")
Back to Journals » Journal of Inflammation Research » Volume 15
Role of JAK2/STAT3 Signaling Pathway in the Tumorigenesis, Chemotherapy Resistance, and Treatment of Solid Tumors: A Systemic Review
Authors Mengie Ayele T , Tilahun Muche Z , Behaile Teklemariam A , Bogale Kassie A , Chekol Abebe E
Received 24 December 2021
Accepted for publication 11 February 2022
Published 25 February 2022 Volume 2022:15 Pages 1349—1364
DOI https://doi.org/10.2147/JIR.S353489
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Teklie Mengie Ayele,1 Zelalem Tilahun Muche,2 Awgichew Behaile Teklemariam,3 Achenef Bogale Kassie,1 Endeshaw Chekol Abebe3
1Department of Pharmacy, Debre Tabor University, Debre Tabor, Amhara, Ethiopia; 2Department of Physiology, Debre Tabor University, Debre Tabor, Amhara, Ethiopia; 3Department of Medical Biochemistry, Debre Tabor University, Debre Tabor, Amhara, Ethiopia
Correspondence: Endeshaw Chekol Abebe, Tel +251928428133, Email [email protected]
Abstract: Janus kinase 2/signal transducer and activator of transcription 3 (JAK2/STAT3) pathway is a common signaling pathway used to transduce signals from the extracellular to the intracellular (nucleus) upon the binding of cytokines and growth factors to the extracellular domain of specific cell surface receptors. This signaling pathway is tightly regulated and has a multitude of biological functions such as cell proliferation, differentiation, and apoptosis. Besides, the regulated JAK2/STAT3 signaling plays a crucial role in embryonic development, hemopoiesis, and controlling the immune system. Conversely, aberrantly activated JAK2/STAT3 is frequently detected in varieties of tumors and involved in oncogenesis, angiogenesis, and metastasis of many cancer diseases that are usually refractory to the standard chemotherapy. However, the JAK3/STAT3 pathway recently emerged interestingly as a new site for the development of novel anti-tumor agents and becomes a promising therapeutic target in the treatment of many solid malignancies. Herein, this review aimed to provide insight into the JAK2/STAT3 pathway, in the hope to gain an understanding of its potential role in the pathogenesis, progression, chemotherapy resistance, and cancer therapy of solid tumors.
Keywords: solid tumors, JAK2/STAT3 signaling, tumorigenesis, JAK2/STAT3 targeted therapy
Introduction
Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway is an essential intracellular signaling network that was discovered nearly three decades ago. Many cytokines and growth factors use this signaling pathway to transduce signals from the cell membrane to nucleus, where they activate genes through transcription in animals, from humans to fruit flies.1
The receptor tyrosine kinase (RTK), JAK and STAT proteins are the key components of the JAK/STAT pathway.2–4 The RTKs are tyrosine kinases (TYK) linked membrane receptors that belong to the cytokine receptor superfamily and bind with many cytokines and growth factor signals in the JAK/STAT signal pathway. Each RTK has an extracellular domain to bind with ligands, a transmembrane domain, and an intracellular domain for downward cascading. The RTK does not have kinase activity by itself but has a binding site for JAK in its intracellular domain.4
The Janus kinase (JAK) protein, originally known as “Just another kinase”, is named after Janus, the Roman god of gates and doors. It is a family of non-membrane receptor TYK possessing catalytic domains that were discovered in 1992. On the other hand, the STAT proteins play a crucial role in signal transduction and activation of gene expression (transcription). The first STAT protein (STAT1) was identified 30 years back in 1988, while the other protein members were purified in the later years.2 Besides these key components, other effector proteins have been identified to contribute to at least a subset of JAK/STAT signaling events such as signal-transducing adapter molecules (STAMs), which are adapter molecules with retained VHS and SH3 domains and interferon (IFN) regulatory factor 9 (IRF-9) in the case of type I IFN activated JAK/STAT signaling.5
The JAK/STAT pathway, particularly JAK2/STAT3, is involved in different biological processes such as immunity, cell division, cell death, and tumor formation.2,6 This review is primarily aimed to provide insight into JAK2/STAT3 signaling, in the hope to gain an understanding of its potential role in the pathogenesis, progression, chemotherapy resistance, and cancer therapy of solid tumors.
The Structure of JAK and STAT Proteins
The JAK protein family consists of JAK1, JAK2, JAK3, and TYK2, which share structural and functional homologies. The JAK proteins have seven JAK homology (JH) domains, containing two adjacent kinase domains at the carboxyl (C)-terminus, named JH1 domain that is involved in phosphorylation of STATs and receptors, and the pseudokinase (JH2) domain that lacks catalytic activity but regulates JH1.7 Besides, JAKs also contain an Src homology 2 (SH2)-like domain (JH3-4) that interacts with phosphorylated tyrosine residues, 4.1 protein, ezrin, radixin, and moesin (FERM) domain (JH5-7) at the amino (N)-terminus that is responsible for binding with receptors and regulating catalytic activity8 (Figure 1A).
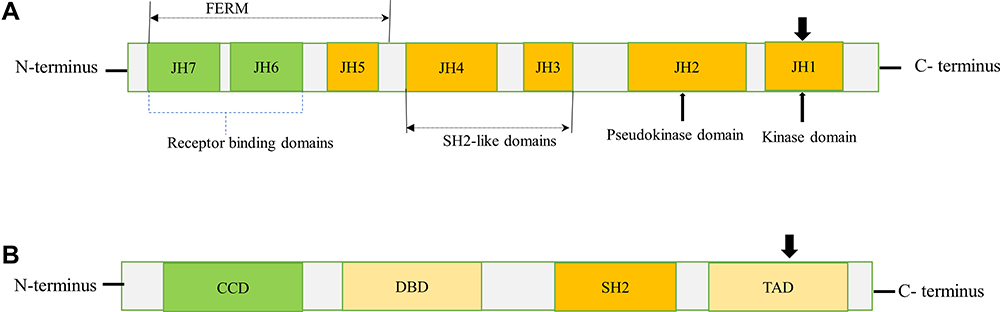 |
Figure 1 Schematic diagram of JAK and STAT structures. (A) JAKs contain a FERM domain (400 amino acid residues) that associates with receptors, a SH2 domain (100 amino acid residues) that binds phosphorylated tyrosine residues, and a kinase (JH1) domain (250 amino acid residues) and pseudo kinase (JH2) domain (300 amino acid residues). The arrowhead pointed downward (JH1) indicates phosphorylation sites (tyrosine residues) needed for JAK activation. (B) STATs contain a CCD for dimerization, a DBD, a SH2 domain, and a TAD for transcriptional activation of target genes. The arrowhead pointed downward (TAD) indicates the conserved tyrosine residue required to be phosphorylated for STAT activation. N and C represents the amino- and carboxy-terminal ends respectively. Abbreviations: CCD, coiled-coil domain; DBD, DNA-binding domain; FERM, four-point-one, ezrin, radixin, moesin; TAD, transactivation domain. |
The STAT protein family comprises seven members, namely STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. They range in size from 750 to 850 amino acids and share several functional domains, including coiled-coil domain (CCD), DNA-binding domain (DBD), SH2 domain, and transcriptional activation domain (TAD) (Figure 1B). The CCD is the functional domain present at the amino end that enables dimerization of activated STATs and interacts with other proteins.8 A central DBD is involved in DNA binding and nuclear translocation of STAT dimers. These domains are followed by a linker domain called SH2 domain containing two α-helices and a β-pleated sheet structure that consist of 575–680 amino acid residues. The SH2 domain recognizes phosphorylated tyrosine residues on the receptors and activates STATs. STATs also have the carboxyl-terminal TAD that serves as an interaction domain for coactivators and co-repressors necessary for regulation of transcription.3,9
The JAK2/STAT3 Signaling Pathway in Normal Cells
Activation of JAK2/STAT3 Pathway
Normally, the JAK2/STAT3 pathway is activated upon the binding of hormones like prolactin, growth factors such as epidermal growth factor (EGF), and cytokines mainly interleukin-6 (IL-6) family to the extracellular domain of specific cellular receptors (RTK).1 In particular, the JAK2 mediates signaling via multiple cytokine receptors, such as the IL-6, erythropoietin, leptin, and IFN-γ receptors.1 This interaction leads to dimerization/multimerization of the receptor subunits, which brings JAK2 that are non-covalently associated with the intracellular domains of the receptors in close proximity of each other. This brings phosphorylation of JAK2 to each other on their tyrosine residues (known as autophosphorylation) and activates their kinase domain as shown in Figure 2. Tyrosine 221 and 570 in murine JAK2 have been found to be the sites of autophosphorylation from in vitro assay. Tyrosine 570 in JAK2 is conserved in humans, rats, mice, pigs, and fish. However, the corresponding tyrosine at 221 and 570 in JAK2 are not found in JAK1, JAK3, or TYK2, and hence phosphorylation of tyrosine 221 and 570 in JAK2 may initiate functions unique to JAK2.10 Once the JAK2 proteins are phosphorylated, then their activated kinase domain, in turn, enables phosphorylation of the tyrosine residues on the receptor’s intracellular domains to create docking sites for SH2 domain possessing proteins. This allows the binding of cytoplasmic STAT3 proteins to the phosphorylated tyrosine residues on the receptor using their SH2 domains. Then STAT3 undergoes phosphorylation at Tyr705 by JAKs, causing the STATs dimerization, dissociation from the receptor, and then translocation to the nucleus where they bind to specific DNA sequences and induce transcription of their target genes.1,2 It has been documented that the JAK2/STAT3 signaling can interact with other pathways such as MAPK/ERK and PI3K/AKT/mTOR signaling pathway to integrate certain cellular activities.8
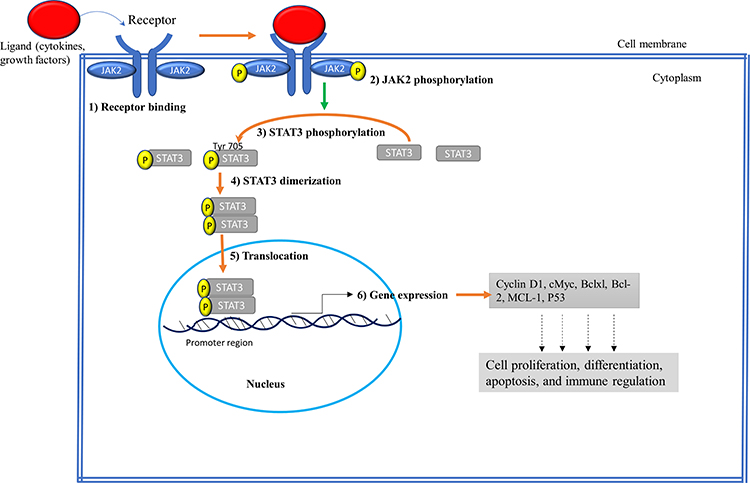 |
Figure 2 Schematic representation of the JAK2/STAT3 signaling pathway. (1) Receptor binding: Ligands such as cytokines and growth factors bind to extracellular domains of the receptors. (2) JAK2 phosphorylation: Following ligand binding receptor-associated JAK2 undergo phosphorylation to each other. (3) STAT3 phosphorylation: Activated JAK2 kinase domain (JH1) phosphorylates cytoplasmic tails of receptors, recruiting STAT3 to the receptor, and becomes phosphorylated at Tyr 705. (4) Dimerization: Phosphorylated STAT3 dimerizes at SH2 domain. (5) Translocation: Dimerized STAT3 translocates into the nucleus where it binds to the promoter region of DNA. (6) Gene Expression: Dimerized STAT3 acts as transcription factor and activates the transcription of target genes involved in regulating cell growth. |
Regulation of JAK2/STAT3 Pathway
While tyrosine kinases play a crucial role in activating JAK2 and STAT3, numerous regulatory mechanisms are engaged in tightly regulating JAK2/STAT3 signaling. This allows fine-tuning of cellular effects ensuring that it functions optimally under normal physiological conditions, and prevents the inappropriate activity linked with disease (eg, tumor) development.
Primarily, three main regulatory mechanisms have evolved to control the magnitude and duration of signaling. Firstly, by removing phosphate group using the enzyme tyrosine phosphatases, including classical protein tyrosine phosphatases, dual-specificity phosphatases, and low-molecular-weight phosphatases to terminate JAK2/STAT3 signaling and action through dephosphorylation/deactivation of JAK2 and/or STAT3.11 Secondly, by adding a small ubiquitin-like modifier by the protein inhibitor of activated STAT (PIAS) onto proteins such as JAKs and STATs, which may block their phosphorylation and modify their function.12 Thirdly, by forming a protein complex through the interaction between suppressors of cytokine signaling (SOCS) with different proteins, which can then cause the breakdown of JAK2 and the receptors themselves, therefore inhibiting JAK2/STAT3 signaling.13
Additionally, the JAK2 activity can also be tightly controlled through the self-inhibitory effect of its JH2 domain, which restricts the strength and duration of JAK2/STAT3 signaling under normal conditions.14 Similarly, JAK2/STAT3 signaling is tightly regulated at the STAT level in normal healthy conditions to be involved in myriads of functions.2
Role of JAK2/STAT3 Signaling in Normal Cells
JAK2/STAT3 signaling is a universal intracellular pathway that has significant functions in many biological processes such as cell proliferation, differentiation, and apoptosis, and immune regulation. The JAK2/STAT3 signaling has also been reported to play prominent roles in embryonic development, for instance in the maintenance of the self-renewal capacity of embryonic stem cells and FMS-like tyrosine kinase 3 (Flt3) ligand-dependent dendritic cell (DC) differentiation, as well as in the control of processes such as hematopoiesis, inflammatory response, and immune system.15
Available evidence has documented the involvement of JAK2 in the progression of apoptosis, autophagy, and proliferation of normal cells.2 It is also responsible for receptor signaling from IL-3 receptor or granulocyte-macrophage colony-stimulating factor, both used in hematopoietic cell development.16 On the other hand, STAT3 is thought to be an oncogenic transcription factor widely expressed in many tissues and plays a key role in controlling cellular activities, such as angiogenesis via the transcription of vascular endothelial growth factor (VEGF).17 Although STAT3 is a well-known transcriptional activator for many genes such as the VEGF gene, it has also recently been documented to inhibit gene expression via tumor-suppressor gene promoter methylation. The STAT3 acetylation thus influences tumor growth, DNA methylation, and silencing of the tumor-suppressor gene.18 Altogether, the JAK2/STAT3 pathway regulates the expression of genes related to cell survival, proliferation, angiogenesis, and immune evasion.
Potential Roles of JAK2/STAT3 Pathway in Solid Tumors
In normal conditions, the activation of JAK2/STAT3 is tightly regulated and transient. Nonetheless, the persistent activation of JAK2/STAT3 signaling is found in many human solid tumors and plays essential biological roles in the development of malignancies.19
Human cancer is characterized by an abnormal and unregulated cell growth in the body. Thus, since the JAK2/STAT3 pathway can allow the expression of genes involved in cell division, over-regulation of JAK2/STAT3 signaling potentially results in cancer formation.7 Recently, ample evidence indicated that the overly activated JAK2/STAT3 pathway promotes tumorigenesis, tumor growth, cancer cell survival, and metastasis of solid tumors.19 Hence, these findings suggest that targeting the abnormal JAK2/STAT3 pathway in carcinoma may hold great potential as a novel therapeutic target for cancer therapy. Currently, drugs targeting the JAK2/STAT3 cascade are developed for solid tumors. This section of the review discusses the role of the JAK2/STAT3 pathway in the pathogenesis and progression of human solid malignancies as well as in chemotherapy resistance and cancer therapy.
Roles of JAK2/STAT3 Pathway in Tumorigenesis
Evidence suggests that constitutive activation of the JAK2/STAT3 pathway is frequently observed in cancer and plays major functions in many solid tumors. Persistent activation and defects of JAK2/STAT3 have been linked with survival, proliferation, angiogenesis, host immune evasion, resistance to apoptosis, carcinogenesis, and metastasis in many human cancer cells.18,20–22 This oncogenic signaling is primarily attributed to the disruption of the tightly regulated JAK2/STAT3 signaling in neoplasia as well as being due to the secretion of ligands by cancer cells and/or by cells from the tumor microenvironment, which allows the JAK2/STAT3 pathway to be activated constitutively.23
Mutations of JAK2 and/or STAT3 were found to be associated with sustained activation of the JAK2/STAT3 pathway. All JAK2 domains have shown mutations that have considerable effects on JAK expression, mostly on their kinase activity, receptor binding, and intracellular transport.19 Multiple mutations like deletion of JAK2 and gain-of-function mutations within JAK2 in mice were shown to be lethal as they abrogate the function of JH2 and sustain JAK2 activation. However, how cancer cells circumvent the autoinhibitory effect of the JH2 domain to sustain constitutive activation of JAK2/STAT3 signaling remains puzzling.24,25 Evidence showed that the JAK2 mutations are widely observed in hematological malignancies such as leukemia, polycythemia vera, essential thrombocythemia, and myeloproliferative diseases, but comparable mutations have not been detected in solid tumors.18
The potential oncogenic role of STAT3 has been established by the expression of constitutively activated STAT3 in many solid tumor cell lines including breast, colon, gastric, lung, head and neck, skin, and prostate cancers.26 Persistently activated STAT3 promotes the survival, proliferation, motility, and progression of cancer cells, elicits angiogenesis, and suppresses the immune response to tumors. The STAT3 activation in immune cells promotes tumor immune evasion and oncogenesis, which is in part mediated by tumor-derived factors, such as hypoxia-inducible factor 1 (HIF-1), VEGF, IL-10, and IL-6. A strong unregulated STAT3 isoform activity involving JAK/STAT signaling inhibition has been documented in tumorigenesis from in vivo and in vitro models.27 Conversely, STAT3 ablation in immune cells leads to induction of Th1 mediators involved in both innate and T-cell-mediated adaptive immunity. In turn, this causes the increased anti-tumor activity of immune cells that impedes tumor progression.28 It has been demonstrated that IL-6 signaling via STAT3 promotes the maintenance of Th17 in inflamed tissue and plays a regulatory role in acute inflammation in various immune cells.29 The release of proinflammatory mediators, including cytokines and nitric oxide, is dramatically enhanced in STAT3-deficient macrophages and neutrophils on lipopolysaccharide stimulation.30
The aberrant activation of the JAK2/STAT3 pathway could be ascribed to the downregulation of a tumor suppressor gene, called p53-inducible cancer-associated RNA transcript 1 (PICART1), that affects cell apoptosis and migration. Thus, the knockdown of PICART1 in animal model promotes tumor formation, which is verified from the anti-osteosarcoma effect of pterostilbene that produces its effect through downregulation of JAK2/STAT3.31,32 JAK2/STAT3 has also been involved in controlling the expression of genes linked with metastasis. Thus, anomalous JAK2/STAT3 enhances the metastatic ability of solid tumors, such as lung cancer by IL-6/STAT3 signaling.33 Several in vitro and in vivo experiments demonstrated that autophagy preserves the metastatic potential of cancer cells via JAK2/STAT3 activation.19,22 This is further evident from recent studies showing that autophagy regulates JAK2/STAT3 signaling in lung cancer, hepatocarcinoma, pancreatic cancer, and glioblastoma.33,34
Roles of JAK2/STAT3 in Mediating Chemotherapy Resistance
In addition, the unabated JAK2/STAT3 activation has been identified to mediate resistance to standard chemotherapy or radioresistance.6,35,36 Studies on non-small cell lung cancer (NSCLC) and other cancers have found that resistance to anti-EGFR agents is common and occurs through several suggested mechanisms. The de novo resistance was reported due to genetic alteration of receptors or downstream signaling molecules in JAK2/STAT3, while acquired resistance was demonstrated to be due to the activation of the alternative signaling pathways.37 Park and co-workers demonstrated that the JAK2/STAT3/CCND2 pathway is a key mediator of radioresistance.6 The standard chemotherapy resistance or radioresistance in turn is associated with poor prognosis and less survival chance of cancer patients.6,35 For this reason, scholars suggested that combining cancer therapy with JAK2/STAT3 inhibitors could have clinical benefits, and thus JAK2/STAT3 target-specific drug therapies are under clinical trials and some are now approved for clinical use.
Roles of JAK2/STAT3 Pathway in Cancer Treatment
Recently, the JAK2/STAT3 pathway is becoming a potentially valuable therapeutic target for cancer treatment, and it will resolve the problem of chemotherapy resistance associated with an excessively stimulated JAK2/STAT3 pathway.38 A constitutively activated STAT3 has been reported to be a critical oncogene to many types of cancer cells, and targeting it is leading to the discovery of novel therapeutic approaches for human cancers.39,40 For instance, available evidence has suggested that quinazolines block epidermal growth factor receptor (EGFR) activity by blocking JAK2/STAT3 as a potential target of their action.41 Besides, studies have shown that the newly defined STAT3 inhibitor, dihydroartemisinin (DHA), can be employed for chemoprevention, direct tumor-killing, and therapeutic sensitization in the treatment of different malignancies harboring STAT3 phosphorylation.42 Research has also shown that tannic acid induces G1 arrest and apoptosis in human gingival cancer cells primarily by inhibiting the JAK2/STAT3 pathway, particularly by targeting STAT3.43 Moreover, the JAK2/STAT3 targeted disruption with anti-tumor agents has been observed to downregulate the expression of angiogenic factors which leads to suppression of angiogenesis and cancer cell metastasis.26,32 The effect of pantoprazole on cancer cachexia-induced muscle wasting has also been found to be partially associated with the inactivation of the JAK2/STAT3 pathway.44
These drugs/inhibitors (whether approved or under clinical trials) are designed to target ligands, receptors, JAK2, and STAT3 of the JAK2/STAT3 pathway for cancer therapy. The JAK2/STAT3 targeted antitumor agents have been proposed and are being used in combination with the standard treatment of cancer. Overall, the JAK2/STAT3 pathway appears to be a promising therapeutic target for solid tumors, while more investigations are needed to fully understand the molecular mechanisms, efficacy, and side effects to improve clinical outcomes and possible personalized treatments. The details of the JAK2/STAT3 targeted therapies in different human solid cancers are discussed separately in the next section.
Roles of JAK2/STAT3 in Tumorigenesis and Treatment of Different Solid Tumors
This section of the review focuses on the current understanding of the molecular mechanisms of how JAK2/STAT3 plays an intricate role in the progression of various solid tumors and how its disruption serves as a novel therapeutic target that could pave the way for improved cancer therapies (Table 1).
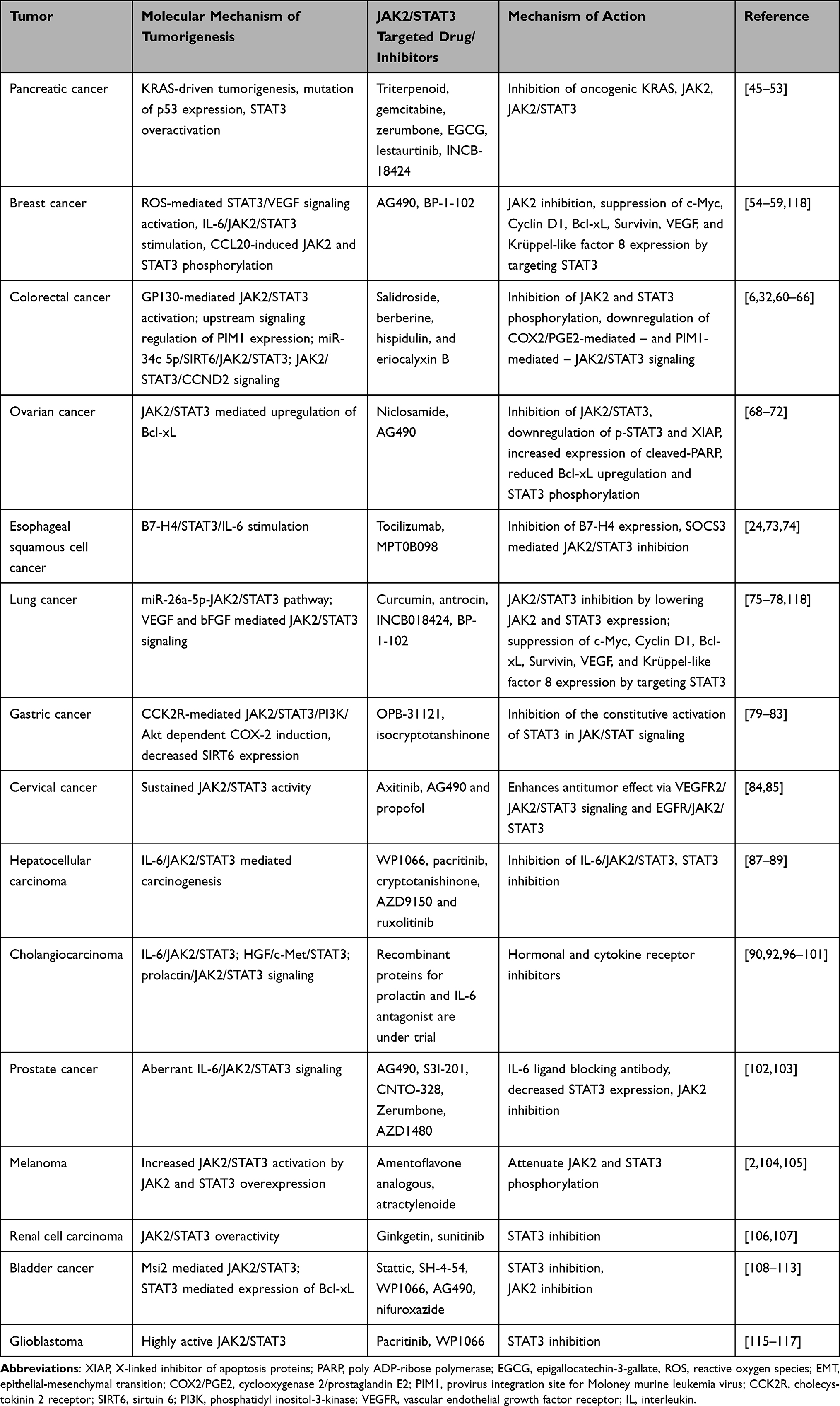 |
Table 1 Table Summary on the Role of JAK2/STAT3 Signaling Pathway in Tumorigenesis and Treatment of Different Solid Tumors |
Pancreatic Cancer
STAT3 is a latent cytosolic transcription factor that is phosphorylated by JAK2, leading to STAT3 dimerization and nuclear localization to activate downstream genes. Recent studies have shown that STAT3 plays a critical role in KRAS-driven pancreatic tumorigenesis as well as in enhancing matrix remodeling and stiffening in pancreatic cancer.45,46 In the animal model, the STAT3-signaled oncogenic activities inhibit p53 expression, and, in turn, the P53 functional loss or mutation in pancreatic tumor cells activates JAK2/STAT3 and constitutively phosphorylates STAT3.47 JAK2 and phospho-STAT3 play a part in facilitating the transition from chronic pancreatitis to pancreatic cancer on pancreatic ductal adenocarcinoma.48
Targeting JAK2/STAT3 recently offered innovative insight into pancreatic cancer therapy. A promising result was observed from the novel triterpenoid derived from Amoora rohituka stem, called aphanin, and it was seen to inhibit oncogenic KRAS and thus exhibit antiproliferative effect in KRAS mutant pancreatic cancer by causing G0-G1 cell cycle arrest.49 Besides, agents like gemcitabine, JAK2 inhibitors, and zerumbone (a phytochemical of Asian ginger) have been shown to possess a novel inhibitory activity against JAK2/STAT3 and significantly block major pro-metastatic gene expression in pancreatic cancer cells.50,51 Moreover, epigallocatechin-3-gallate (EGCG) inhibits the JAK3/STAT3 pathway in pancreatic cancer, phosphorylation of STAT3 and JAK2, and nuclear expression of phospho-STAT3 in both AsPC-1 and PANC-1 cells, thereby enhancing the therapeutic potential of gemcitabine by inhibiting the cell cycle and inducing apoptosis in pancreatic cancer.52 Further novel pancreatic cancer therapy targeting the JAK2/STAT3 pathway, such as lestaurtinib and INCB-18424, has been used in clinical trials to act on JAK2.53
Breast Cancer
Findings have demonstrated that the breast cancer cell line exhibits constitutively active JAK2/STAT3 signaling.54 In line with this, another study has proved that JAK2/STAT3 plays a key role in the survival and progression of breast cancer. The inhibition of this pathway reduces cell viability, invasion, and migration, and also induces cell apoptosis in breast cancer.55 The molecular mechanism by which the JAK2/STAT3 signaling cascade ultimately regulates apoptosis and breast tumor growth has been shown by abnormal STAT3 signaling in malignant cells.56 The reactive oxygen species (ROS)-mediated activation of STAT3/VEGF signaling was also involved in the 27-hydroxycholesterol (27HC)-induced angiogenesis in human breast cancer cells, thus either blocking the generation of ROS or knocking down of STAT3 led to attenuation of the 27HC-induced autocrine activity of VEGF and angiogenesis.57
There is an accumulation of evidence that the activation of the IL-6/JAK2/STAT3 pathway and stem cell-like characteristics lead to poor outcomes of metastatic breast cancers.56 The IL-6/JAK2/STAT3 pathway was preferentially active in CD44+CD24– breast cancer cells compared with other tumor cell types, and inhibition of JAK2 reduced their number and blocked xenograft growth. Distinction has also been revealed between various breast cancer cell types, and targets such as JAK2 and STAT3 have been identified that could lead to more specific and successful therapies for breast cancer.56 A TRAIL agent, known as AG490, exhibits an effective inhibitory activity in cell growth as well as a potent induction agent for cell death in STAT3-active breast cancer cell lines.58 Activation of JAK2/STAT3 is also required for chemokine ligand 20-mediated matrix metalloproteinase-2 expressions by AG490. For example, CCL20-induced phosphorylation of JAK2 and STAT3 may be inhibited by a specific JAK2 inhibitor.59
Colorectal Cancer
Park et al first identified the critical role of the JAK2/STAT3 signaling in promoting tumor initiation and radioresistance by inhibiting apoptosis and enhancing clonogenic potential.6 JAK2 was reported to be predominately overexpressed in the stem cells of colorectal cancer, which leads to STAT phosphorylation, particularly STAT3. The direct binding of STAT3 to the cyclin D2 (CCND2) promoter increased CCND2 expression which is required for persistent growth of cancer stem cells (CSC). This suggests that JAK2/STAT3/CCND2 signaling could serve as potential biomarkers and therapeutic targets for improving outcomes of patients with colonic cancer.6
Numerous reports indicated that gastrin induces epithelial-mesenchymal transition (EMT) in colonic cancer by the activation of JAK2/STAT3, while oncostatin induces EMT in tubular epithelial cells by GP130-mediated activation of the JAK2/STAT3 pathway.32,60,61 Based on the mice model study, a consistently enhanced phosphorylated STAT3 (p-STAT3) and total STAT3 has been shown in colonocytes from infected mice as the STAT pathway is presumed to be activated by infection, which promotes epithelial cancer. There was also a huge variation in the distribution of STAT3 and p-STAT3 in the colon between control and experimental groups.32
Agents such as salidroside, through inhibition of JAK2 and STAT3 phosphorylation and inhibition of metastasis of colon cell lines (SW1116), act as potential therapeutic target for human colonic cancer treatment.32 Similarly, another agent called berberine inhibits invasion and metastasis of colorectal cancer cells via the cyclooxygenase 2/prostaglandin E2 (COX2/PGE2)-mediated JAK2/STAT3 pathway.62 Besides, agents such as hispidulin represses JAK2/STAT3 activation in colorectal cancer via the downregulation of the provirus integration site for Moloney murine leukemia virus (PIM1) expression since JAK2/STAT3 functions as an upstream signaling regulator of PIM1 expression.60 The hispidulin-dependent inhibition of JAK2/STAT3 signaling, which is mediated by downregulation of PIM1 expression, is by generating ROS in colorectal cancer.63
Furthermore, eriocalyxin B has been suggested as a potential therapeutic target for human colonic cancer treatment by inhibiting JAK2/STAT3 pathway in SW116 cells.64 The sirtuin 6 (SIRT6) downregulation, partly due to the upregulation of miR‑34c-5p, also promotes colorectal cancer growth via activation of the JAK2/STAT3 pathway, and SIRT6 was found to be the most likely biomarker for predicting the prognosis of colorectal cancer. Thus, targeting the miR-34c-5p/SIRT6/JAK2/STAT3 regulatory axis could be a promising strategy for colonic cancer treatment.65 A more recent in vitro study has confirmed that a novel selective compound NSC13626 can be used for the treatment of colorectal cancer cells by inhibiting cell growth, arresting cell cycle in the S phase, and downregulating phosphorylation of STAT3 in the JAK2/STAT2 pathway.66
Ovarian Cancer
Three ovarian cancer cell lines, including invasive CAOV-3 cells and the more invasive, aggressive OVCAR-3, and SKOV-3 cells, have been identified.67 It has been suggested that JAK2 might be one of the key upstream activators of STAT3 which upregulates Bcl-xL in CAOV-3 ovarian cancer cells. The phosphorylation of JAK2 leads to the upregulation of Bcl-xL and phosphorylation of STAT3 in ovarian cancer cells, particularly in CAOV-3 cancer cell lines. Thus, aberrant activation of JAK2/STAT3 signaling shows a significant impact on the survival of ovarian cancer patients.68
Although further analysis is required, suppression of the JAK2/STAT3 pathway is thought to be an approach worth considering in the treatment of ovarian cancer, especially chemotherapy-resistant clear cell type.69,70 Agents such as niclosamide are known to have an antitumor effect by inhibiting the JAK2/STAT3 pathway. In addition, Western blot analysis revealed the downregulation of p-STAT3 and x-linked inhibitor of apoptosis (XIAP) proteins, and increased expression of cleaved-poly ADP-ribose polymerase (PARP) by treating with niclosamide in a dose-dependent manner.69,70 It has also been observed that AG490 alone, and with cisplatin, reduces the upregulation of Bcl-xL as well as the phosphorylation of STAT3, which ultimately induces apoptosis in ovarian cancer cell lines.71,72
Esophageal Squamous Cell Cancer (ESCC)
JAK2/STAT3 signaling is required for the cancer stem cell (CSC)-promoting effect of acylglycerol kinase (AGK). A study has reported that JAK2/STAT3 activation is responsible for the enhancing effect of AGK on the CSC population (tumorigenicity) in ESCC in vivo.24 B7-H4, which is highly expressed in three ESCC cell lines, Eca109, TE1, and TE13, plays a critical role in promoting cell proliferation through B7-H4/STAT3/IL-6 positive feedback stimulation. The B7-H4 silence dampened IL-6 secretion through JAK2/STAT3 pathway inactivation, which accounted for ESCC cell proliferation inhibition as well as apoptosis induction. An agent called tocilizumab has been shown to be a novel inhibitor of B7-H4 expression.73 Furthermore, it has been identified that a unique microtubule inhibitor, named MPT0B098, displayed a novel mechanism to inhibit JAK2/STAT3 activity through SOCS3. The combination of MPT0B098 with STAT3 inhibitors, 5-FU, or cisplatin offers a synergistic effect to become a useful therapeutic strategy for ESCC.74
Lung Cancer
Aberrant expression of the JAK2/STAT3 pathway has also been involved in lung CSCs and can promote cancer initiation. JAK2/STAT3 signaling mediates lung cancer cell metastasis through the activation of the miR-26a-5p-JAK2/STAT3 pathway.75 STAT3 is predominantly activated by JAK2-dependent phosphorylation of Y705 in non-small cell lung cell lines (NSCLC) possibly by mutations and inhibition of mitogen-activated protein kinase kinase (MEK) and EGFR. Besides, the JAK2/STAT3 activation promotes angiogenesis through VEGF and basic fibroblast growth factor (bFGF) expression in NSCLC.76 The JAK2/STAT3 pathway might be a promising therapeutic target for lung cancer treatment. Agents such as curcumin and antrocin are targeted on the suppression of stem-like traits of lung cancer cells via inhibition of JAK2/STAT3.77 Moreover, a study done by Liu and co-workers reported that INCB018424 reversed the increased p-JAK2 and p-STAT3Tyr705 protein levels in lung cell lines.78
Gastric Cancer
Anomalous STAT3 activation has been linked with the development, progression, and angiogenesis of gastric cancer.79 It has been reported that JAK2 and STAT3 proteins are expressed at higher levels in all gastric cancer cell lines, and the JAK2/STAT3 axis was demonstrated as a novel signaling pathway.80 The G17 promotes gastric cancer cell proliferation via CCK2R-mediated JAK2/STAT3/PI3K/Akt-dependent COX-2 induction.81 Based on the findings in animal studies, SIRT6 inhibits JAK2/STAT3 pathway activation in gastric cancer cells in addition to their suppressive role on the growth of SGC7901 cells. Hence, SIRT6 can serve as a promising biomarker and therapeutic target for gastric cancer patients.79 A novel small, potent molecular agent, called OPB-31121, has been reported to selectively inhibit constitutive activation of STAT3 in the JAK/STAT signaling pathway in gastric cancer cells.82 Moreover, isocryptotanshinone has been reported to have a more potent inhibitory effect on STAT3 in gastric cancer, which inhibits the proliferation, expression of cell cycle, and apoptosis of gastric cancer cells in a dose- and time-dependent manner.83
Cervical Cancer
Besides its involvement in carcinogenesis, the JAK2/STAT3 signaling that mediates growth arrest can be essential in the clinical development of a new therapeutic approach in chemotherapy of cervical cancer. A study by Zhang and colleagues showed that the synergistic action of axitinib and AG490 enhances the antitumor effect via VEGFR2/JAK2/STAT3 signaling-mediated EMT in cervical cancer in vitro.84 Another study demonstrated that propofol inhibits cervical cancer cell viability, potentiates the inhibitory effect of cisplatin to cervical cancer cell growth, while it enhances the anti-tumor effect of cisplatin via EGFR/JAK2/STAT3 pathway-targeted disruption.85
Hepatocellular Carcinoma
Hepatocellular carcinoma (HCC) is the most prevalent malignant tumor of the liver, accounting for more than 80% of all liver cancers, and the fourth leading cause of cancer-related death globally.86 In vivo and in vitro experimental studies were done on JAK2 knockout liver cancer cell lines and determined the key signaling pathway regulated by JAK2 for liver cancer progression. It has been observed that activation of the JAK2/STAT3 pathway is a common mechanism by which inflammation leads to liver carcinogenesis and serves as a potent promoting factor driving liver tumor formation.87 Particularly, the IL-6/JAK2/STAT3 pathway is aberrantly activated and leads to the dysregulation of downstream target genes that control survival, angiogenesis, stemness, immune surveillance, invasion, and metastasis. Pumpkin polysaccharide-induced apoptosis in HepG2 cells is mediated by the JAK2/STAT3 signal transduction pathways, and therefore this pathway may be considered one of the therapeutic options for HCC.88 Thus, JAK2/STAT3 signaling pathways play a direct role in the progression of liver cancer and is possibly a potential target for future therapeutic clinical approach. Recently, it has been reported that WP1066, pacritinib, cryptotanishinone, and ruxolitinib are JAK2/STAT3 inhibitors under clinical trials for their relevance in HCC treatment.89
Cholangiocarcinoma
Cholangiocarcinoma, which comprises intrahepatic and extrahepatic cholangiocarcinoma, is one of the aggressive malignancies of the biliary tract.90,91 In different experimental models, prolactin, IL-6, EGF, and HGF mediated activation of the JAK2/STAT3 cascade and were found to induce carcinogenesis in cholangiocarcinoma tissues and cell lines.90,92 Prolactin participates in the stimulation of rat bile duct cell proliferation and cholangiocyte regulation of bile water–salt balance, using JAK2/STAT3 as the main signaling pathway.93,94 Prolactin receptor expression in human cholangiocarcinoma samples is much higher than in normal cholangiocytes, indicating that the prolactin/JAK2/STAT3 pathway is involved in its development.92 Furthermore, leptin-mediated induction of JAK2/STAT3 signaling may be involved in cholangiocarcinoma development, although further investigation is needed.95
Cholangiocarcinoma development is also associated with elevation of the systemic plasma IL-6 level, which activates STAT3 proteins forming homodimers.96 In cholangiocarcinoma, sustained IL-6/JAK2/STAT3 signaling and enhanced Mcl-1 expressions are linked with at least partial epigenetic SOCS3 silencing.97 The hepatocyte growth factor (HGF)-induced receptor tyrosine kinase c-Met signaling activates STAT3 phosphorylation, which enhances matrix dissociation, motility of epithelial cells, and invasiveness of tumor cells. In cholangiocarcinoma patients, serum HGF levels are elevated, suggesting that this may increase the invasiveness of tumor cells.98,99 Recent breakthroughs in the development of hormonal and cytokine inhibitors like prolactin and IL-6 suggest that they could be useful in the treatment of cholangiocarcinoma.100,101
Prostate Cancer
Recent Western blotting data revealed that p-STAT3 protein expression decreases significantly with AG490 and S3I-201 treatment and thus suppressed prostate cancer cells growth.102 Zerumbone exerts anticancer effects against hormone-refractory DU145 prostate cancer cells, possibly mediated through inhibition of aberrant IL-6/JAK2/STAT3 signaling axis, and improves the sensitivity of paclitaxel and other anticancer drugs in DU145 cells.103
Melanoma
The JAK2/STAT3 signaling pathway also plays a critical role in the tumorigenesis of human melanoma. There is a strikingly increased STAT3 acetylation in melanoma tissues when compared with normal skin specimens. Immunohistochemical analyses of human skin cancer tissues also showed that STAT3 acetylation was elevated in malignant areas compared with normal tissue areas.2 The JAK2/STAT3 pathway was observed to serve as a potential molecular target for potent anti-melanoma therapeutics by inducing cell apoptosis. Amentoflavone analogues and atractylenolide were tested and reported to inhibit the phosphorylation of JAK2 and STAT3 in human melanoma cells but had no discernible effect on total JAK2 and STAT3 levels.104,105
Renal Cell Carcinoma
Renal cell carcinoma (RCC) is a common malignant disease of the human urinary system and constitutes 90% of all kidney cancers. An experimental study done by Hong et al showed that reduced JAK2/STAT3 activity contributes to the ginkgetin-induced apoptosis in 786-O cells, which confirmed that ginkgetin is a promising therapeutic drug for RCC patients.106 The novel multitargeted tyrosine kinase inhibitor, sunitinib, is used as an antiangiogenic agent for the treatment of several types of cancer, including metastatic RCC. Sunitinib inhibition of STAT3 has been shown to induce tumor cell apoptosis and growth arrest in RCC while reducing immunosuppressive cells such as DCs and myeloid-derived suppressor cells (MDSCs) in the animal model.107
Bladder Cancer
Furthermore, studies have documented that Msi2 has an oncogenic role in the progression of bladder cancer, likely through the activation of several signaling pathways but mainly by activating JAK2/STAT3 in bladder cancer cells, driving EMT and tumor invasion.108–110 Hindupur et al indicated that high STAT3 expression was detected in more than half of the specimens from patients with invasive bladder cancer.109 The STAT3 inhibitors Stattic, SH-4-54, WP1066, and nifuroxazide were observed to reduce the proliferation and increase the apoptosis of bladder cancer cells, possibly through STAT3-mediated expression of anti-apoptotic proteins Bcl-xL.111–113 In addition to its promising results as a novel monotherapy against bladder cancer, STAT3 inhibition has an additive effect when combined with the most commonly used chemotherapeutic agents approved for bladder cancer, suggesting that this combination therapy could be helpful in patients with STAT3-mediated chemoresistance.109 The JAK2 inhibitor AG490 has also been investigated in bladder cancer with promising outcomes; however, Hindupur et al observed conflicting findings with the JAK inhibitors ruxolitinib and BSK-805.109,110
Glioblastoma
Glioblastoma, also called glioblastoma multiforme (GBM), is an aggressive type of cancer that starts in the astrocytes of the spinal cord and brain.114 The JAK2/STAT3 pathway regulates many cellular processes in GBM, for instance, in the survival, proliferation, invasion, anti-apoptosis, and immune evasion of the cells.115 In the central nervous system, the JAK2/STAT3 pathway is highly active during embryonic development. One mechanism through which the JAK2/STAT3 pathway is activated in GBM is through the upstream receptor of tyrosine kinases such as EGFR, which itself is highly mutated in GBMs.116 The JAK2/STAT3 inhibitor, pacritinib, effectively suppresses patient-derived GBM brain tumor-initiating cells in vitro and improves survival in an orthotopic xenograft model when used in combination with temozolomide.117
Discussion
The field of JAK/STAT has developed from the discovery of the individual components three decades ago to high-resolution molecular structures of both JAK and STAT, and an understanding of the complexities of the molecular activation and inhibition of the pathway. This signaling pathway interacts with numerous cytokines and growth factors that result in complex, yet highly specific and regulated cellular responses. This article therefore comprehensively reviews several recent publications to ease the understanding of this complex topic to readers.
Based on the existing evidence, our review appreciated how disruption and dysregulation of the JAK2/STAT3 pathway contribute to the development of a variety of solid tumors, although more research detailing the molecular mechanism is required. Besides, the accumulated evidence indicated that the unabated JAK2/STAT3 activation mediates standard chemotherapy resistance and may be associated with poor prognosis and less chance of survival in cancer patients, urging a novel and better therapeutic intervention. Thus, the JAK2/STAT3 target-specific pharmacological therapies are currently under clinical trials, with several already licensed for clinical use. Recently, there are some approved JAK2/STAT3 targeted drugs that could be used as an alternative therapy and hold great promise for patients with solid tumors. Notably, the JAK2/STAT3 inhibitors may offer more clinical benefits when they are considered in conjunction with traditional cancer therapy, although their detailed antitumorigenic roles and efficacy in solid tumors need to be further elucidated.
Current JAK2/STAT3 inhibitors, however, are not selective and may produce unwanted side effects due to the extensive tissue expression and multifunctionality of the JAK2/STAT3 pathway. Targeting this pathway has pleiotropic effects, making proof-of-concept difficult and increasing the likelihood of adverse outcomes. Hence, close monitoring of side effects and toxicity for patients under JAK2/STAT3 targeted therapy may be required. Besides, it is believed that increasing the selectivity of these inhibitors may reduce the side effects observed with current therapeutic options. We look forward to the adoption of the next generation of JAK2/STAT3 targeted inhibitors which are more selective and have fewer side effects (toxicity) in routine clinical treatment. In addition, we suggest that further investigations are needed to improve clinical outcomes of the JAK/STAT targeted therapy and possible personalized treatments. Despite our extensive effort to provide a comprehensive and up-to-date review regarding the role of JAK2/STAT3 signaling in different human malignancies, this review covers only solid tumors, while this signaling pathway is critical in the development and treatments of hematological malignancies.
Conclusion
In conclusion, a plethora of studies have demonstrated that the constitutively activated JAK2/STAT3 pathway is involved in pathologies such as cancer and inflammation and that it promotes tumorigenesis, tumor growth, cancer cell survival, and metastasis of solid tumors. Besides, there is considerable evidence that has revealed that the aberrantly active JAK2/STAT3 pathway could also be implicated in mediating resistance to standard cancer therapies. This intriguing linkage of the JAK2/STAT3 pathway to tumorigenesis confers novel targets in advance of chemotherapy and paves the way to improved cancer therapy. Designing specific inhibitors against JAK and STAT proteins are showing promising results that will undoubtedly bring new hope for the treatment of solid tumors. However, further in-depth investigations need to be done for a detailed description of the molecular mechanism of JAK2/STAT3 in the progression of a wide range of human malignancies and in resistance to the standard chemotherapy. Moreover, intensive researches have to be conducted to demonstrate the efficacy of the JAK2/STAT3 targeted cancer therapy in combination with other standard therapies prior to their clinical application as well as to discover additional novel anti-tumor agents targeting JAK2/STAT3.
Abbreviations
AGK, acylglycerol kinase; CSC, cancer stem cell; COX2/PGE2, cyclooxygenase 2/prostaglandin E2; DC, dendritic cells; DHA, dihydroartemisinin; EGFR, epidermal growth factor receptor; EGCG, epigallocatechin-3-gallate; EMT, epithelial-mesenchymal transition; ESCC, esophageal squamous cell cancer; Flt3, FMS-like tyrosine kinase 3; GBM, glioblastoma multiforme; 27HC, 27-hydroxycholesterol; HCC, hepatocellular carcinoma; HIF1, hypoxia inducible factor 1, IFN, interferon; JAK, Janus kinase; MDSC, myeloid-derived suppressor cells; NSCLC, non-small cell lung cancer; PARP, poly ADP-ribose polymerase; PICART1, p53-inducible cancer-associated RNA transcript 1; PIM1, provirus integration site for Moloney murine leukemia virus; RCC, renal cell carcinoma; ROS, reactive oxygen species; SIRT6, sirtuin 6; STAM, signal-transducing adapter molecules; STAT, signal transducer and activator of transcription; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; VEGF, vascular endothelial growth factor; VEGFR2, vascular endothelial growth factor receptor 2; XIAP, X-linked inhibitor of apoptosis.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors report no competing interests.
References
1. Wu H, Huang M, Cao P, et al. MiR-135a targets JAK2 and inhibits gastric cancer cell proliferation. Cancer Biol Ther. 2012;13(5):281–288. doi:10.4161/cbt.18943
2. Kiu H, Nicholson SE. Biology and significance of the JAK/STAT signalling pathways. Growth Factors. 2012;30(2):88–106. doi:10.3109/08977194.2012.660936
3. Siveen KS, Sikka S, Surana R, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta Rev Cancer. 2014;1845(2):136–154.
4. Vigneswara V, Kundi S, Ahmed Z. Receptor tyrosine kinases: molecular switches regulating CNS axon regeneration. J Signal Transduct. 2012;2012:1–14
5. Lohi O, Lehto V-P. STAM/EAST/Hbp adapter proteins–integrators of signalling pathways. FEBS Lett. 2001;508(3):287–290. doi:10.1016/S0014-5793(01)03079-4
6. Park S-Y, Lee C-J, Choi J-H, et al. The JAK2/STAT3/CCND2 Axis promotes colorectal Cancer stem cell persistence and radioresistance. J Exp Clin Cancer Res. 2019;38(1):1–18. doi:10.1186/s13046-019-1405-7
7. Thomas S, Snowden J, Zeidler M, et al. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br J Cancer. 2015;113(3):365–371. doi:10.1038/bjc.2015.233
8. Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178(5):2623–2629. doi:10.4049/jimmunol.178.5.2623
9. von Manstein V, Min Yang C, Richter D, et al. Resistance of cancer cells to targeted therapies through the activation of compensating signaling loops. Curr Signal Transduct Ther. 2013;8(3):193–202. doi:10.2174/1574362409666140206221931
10. Argetsinger LS, Kouadio J-LK, Steen H, et al. Autophosphorylation of JAK2 on tyrosines 221 and 570 regulates its activity. Mol Cell Biol. 2004;24(11):4955–4967. doi:10.1128/MCB.24.11.4955-4967.2004
11. Liu Y, Wang L, Wu Y, et al. Pterostilbene exerts antitumor activity against human osteosarcoma cells by inhibiting the JAK2/STAT3 signaling pathway. Toxicology. 2013;304:120–131. doi:10.1016/j.tox.2012.12.018
12. Shuai K, Liu B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol. 2005;5(8):593–605. doi:10.1038/nri1667
13. Krebs DL, Hilton DJ. SOCS proteins: negative regulators of cytokine signaling. Stem Cells. 2001;19(5):378–387. doi:10.1634/stemcells.19-5-378
14. Shuai K, Liu B. Regulation of JAK–STAT signalling in the immune system. Nat Rev Immunol. 2003;3(11):900–911. doi:10.1038/nri1226
15. Laouar Y, Welte T, Fu X-Y, et al. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19(6):903–912. doi:10.1016/S1074-7613(03)00332-7
16. Huang LJ-S, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001;8(6):1327–1338. doi:10.1016/S1097-2765(01)00401-4
17. Zhang Z, Mao H, Du X, et al. Correction: a novel small molecule agent displays potent anti-myeloma activity by inhibiting the JAK2-STAT3 signaling pathway. Oncotarget. 2016;7(49):81972. doi:10.18632/oncotarget.13803
18. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi:10.1038/nrc2734
19. Groner B, von Manstein V. Jak Stat signaling and cancer: opportunities, benefits and side effects of targeted inhibition. Mol Cell Endocrinol. 2017;451:1–14. doi:10.1016/j.mce.2017.05.033
20. Harada D, Takigawa N, Kiura K. The role of STAT3 in non-small cell lung cancer. Cancers. 2014;6(2):708–722. doi:10.3390/cancers6020708
21. Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer Lett. 2007;251(2):199–210. doi:10.1016/j.canlet.2006.10.017
22. Wang H, Wang L, Cao L, et al. Inhibition of autophagy potentiates the anti‐metastasis effect of phenethyl isothiocyanate through JAK2/STAT3 pathway in lung cancer cells. Mol Carcinog. 2018;57(4):522–535. doi:10.1002/mc.22777
23. Bissell MJ, Radisky D. Putting tumours in context. Nat Rev Cancer. 2001;1(1):46–54. doi:10.1038/35094059
24. Chen X, Ying Z, Lin X, et al. Acylglycerol kinase augments JAK2/STAT3 signaling in esophageal squamous cells. J Clin Invest. 2013;123(6):2576–2589. doi:10.1172/JCI68143
25. Alicea-Velazquez NL, Boggon TJ. The use of structural biology in Janus kinase targeted drug discovery. Curr Drug Targets. 2011;12(4):546–555. doi:10.2174/138945011794751528
26. Liu JF, Deng WW, Chen L, et al. Inhibition of JAK2/STAT3 reduces tumor‐induced angiogenesis and myeloid‐derived suppressor cells in head and neck cancer. Mol Carcinog. 2018;57(3):429–439. doi:10.1002/mc.22767
27. Jing N, Tweardy DJ. Targeting Stat3 in cancer therapy. Anticancer Drugs. 2005;16(6):601–607. doi:10.1097/00001813-200507000-00002
28. Lee H, Pal SK, Reckamp K, et al. STAT3: a target to enhance antitumor immune response. Cancer Immunol Immunother. 2010;2010:41–59.
29. McLoughlin RM, Jenkins BJ, Grail D, et al. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc Nat Acad Sci. 2005;102(27):9589–9594. doi:10.1073/pnas.0501794102
30. Dang CV. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19(1):1–11. doi:10.1128/MCB.19.1.1
31. Munoz J, Dhillon N, Janku F, et al. STAT3 inhibitors: finding a home in lymphoma and leukemia. Oncologist. 2014;19(5):536. doi:10.1634/theoncologist.2013-0407
32. Sun K-X, Xia H-W, Xia R-L. Anticancer effect of salidroside on colon cancer through inhibiting JAK2/STAT3 signaling pathway. Int J Clin Exp Pathol. 2015;8(1):615.
33. Wu J, Guo J, Cao Q, et al. Autophagy impacts on oxaliplatin-induced hepatocarcinoma apoptosis via the IL-17/IL-17R-JAK2/STAT3 signaling pathway. Oncol Lett. 2017;13(2):770–776. doi:10.3892/ol.2016.5476
34. Sun L, Hu L, Cogdell D, et al. MIR506 induces autophagy-related cell death in pancreatic cancer cells by targeting the STAT3 pathway. Autophagy. 2017;13(4):703–714. doi:10.1080/15548627.2017.1280217
35. Pokhriyal R, Hariprasad R, Kumar L, et al. Chemotherapy resistance in advanced ovarian cancer patients. Biomark Cancer. 2019;11:1179299X19860815. doi:10.1177/1179299X19860815
36. Tan HF, Putoczki LT, Stylli SS, et al. The role of STAT3 signaling in mediating tumor resistance to cancer therapy. Curr Drug Targets. 2014;15(14):1341–1353. doi:10.2174/1389450115666141120104146
37. Real PJ, Sierra A, de Juan A, et al. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene. 2002;21(50):7611–7618. doi:10.1038/sj.onc.1206004
38. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–170. doi:10.1056/NEJMra1202117
39. Mukthavaram R, Ouyang X, Saklecha R, et al. Effect of the JAK2/STAT3 inhibitor SAR317461 on human glioblastoma tumorspheres. J Transl Med. 2015;13(1):1–10. doi:10.1186/s12967-015-0627-5
40. Jin Y, Kim Y, Lee Y-J, et al. Natural products targeting STAT3 signaling pathways in cancer cells. Biodesign. 2016;4:1–17.
41. Qiao H, Zhao D, Shi H, et al. Novel quinazoline derivatives exhibit antitumor activity by inhibiting JAK2/STAT3. Oncol Rep. 2015;34(4):1875–1882. doi:10.3892/or.2015.4140
42. Li X, Jia L, Lu X, et al. Dihydroartemisinin is a newly defined STAT3 inhibitor that may be of multiple potential uses in cancer treatment. Cancer Cell Microenviron. 2016;3:1.
43. Darvin P, Baeg SJ, Joung YH, et al. Tannic acid inhibits the Jak2/STAT3 pathway and induces G1/S arrest and mitochondrial apoptosis in YD-38 gingival cancer cells. Int J Oncol. 2015;47(3):1111–1120. doi:10.3892/ijo.2015.3098
44. Guo D, Wang C, Wang Q, et al. Pantoprazole blocks the JAK2/STAT3 pathway to alleviate skeletal muscle wasting in cancer cachexia by inhibiting inflammatory response. Oncotarget. 2017;8(24):39640. doi:10.18632/oncotarget.17387
45. Laklai H, Miroshnikova YA, Pickup MW, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22(5):497–505. doi:10.1038/nm.4082
46. Corcoran RB, Contino G, Deshpande V, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Res. 2011;71(14):5020–5029. doi:10.1158/0008-5472.CAN-11-0908
47. Niu G, Wright KL, Ma Y, et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. 2005;25(17):7432–7440. doi:10.1128/MCB.25.17.7432-7440.2005
48. Loncle C, Bonjoch L, Folch-Puy E, et al. IL17 functions through the novel REG3β–JAK2–STAT3 inflammatory pathway to promote the transition from chronic pancreatitis to pancreatic cancer. Cancer Res. 2015;75(22):4852–4862. doi:10.1158/0008-5472.CAN-15-0896
49. Rabi T, Catapano CV. Aphanin, a triterpenoid from Amoora rohituka inhibits K-Ras mutant activity and STAT3 in pancreatic carcinoma cells. Tumor Biol. 2016;37(9):12455–12464. doi:10.1007/s13277-016-5102-2
50. Wörmann SM, Song L, Ai J, et al. Loss of P53 function activates JAK2–STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology. 2016;151(1):180–93. e12. doi:10.1053/j.gastro.2016.03.010
51. Girisa S, Shabnam B, Monisha J, et al. Potential of zerumbone as an anti-cancer agent. Molecules. 2019;24(4):734. doi:10.3390/molecules24040734
52. Shankar S, Suthakar G, Srivastava RK. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Front Biosci. 2007;12(12):5039–5051. doi:10.2741/2446
53. Wang S, Chen X, Tang M. MicroRNA-216a inhibits pancreatic cancer by directly targeting Janus kinase 2. Oncol Rep. 2014;32(6):2824–2830. doi:10.3892/or.2014.3478
54. Garcia R, Bowman TL, Niu G, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20(20):2499–2513. doi:10.1038/sj.onc.1204349
55. Kim MS, Lee WS, Jeong J, et al. Induction of metastatic potential by TrkB via activation of IL6/JAK2/STAT3 and PI3K/AKT signaling in breast cancer. Oncotarget. 2015;6(37):40158. doi:10.18632/oncotarget.5522
56. Marotta LL, Almendro V, Marusyk A, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44+ CD24–stem cell–like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–2735. doi:10.1172/JCI44745
57. Zhu D, Shen Z, Liu J, et al. The ROS-mediated activation of STAT-3/VEGF signaling is involved in the 27-hydroxycholesterol-induced angiogenesis in human breast cancer cells. Toxicol Lett. 2016;264:79–86. doi:10.1016/j.toxlet.2016.11.006
58. Siddiquee KAZ, Turkson J. STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res. 2008;18(2):254–267. doi:10.1038/cr.2008.18
59. Li GX, Tiulim JW, Lang JE, et al. Inflammatory breast cancer: diagnostic, molecular and therapeutic considerations. Curr Breast Cancer Rep. 2019;11(4):335–346. doi:10.1007/s12609-019-00337-z
60. Ferrand A, Kowalski-Chauvel A, Bertrand C, et al. Involvement of JAK2 upstream of the PI 3-kinase in cell–cell adhesion regulation by gastrin. Exp Cell Res. 2004;301(2):128–138. doi:10.1016/j.yexcr.2004.07.037
61. Nightingale J, Patel S, Suzuki N, et al. Oncostatin M, a cytokine released by activated mononuclear cells, induces epithelial cell-myofibroblast transdifferentiation via Jak/Stat pathway activation. J Am Soc Nephrol. 2004;15(1):21–32. doi:10.1097/01.ASN.0000102479.92582.43
62. Liu X, Ji Q, Ye N, et al. Berberine inhibits invasion and metastasis of colorectal cancer cells via COX-2/PGE 2 mediated JAK2/STAT3 signaling pathway. PLoS One. 2015;10(5):e0123478. doi:10.1371/journal.pone.0123478
63. Liu K, Gao H, Wang Q, et al. Hispidulin suppresses cell growth and metastasis by targeting PIM 1 through JAK 2/STAT 3 signaling in colorectal cancer. Cancer Sci. 2018;109(5):1369–1381. doi:10.1111/cas.13575
64. Lu YM, Chen W, Zhu JS, et al. Eriocalyxin B blocks human SW1116 colon cancer cell proliferation, migration, invasion, cell cycle progression and angiogenesis via the JAK2/STAT3 signaling pathway. Mol Med Rep. 2016;13(3):2235–2240. doi:10.3892/mmr.2016.4800
65. Li N, Mao D, Cao Y, et al. Downregulation of SIRT6 by miR-34c-5p is associated with poor prognosis and promotes colon cancer proliferation through inhibiting apoptosis via the JAK2/STAT3 signaling pathway. Int J Oncol. 2018;52(5):1515–1527. doi:10.3892/ijo.2018.4304
66. Lin TE, HuangFu W-C, Chao M-W, et al. A novel selective JAK2 inhibitor identified using pharmacological interactions. Front Pharmacol. 2018;9:1379. doi:10.3389/fphar.2018.01379
67. Seo J-M, Park S, Kim J-H. Leukotriene B4 receptor-2 promotes invasiveness and metastasis of ovarian cancer cells through signal transducer and activator of transcription 3 (STAT3)-dependent up-regulation of matrix metalloproteinase 2. J Biol Chem. 2012;287(17):13840–13849. doi:10.1074/jbc.M111.317131
68. Chen M-W, Yang S-T, Chien M-H, et al. The STAT3-miRNA-92-Wnt signaling pathway regulates spheroid formation and malignant progression in ovarian cancer. Cancer Res. 2017;77(8):1955–1967. doi:10.1158/0008-5472.CAN-16-1115
69. Pan J-X, Ding K, Wang C-Y. Niclosamide, an old antihelminthic agent, demonstrates antitumor activity by blocking multiple signaling pathways of cancer stem cells. Chin J Cancer. 2012;31(4):178. doi:10.5732/cjc.011.10290
70. YoshikawaT,MiyamotoM,AoyamaT,etal.JAK2/STAT3 pathway as a therapeutic target in ovarian cancers. Oncol Lett. 2018; 15(4):5772–5780. doi: 10.3892/ol.2018.8028
71. Yoshikawa T, Miyamoto M, Aoyama T, et al. JAK2/STAT3 pathway as a therapeutic target in ovarian cancers. Oncol Lett. 2018;15(4):5772–5780. doi:10.3892/ol.2018.8028
72. Benabbou N, Mirshahi P, Cadillon M, et al. Hospicells promote upregulation of the ATP-binding cassette genes by insulin-like growth factor-I via the JAK2/STAT3 signaling pathway in an ovarian cancer cell line. Int J Oncol. 2013;43(3):685–694. doi:10.3892/ijo.2013.2017
73. Zhang W, Lei C, Fan J, et al. miR-18a promotes cell proliferation of esophageal squamous cell carcinoma cells by increasing cyclin D1 via regulating PTEN-PI3K-AKT-mTOR signaling axis. Biochem Biophys Res Commun. 2016;477(1):144–149. doi:10.1016/j.bbrc.2016.06.034
74. Peng H-Y, Cheng Y-C, Hsu Y-M, et al. MPT0B098, a microtubule inhibitor, suppresses JAK2/STAT3 signaling pathway through modulation of SOCS3 stability in oral squamous cell carcinoma. PLoS One. 2016;11(7):e0158440. doi:10.1371/journal.pone.0158440
75. Song Q, Liu B, Li X, et al. miR-26a-5p potentiates metastasis of human lung cancer cells by regulating ITGβ8-JAK2/STAT3 axis. Biochem Biophys Res Commun. 2018;501(2):494–500. doi:10.1016/j.bbrc.2018.05.020
76. Looyenga BD, Hutchings D, Cherni I, et al. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS One. 2012;7(2):e30820. doi:10.1371/journal.pone.0030820
77. Wu L, Guo L, Liang Y, et al. Curcumin suppresses stem-like traits of lung cancer cells via inhibiting the JAK2/STAT3 signaling pathway. Oncol Rep. 2015;34(6):3311–3317. doi:10.3892/or.2015.4279
78. Liu X, Chen B, You W, et al. The membrane bile acid receptor TGR5 drives cell growth and migration via activation of the JAK2/STAT3 signaling pathway in non-small cell lung cancer. Cancer Lett. 2018;412:194–207. doi:10.1016/j.canlet.2017.10.017
79. Zhou J, Wu A, Yu X, et al. SIRT6 inhibits growth of gastric cancer by inhibiting JAK2/STAT3 pathway. Oncol Rep. 2017;38(2):1059–1066. doi:10.3892/or.2017.5753
80. Katsha A, Arras J, Soutto M, et al. AURKA regulates JAK2–STAT3 activity in human gastric and esophageal cancers. Mol Oncol. 2014;8(8):1419–1428. doi:10.1016/j.molonc.2014.05.012
81. Xu W, Chen G-S, Shao Y, et al. Gastrin acting on the cholecystokinin2 receptor induces cyclooxygenase-2 expression through JAK2/STAT3/PI3K/Akt pathway in human gastric cancer cells. Cancer Lett. 2013;332(1):11–18. doi:10.1016/j.canlet.2012.12.030
82. Kim M-J, Nam H-J, Kim H-P, et al. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013;335(1):145–152. doi:10.1016/j.canlet.2013.02.010
83. Chen Z-M, Huang L, Li -M-M, et al. Inhibitory effects of isocryptotanshinone on gastric cancer. Sci Rep. 2018;8(1):1–10. doi:10.1038/s41598-017-17765-5
84. Zhang -R-R, Wang H, Hui N, et al. Enhanced antitumor effect of axitinib synergistic interaction with AG490 via VEGFR2/JAK2/STAT3 signaling mediated epithelial-mesenchymal transition in cervical cancer in vitro. Asian Biomed. 2013;7(1):39–49.
85. Li H, Lu Y, Pang Y, et al. Propofol enhances the cisplatin-induced apoptosis on cervical cancer cells via EGFR/JAK2/STAT3 pathway. Biomed Pharmacother. 2017;86:324–333. doi:10.1016/j.biopha.2016.12.036
86. Rizzo A, Brandi G. Biochemical predictors of response to immune checkpoint inhibitors in unresectable hepatocellular carcinoma. Cancer Treat Res Commun. 2021;27:100328. doi:10.1016/j.ctarc.2021.100328
87. Niu G, Wright KL, Huang M, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21(13):2000–2008. doi:10.1038/sj.onc.1205260
88. Shen W, Chen C, Guan Y, et al. A pumpkin polysaccharide induces apoptosis by inhibiting the JAK2/STAT3 pathway in human hepatoma HepG2 cells. Int J Biol Macromol. 2017;104:681–686. doi:10.1016/j.ijbiomac.2017.06.078
89. Tang JJH, Thng DKH, Lim JJ, et al. JAK/STAT signaling in hepatocellular carcinoma. Hepat Onco. 2020;7:1.
90. Smirnova OV, Ostroukhova TY, Bogorad RL. JAK-STAT pathway in carcinogenesis: is it relevant to cholangiocarcinoma progression. World J Gastroenterol. 2007;13(48):6478. doi:10.3748/wjg.v13.i48.6478
91. Rizzo A, Ricci AD, Brandi G. PD-L1, TMB, MSI, and other predictors of response to immune checkpoint inhibitors in biliary tract cancer. Cancers. 2021;13(3):558. doi:10.3390/cancers13030558
92. Ostroukhova TY, Kulikov A, Rozenkrants A, et al. Overexpression of prolactin receptors during intrahepatic transplantation of RS1 rat cholangiocellular carcinoma cells. Bull Exp Biol Med. 2006;141(3):364–367. doi:10.1007/s10517-006-0172-6
93. Orlova A, Smirnova O, Turovetskiĭ V, et al. Effect of bromocriptin on prolactin receptor expression in liver cells after the common bile duct ligation. Biull Eksp Biol Med. 1998;126(7):52–55.
94. Orlova A, Smirnov A, Smirnova O. The role of prolactin in the functional regulation of liver cells after the common bile duct ligation. Biull Eksp Biol Med. 1999;127(5):573–575.
95. Frühbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393(1):7–20. doi:10.1042/BJ20051578
96. Fernández-Martínez E, Pérez-álvarez V, Tsutsumi V, et al. Chronic bile duct obstruction induces changes in plasma and hepatic levels of cytokines and nitric oxide in the rat. Exp Toxicol Pathol. 2006;58(1):49–58. doi:10.1016/j.etp.2006.03.002
97. Meng F, Yamagiwa Y, Ueno Y, et al. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J Hepatol. 2006;44(6):1055–1065. doi:10.1016/j.jhep.2005.10.030
98. Wu F, Wu L, Zheng S, et al. The clinical value of hepatocyte growth factor and its receptor—c-met for liver cancer patients with hepatectomy. Dig Liver Dis. 2006;38(7):490–497. doi:10.1016/j.dld.2006.03.007
99. Imai Y, Kubota Y, Yamamoto S, et al. Neutrophils enhance invasion activity of human cholangiocellular carcinoma and hepatocellular carcinoma cells: an in vitro study. J Gastroenterol Hepatol. 2005;20(2):287–293. doi:10.1111/j.1440-1746.2004.03575.x
100. Bernichtein S, Kayser C, Dillner K, et al. Development of pure prolactin receptor antagonists. J Biol Chem. 2003;278(38):35988–35999. doi:10.1074/jbc.M305687200
101. Kirchhofer D, Lipari MT, Santell L, et al. Utilizing the activation mechanism of serine proteases to engineer hepatocyte growth factor into a Met antagonist. Proc Nat Acad Sci. 2007;104(13):5306–5311. doi:10.1073/pnas.0700184104
102. Gurbuz V, Konac E, Varol N, et al. Effects of AG490 and S3I‑201 on regulation of the JAK/STAT3 signaling pathway in relation to angiogenesis in TRAIL‑resistant prostate cancer cells in vitro. Oncol Lett. 2014;7(3):755–763. doi:10.3892/ol.2014.1795
103. Jorvig JE, Chakraborty A. Zerumbone inhibits growth of hormone refractory prostate cancer cells by inhibiting JAK2/STAT3 pathway and increases paclitaxel sensitivity. Anticancer Drugs. 2015;26(2):160–166. doi:10.1097/CAD.0000000000000171
104. Wu K-J, Huang J-M, Zhong H-J, et al. A natural product-like JAK2/STAT3 inhibitor induces apoptosis of malignant melanoma cells. PLoS One. 2017;12(6):e0177123. doi:10.1371/journal.pone.0177123
105. Fu XQ, Chou JY, Li T, et al. The JAK 2/STAT 3 pathway is involved in the anti‐melanoma effects of atractylenolide I. Exp Dermatol. 2018;27(2):201–204. doi:10.1111/exd.13454
106. Ren Y, Huang S-S, Wang X, et al. Ginkgetin induces apoptosis in 786-O cell line via suppression of JAK2-STAT3 pathway. Iran J Basic Med Sci. 2016;19(11):1245.
107. Hao Z, Sadek I. Sunitinib: the antiangiogenic effects and beyond. Onco Targets Ther. 2016;9:5495. doi:10.2147/OTT.S112242
108. Yang C, Zhang W, Wang L, et al. Musashi-2 promotes migration and invasion in bladder cancer via activation of the JAK2/STAT3 pathway. Lab Investig. 2016;96(9):950–958. doi:10.1038/labinvest.2016.71
109. Hindupur SV, Schmid SC, Koch JA, et al. STAT3/5 inhibitors suppress proliferation in bladder cancer and enhance oncolytic adenovirus therapy. Int J Mol Sci. 2020;21(3):1106. doi:10.3390/ijms21031106
110. Joung YH, Na YM, Yoo YB, et al. Combination of AG490, a Jak2 inhibitor, and methylsulfonylmethane synergistically suppresses bladder tumor growth via the Jak2/STAT3 pathway. Int J Oncol. 2014;44(3):883–895. doi:10.3892/ijo.2014.2250
111. Zhang H, Ye Y, Li M, et al. CXCL2/MIF-CXCR2 signaling promotes the recruitment of myeloid-derived suppressor cells and is correlated with prognosis in bladder cancer. Oncogene. 2017;36(15):2095–2104. doi:10.1038/onc.2016.367
112. Tsujita Y, Horiguchi A, Tasaki S, et al. STAT3 inhibition by WP1066 suppresses the growth and invasiveness of bladder cancer cells. Oncol Rep. 2017;38(4):2197–2204. doi:10.3892/or.2017.5902
113. Chen C-L, Cen L, Kohout J, et al. Signal transducer and activator of transcription 3 activation is associated with bladder cancer cell growth and survival. Mol Cancer. 2008;7(1):1–12. doi:10.1186/1476-4598-7-78
114. Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35. doi:10.1007/s00401-009-0619-8
115. Network CGAR. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061.
116. Wang Y, Wong CW, Yan M, et al. Differential regulation of the pro-inflammatory biomarker, YKL-40/CHI3L1, by PTEN/Phosphoinositide 3-kinase and JAK2/STAT3 pathways in glioblastoma. Cancer Lett. 2018;429:54–65. doi:10.1016/j.canlet.2018.04.040
117. Jensen KV, Cseh O, Aman A, et al. The JAK2/STAT3 inhibitor pacritinib effectively inhibits patient-derived GBM brain tumor initiating cells in vitro and when used in combination with temozolomide increases survival in an orthotopic xenograft model. PLoS One. 2017;12(12):e0189670. doi:10.1371/journal.pone.0189670
118. Zhang X, Yue P, Page BD, et al. Orally bioavailable small-molecule inhibitor of transcription factor Stat3 regresses human breast and lung cancer xenografts. Proc Nat Acad Sci. 2012;109(24):9623–9628. doi:10.1073/pnas.1121606109
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.