3(8):e2888. | |
Sarrió D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68(4):989–997. | |
Gregory PA, Bracken CP, Smith E, et al. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22(10):1686–1698. | |
Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell. 2010;39(5):761–772. | |
Korpal M, Ell BJ, Buffa FM, et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat Med. 2011;17(9):1101–1108. | |
Saydam O, Shen Y, Würdinger T, et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/beta-catenin signaling pathway. Mol Cell Biol. 2009;29(21):5923–5940. | |
Li XL, Hara T, Choi Y, et al. A p21-ZEB1 Complex Inhibits Epithelial-Mesenchymal Transition through the MicroRNA 183-96-182 Cluster. Mol Cell Biol. 2014;34(3):533–550. | |
Kato M, Zhang J, Wang M, et al. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc Natl Acad Sci U S A. 2007;104(9):3432–3437. | |
Liu X, Wang C, Chen Z, et al. MicroRNA-138 suppresses epithelial-mesenchymal transition in squamous cell carcinoma cell lines. Biochem J. 2011;440(1):23–31. | |
Dong P, Karaayvaz M, Jia N, et al. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene. 2013;32(27):3286–3295. | |
Stinson S, Lackner MR, Adai AT, et al. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci Signal. 2011;4(177):ra41. | |
Siemens H, Jackstadt R, Hünten S, et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10(24):4256–4271. | |
Moes M, Le Béchec A, Crespo I, et al. A novel network integrating a miRNA-203/SNAI1 feedback loop which regulates epithelial to mesenchymal transition. PLoS One. 2012;7(4):e35440. | |
Ru P, Steele R, Newhall P, Phillips NJ, Toth K, Ray RB. miRNA-29b suppresses prostate cancer metastasis by regulating epithelial-mesenchymal transition signaling. Mol Cancer Ther. 2012;11(5):1166–1173. | |
Zhang J, Zhang H, Liu J, et al. miR-30 inhibits TGF-β1-induced epithelial-to-mesenchymal transition in hepatocyte by targeting Snail1. Biochem Biophys Res Commun. 2012;417(3):1100–1105. | |
Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17(1):19–36. | |
Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM. Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev. 2006;20(16):2202–2207. | |
Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174(2):175–183. | |
Watanabe S, Ueda Y, Akaboshi S, Hino Y, Sekita Y, Nakao M. HMGA2 maintains oncogenic RAS-induced epithelial-mesenchymal transition in human pancreatic cancer cells. Am J Pathol. 2009;174(3):854–868. | |
Qi J, Rice SJ, Salzberg AC, et al. MiR-365 regulates lung cancer and developmental gene thyroid transcription factor 1. Cell Cycle. 2012;11(1):177–186. | |
Yu F, Yao H, Zhu P, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131(6):1109–1123. | |
Yun J, Frankenberger CA, Kuo WL, et al. Signalling pathway for RKIP and Let-7 regulates and predicts metastatic breast cancer. EMBO J. 2011;30(21):4500–4514. | |
Liu YN, Yin JJ, Abou-Kheir W, et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene. 2013;32(3):296–306. | |
Wang FE, Zhang C, Maminishkis A, et al. MicroRNA-204/211 alters epithelial physiology. FASEB J. 2010;24(5):1552–1571. | |
Zhang Z, Zhang B, Li W, et al. Epigenetic Silencing of miR-203 Upregulates SNAI2 and Contributes to the Invasiveness of Malignant Breast Cancer Cells. Genes Cancer. 2011;2(8):782–791. | |
Liu YN, Abou-Kheir W, Yin JJ, et al. Critical and reciprocal regulation of KLF4 and SLUG in transforming growth factor β-initiated prostate cancer epithelial-mesenchymal transition. Mol Cell Biol. 2012;32(5):941–953. | |
Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, Yang J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011;71(1):245–254. | |
Cubillo E, Diaz-Lopez A, Cuevas EP, et al. E47 and Id1 interplay in epithelial-mesenchymal transition. PLoS One. 2013;8(3):e59948. | |
Dave N, Guaita-Esteruelas S, Gutarra S, et al. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J Biol Chem. 2011;286(14):12024–12032. | |
Sobrado VR, Moreno-Bueno G, Cubillo E, et al. The class I bHLH factors E2-2A and E2-2B regulate EMT. J Cell Sci. 2009;122(Pt 7):1014–1024. | |
Tavazoie SF, Alarcón C, Oskarsson T, et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature. 2008;451(7175):147–152. | |
Png KJ, Halberg N, Yoshida M, Tavazoie SF. A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature. 2012;481(7380):190–194. | |
Valastyan S, Chang A, Benaich N, Reinhardt F, Weinberg RA. Concurrent suppression of integrin alpha5, radixin, and RhoA phenocopies the effects of miR-31 on metastasis. Cancer Res. 2010;70(12):5147–5154. | |
Valastyan S, Weinberg RA. miR-31: a crucial overseer of tumor metastasis and other emerging roles. Cell Cycle. 2010;9(11):2124–2129. | |
Ma L. Role of miR-10b in breast cancer metastasis. Breast Cancer Res. 2010;12(5):210. | |
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–688. | |
Dong P, Kaneuchi M, Watari H, Sudo S, Sakuragi N. MicroRNA-106b modulates epithelial-mesenchymal transition by targeting TWIST1 in invasive endometrial cancer cell lines. Mol Carcinog. Epub September 3, 2013. | |
Huang Q, Gumireddy K, Schrier M, et al. The microRNAs miR-373 and miR-520c promote tumour invasion and metastasis. Nat Cell Biol. 2008;10(2):202–210. | |
Yang K, Handorean AM, Iczkowski KA. MicroRNAs 373 and 520c are downregulated in prostate cancer, suppress CD44 translation and enhance invasion of prostate cancer cells in vitro. Int J Clin Exp Pathol. 2009;2(4):361–369. | |
Pencheva N, Tran H, Buss C, et al. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell. 2012;151(5):1068–1082. | |
Gaziel-Sovran A, Segura MF, Di Micco R, et al. miR-30b/30d regulation of GalNAc transferases enhances invasion and immunosuppression during metastasis. Cancer Cell. 2011;20(1):104–118. | |
Kim NH, Kim HS, Li XY, et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011;195(3):417–433. | |
Jackstadt R, Röh S, Neumann J, et al. AP4 is a mediator of epithelial-mesenchymal transition and metastasis in colorectal cancer. J Exp Med. 2013;210(7):1331–1350. | |
Shi L, Jackstadt R, Siemens H, Li H, Kirchner T, Hermeking H. p53-Induced miR-15a/16-1 and AP4 Form a Double-Negative Feedback Loop to Regulate Epithelial-Mesenchymal Transition and Metastasis in Colorectal Cancer. Cancer Res. 2014;74(2):532–542. | |
Ren D, Wang M, Guo W, et al. Wild-type p53 suppresses the epithelial-mesenchymal transition and stemness in PC-3 prostate cancer cells by modulating miR145. Int J Oncol. 2013;42(4):1473–1481. | |
Vogt M, Munding J, Grüner M, et al. Frequent concomitant inactivation of miR-34a and miR-34b/c by CpG methylation in colorectal, pancreatic, mammary, ovarian, urothelial, and renal cell carcinomas and soft tissue sarcomas. Virchows Arch. 2011;458(3):313–322. | |
Davalos V, Moutinho C, Villanueva A, et al. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene. 2012;31(16):2062–2074. | |
Wee EJ, Peters K, Nair SS, et al. Mapping the regulatory sequences controlling 93 breast cancer-associated miRNA genes leads to the identification of two functional promoters of the Hsa-mir-200b cluster, methylation of which is associated with metastasis or hormone receptor status in advanced breast cancer. Oncogene. 2012;31(38):4182–4195. | |
Martello G, Rosato A, Ferrari F, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141(7):1195–1207. | |
Karube Y, Tanaka H, Osada H, et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005;96(2):111–115. | |
Pampalakis G, Diamandis EP, Katsaros D, Sotiropoulou G. Down-regulation of dicer expression in ovarian cancer tissues. Clin Biochem. 2010;43(3):324–327. | |
Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature. 2009;460(7254):529–533. | |
Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–10518. | |
Ng EK, Wong CL, Ma ES, Kwong A. MicroRNAs as New Players for Diagnosis, Prognosis, and Therapeutic Targets in Breast Cancer. J Oncol. 2009;2009:305420. | |
Huang Z, Huang D, Ni S, Peng Z, Sheng W, Du X. Plasma microRNAs are promising novel biomarkers for early detection of colorectal cancer. Int J Cancer. 2010;127(1):118–126. | |
Roth C, Rack B, Müller V, Janni W, Pantel K, Schwarzenbach H. Circulating microRNAs as blood-based markers for patients with primary and metastatic breast cancer. Breast Cancer Res. 2010; 12(6):R90. | |
Schetter AJ, Leung SY, Sohn JJ, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299(4):425–436. | |
Camps C, Buffa FM, Colella S, et al. hsa-miR-210 Is induced by hypoxia and is an independent prognostic factor in breast cancer. Clin Cancer Res. 2008;14(5):1340–1348. | |
Fu X, Han Y, Wu Y, et al. Prognostic role of microRNA-21 in various carcinomas: a systematic review and meta-analysis. Eur J Clin Invest. 2011;41(11):1245–1253. | |
Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov. 2010;9(10):775–789. | |
Huynh C, Segura MF, Gaziel-Sovran A, et al. Efficient in vivo microRNA targeting of liver metastasis. Oncogene. 2011;30(12):1481–1488. | |
Kong YW, Ferland-McCollough D, Jackson TJ, Bushell M. microRNAs in cancer management. Lancet Oncol. 2012;13(6):e249–e258. | |
Mei M, Ren Y, Zhou X, et al. Downregulation of miR-21 enhances chemotherapeutic effect of taxol in breast carcinoma cells. Technol Cancer Res Treat. 2010;9(1):77–86. | |
Iorio MV, Casalini P, Piovan C, et al. microRNA-205 regulates HER3 in human breast cancer. Cancer Res. 2009;69(6):2195–2200. | |
Chen D, Sun Y, Wei Y, et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med. 2012;18(10):1511–1517. | |
Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol. 2013;15(2):201–213. | |
Wu Q, Lu Z, Li H, Lu J, Guo L, Ge Q. Next-generation sequencing of microRNAs for breast cancer detection. J Biomed Biotechnol. 2011;2011:597145. | |
Kumarswamy R, Mudduluru G, Ceppi P, et al. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer. 2012;130(9):2044–2053. | |
Liu C, Kelnar K, Liu B, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17(2):211–215. | |
Cheng H, Zhang L, Cogdell DE, et al. Circulating plasma MiR-141 is a novel biomarker for metastatic colon cancer and predicts poor prognosis. PLoS One. 2011;6(3):e17745. | |
Zhang L, Huang J, Yang N, et al. microRNAs exhibit high frequency genomic alterations in human cancer. Proc Natl Acad Sci U S A. 2006;103(24):9136–9141. | |
Kim T, Veronese A, Pichiorri F, et al. p53 regulates epithelial- mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208(5):875–883. | |
Saini S, Majid S, Yamamura S, et al. Regulatory Role of mir-203 in Prostate Cancer Progression and Metastasis. Clin Cancer Res. 2011;17(16):5287–5298. | |
Matsushima K, Isomoto H, Yamaguchi N, et al. MiRNA-205 modulates cellular invasion and migration via regulating zinc finger E-box binding homeobox 2 expression in esophageal squamous cell carcinoma cells. J Transl Med. 2011;9:30. | |
Romero-Pérez L, Castilla MÁ, López-García MÁ, et al. Molecular events in endometrial carcinosarcomas and the role of high mobility group AT-hook 2 in endometrial carcinogenesis. Hum Pathol. 2013;44(2):244–254. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.