")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Role of inflammatory cells in airway remodeling in COPD
Authors Wang Y , Xu J, Meng Y, Adcock IM , Yao X
Received 2 June 2018
Accepted for publication 20 July 2018
Published 12 October 2018 Volume 2018:13 Pages 3341—3348
DOI https://doi.org/10.2147/COPD.S176122
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Yujie Wang,1,2 Jiayan Xu,1 Yaqi Meng,1 Ian M Adcock,3 Xin Yao1
1Department of Respiratory Medicine, the First Affiliated Hospital of Nanjing Medical University, Nanjing, China; 2Department of Respiratory Medicine, The Second Affiliated Hospital of Hainan Medical University, Haikou, China; 3Cell and Molecular Biology Group, Airways Disease Section, National Heart and Lung Institute, Imperial College London, London, UK
Abstract: COPD is characterized by chronic bronchitis, chronic airway obstruction, and emphysema, leading to a progressive and irreversible decline in lung function. Inflammation is central for the development of COPD. Chronic inflammation in COPD mainly involves the infiltration of neutrophils, macrophages, lymphocytes, and other inflammatory cells into the small airways. The contribution of resident airway structural cells to the inflammatory process is also important in COPD. Airway remodeling consists of detrimental changes in structural tissues and cells including airway wall thickening, epithelial metaplasia, goblet cell hypertrophy, and smooth muscle hyperplasia. Persistent airway inflammation might contribute to airway remodeling and small airway obstruction. However, the underlying mechanisms remain unclear. In this review, we will provide an overview of recent insights into the role of major immunoinflammatory cells in COPD airway remodeling.
Keywords: COPD, airway inflammation, airway remodeling
Introduction
COPD is characterized by chronic bronchitis, chronic airway obstruction, airway remodeling, and emphysema, leading to a progressive and irreversible decline in lung function.1 Inflammation is central for COPD development and the release of inflammatory mediators and destructive enzymes by inflammatory cells particularly infiltrating immune cells, which is implicated in the progressive destruction of the lung in COPD.2,3 However, the role of resident structural cells in this process should not be discounted.
Remodeling has been described in central airways, distal airways, and lung parenchyma. It is a process of structural changes involving hyperplasia of airway epithelial cells, thickening of the reticular basement membrane (RBM), deposition of collagen, peribronchial fibrosis, airway epithelial-to-mesenchymal transition, and bronchial smooth muscle cell hyperplasia.4 In COPD, remodeling of the parenchyma contributes to emphysema, while small airway remodeling largely results in airway obstruction. These changes cause the airflow limitation seen in COPD patients. However, the underlying mechanisms remain unclear.
The chronic inflammation in COPD involves the infiltration of the major inflammatory cells including neutrophils, monocytes/macrophages, and lymphocytes into the airway and lung tissue, and these can be detected in bronchoalveolar fluid and induced sputum.5 It is generally acknowledged that persistent chronic inflammation may contribute to not only bronchial remodeling but also parenchyma remodeling to some extent.6,7 In this review, we will highlight the recent studies that have provided additional insight into the role of these major inflammatory cells in COPD airway remodeling.
Neutrophils
Neutrophils are key inflammatory cells in the pathogenesis of COPD, with sputum and blood neutrophilia being a characteristic feature of all COPD patients. They have also been reported as a marker of COPD severity.8,9 An observational study found that patients with higher sputum neutrophil percentages had a higher dyspnea score across different severities of COPD.10
Neutrophils are recruited to the airways of COPD patients and secrete several serine proteases including neutrophil elastase (NE), matrix metalloproteinase (MMP), as well as myeloperoxidase (MPO) all of which contribute to alveolar destruction.11,12 In addition, some neutrophil-derived chemokines such as IL-1 and CXCL8/IL-8 are proven to be involved in tissue injury and remodeling in a mouse model.13
MMPs are a family of zinc-dependent proteases that can be secreted by stromal cells, neutrophils, and macrophages. They are commonly classified according to the substrates they degrade. The majority of MMPs implicated in emphysema pathogenesis include the collagenase MMP-1, the gelatinase MMP-9, and the metalloelastase MMP-12.14 Among those, the gelatinase MMP-9 is synthesized by mature neutrophils and is mainly stored in intracellular granules of neutrophils and is secreted extracellular after activation.15
MMP-9 activity is countered by the tissue inhibitors of metalloproteinases, and any changes in the activity of this enzyme will alter this balance.13 Most studies have shown increased MMPs in bronchoalveolar lavage fluid (BALF) and plasma of emphysema patients and contribute to airway obstruction by destroying the structural components of extracellular matrix (ECM).16,17 Moreover, as MMP-9 is a known target of Wnt/β-catenin signaling, it has been proved to be induced by transforming growth factor-β (TGF-β) + poly(I:C) treatment through the β-catenin pathway.18 In animal models of COPD, it demonstrated that dominant-negative MafB suppressed porcine pancreatic elastase-induced emphysema by downregulating MMPs.19 Considering the significant role of MMP-9 in the above studies, it may be worthwhile exploring its role in the function of different primary cells from patients with disease.
NE is a neutrophil-derived serine proteinase that has proven to be involved in tissue damage and remodeling,20 and further a study found that mice deficiency in NE resulted in the protection of mice against emphysema after cigarette smoke (CS) exposure.21 The underlying mechanism(s) may largely depend on the fact that NE has a similar ability as MMPs in causing tissue damage by degrading the structural components of ECM.22 Moreover, NE can cooperate with MMPs and amplify the effect of ECM degradation.23 In addition to matrix degradation, NE can also promote peribronchial fibrosis by enhancing fibroblast proliferation.24 Moreover, NE is a potent stimulant of mucus secretion from submucosal glands and goblet cells, which are involved in airway obstruction.25 The combined effect of NE on matrix degradation, fibroblast proliferation, and mucus metaplasia might accelerate small airway obstruction in disease.
MPO is a product of both neutrophils and macrophages and mainly stored in the primary granules of neutrophils. It is an inflammatory mediator that is upregulated during the inflammatory response and can also accelerate the inflammatory response.26 3-Chlorotyrosine expression is strongly associated with MPO activity in the sputum of COPD patients, suggesting that it might act as a biomarker for MPO-mediated tissue damage in COPD pathogenesis.27 MPO inhibitors prevent the development of emphysema and remodeling of small airways in an animal model of COPD.28 These studies indicated detrimental effects of MPO in the pathogenesis of airway remodeling. However, a study of myocardial infarction in MPO knockout mice observed increased expression of MMPs.29 This finding suggests a possible protective role of MPO in airway remodeling.
Neutrophil extracellular traps (NETs) are released by activated neutrophils and decorated with histones and enzymes such as NE and MPO that ensnare bacteria; however, excessive formation of NETs may contribute to organ damage.30 Large amounts of NETs were observed in the airways of COPD patients and associated with disease severity and exacerbation frequency.31
The high-mobility group box 1 (HMGB1) is a protein released by necrosis of neutrophil cells that warn and activate inflammation. COPD patients express high HMGB1 levels both in sputum and in plasma.32 HMGB1 has a marked effect on epithelial cell repair and restitution via activation of toll-like receptor 4 (TLR4) and/or receptor for advanced glycation end (RAGE) signaling, which may explain, in least in part, the mechanism of airway remodeling.33
Macrophages
Macrophages are mononuclear leukocyte-derived inflammatory cells the numbers of which are increased in the airways, BALF, alveolar areas, and in induced sputum of COPD patients and correlated with the inflammatory response and alveolar wall destruction in COPD.34 They produce a host of inflammatory mediators implicated in COPD such as IL-1β, tumor necrosis factor-α (TNF-α), IL-8, monocyte chemoattractant protein-1 (MCP-1), reactive oxygen species, and MMPs.35
IL-8 is a member of the CXC chemokine family and is secreted by macrophages, epithelial cells, and even endothelial cells. It is a major inflammatory factor in the sputum and BALF in COPD patients. IL-8 can enhance the expression of MUC5AC directly or indirectly by inducing the secretion of NE from neutrophils, leading to mucin overproduction and airway obstruction.36 And the inhibition of IL-8 by azithromycin in stable neutrophilic COPD adults showed a reduced severe exacerbation rate.37 Further studies demonstrated that knockout of CXCR2, a receptor of IL-8, protected mice from cigarette smoke-induced lung inflammation and DNA damage in COPD pathogenesis.38
IL-1β and TNF-α cytokines are proinflammatory cytokines that are mainly secreted by macrophages. Both IL-1β and TNF-α receptor knockout mice are protected from developing emphysema and small airway remodeling when compared with wild-type mice in response to CS.39 However, in a randomized phase II study, MEDI8968, an anti-IL-1R1 antibody, did not protect COPD patients from lung function decline.40 The different outcomes that attribute to the trials are based on measures of lung function, sputum inflammatory cells, or exacerbation rates, which may not reflect the remodeling effect of these drugs. In addition, persistent production of IL-1β upregulates the expression of neutrophilic cytokines and MMPs including MMP-9 and MMP-12 in mice and results in airway inflammation and alveolar enlargement.41,42 Alveolar and airway wall remodeling occurs in SPC-TNF-α mice with increased expression of elastin-degrading enzymes and consequent matrix remodeling.43 This effect was attributed to TNF-α-induced stimulation of MMPs and NE and the activation of CD8+ T lymphocytes all of which contribute to the destruction of lung tissues.
Different TGF-β isoforms exist, including TGF-β1, TGF-β2, and TGF-β3. Among them, TGF-β1 is implicated in the progression of COPD pathogenesis.44,45 In relation to COPD, TGF-β induces the secretion of ECM, proliferation of smooth muscle cells, and transition of epithelial-to-mesenchymal phenotype.46 Exposure to CS increases the production of TGF-β from epithelial cells and inflammatory cells, and this was linked to CS-induced lung injury and airway remodeling in COPD.47 This might partially provide a mechanism for CS-induced small airway obstruction.
Macrophages are just as an important source of MMPs as neutrophils. Activation of macrophages in lungs plays a critical role in MMP production that contributes to alveolar wall destruction. Researchers found that the loss of tyrosine phosphatase 2 in mice, a macrophage activation regulator, resulted in TGF-β activation, thereby upregulating MMP-12 expression in macrophages, leading to progressive emphysema-like injury in the mice lungs.48 Moreover, recently a study suggested that dehydration of airway surface can also activate macrophages to produce MMP-12 and trigger MMP-12-dependent emphysema independent of CS.49 These findings might provide some clues to explore non-CS-induced emphysema.
MCP-1 is a monocyte chemokine that can be produced by macrophages, epithelial cells, smooth muscle cells, and even endothelial cells.50 Although some studies showed that the protein level of MCP-1 was not correlated with the number of macrophages, intratracheal instillation of MCP-1 in macrophage elastase knockout mice resulted in an increased macrophage infiltration into the airway.51 The ability of MCP-1 to accumulate macrophages might help to accelerate the process of airway obstruction.
Autophagy plays a critical role in the development of many inflammatory cells such as macrophages, neutrophils, and lymphocytes, which play critical roles in the development and pathogenesis of COPD inflammation.52 In vivo studies in mice showed that miR-34/449 overexpression leads to decreased ovalbumin-induced airway remodeling by suppressing autophagy-related airway inflammation and fibrosis.53
Overall, macrophages are major inflammatory cells in COPD lung. They are directly involved in the process of airway remodeling by secreting enzymes and inflammatory factors that act directly and indirectly on airway structural cells to modulate epithelial and stromal cell function.
Mast cells
Mast cells are multifunctional immune cells composed of two subsets: mucosal mast cell (MCT) and connective tissue mast cell (MCTC).54 Mast cells have been implicated in asthma for many years where, in addition to the release of lipid mediators and other bronchoconstrictor agents, they promote airway remodeling.55 However, they have been poorly studied in COPD.
Increased numbers of mast cells have been reported in COPD patients with centrilobular emphysema, where they are mainly distributed in the bronchial mucosa, parenchyma, and even smooth muscle.56 The population of mast cells within the lung may also change with disease, with increased MCTC and decreased MCT reported in COPD. Moreover, the increased number of MCTC positively correlated with airway remodeling and poorer lung function.57,58 In addition, perivascular mast cell density is positively correlated with increased angiogenesis in the RBM of COPD airways where they are proposed to contribute to airway remodeling.59,60 Furthermore, IL-17A, which has been reported to be upregulated in COPD,61 can stimulate mast cells to secrete the proangiogenic mediators, basic fibroblast growth factor and vascular endothelial growth factor (VEGF), which both drive vascular remodeling.62
However, analysis of the distribution of tryptase and chymase staining in mast cells indicates that mast cells are positively correlated with lung function,63 and this was particularly the case with chymase-positive mast cells.64 Results from the latter research may be confounded by the presence of pulmonary hypertension. In addition, although mast cells appear to play a role in COPD, the molecular mechanism(s) by which they act are unknown. Mast cells are a rich source of inflammatory cytokines, proteases, VEGF, and mast cell-specific mediators such as histamine and cysteinyl leukotrienes.65 Further investigations into the mechanisms of mast cells involved in the development of airway remodeling are required.
Lymphocytes
The adaptive immune system is activated in COPD, with infiltration of T-cells, B-cells, T-helper type 17 (Th17) cells along with a decrease in regulatory T-cells within the airways.66,67 Mice lacking either B-cells or T-cells fail to elicit airway remodeling illustrating the importance of the adaptive immune response in airway remodeling.68
T-lymphocytes are increased in the lung parenchyma and airways of smokers when compared with never smokers whether they develop COPD or not. There is a greater increase in CD8+ cells compared with CD4+ cells.69 The increased CD8+ cells in the peripheral airways of smokers with COPD have been related to smoking-induced airway limitation.70,71 In addition, T-cells can cause lung tissue destruction directly by T-cell-induced cytotoxicity or indirectly by activating macrophages.72,73 These data suggest that increased CD8+ cells in COPD act as a bridge between smoking and airway obstruction.
CD8+ cells can be divided into TC1 cells and TC2 cells according to the cytokines they secrete. Isolation of CD4+ and CD8+ cells from COPD BALF indicates that CD8+ TC2 cells, which mainly produce IL-4 and IL-5 cytokines, were significantly increased in COPD lungs and might promote tissue damage and the development of emphysema during exacerbations.74
The number of B-cells within lymphoid follicles is greatly increased in advanced stages of COPD patients.75 CXCL13 is a B-cell attractant or chemokine,76 and the reduction of CXCL13 expression attenuated CS-induced BALF inflammatory cell numbers and partially protected alveolar walls from destruction but had no effect on the development of airway remodeling.77
Th17 cells are the major source of the cytokine IL-17.78 IL-17 can enhance airway smooth muscle contraction and proliferation, and Th17-deficient mice are protected from airway remodeling after chronic allergen challenge in an animal model of asthma.79
Innate lymphoid cells
Innate lymphoid cells (ILCs) are a new class of immune cells that can be classified into three groups (ILC1, ILC2, and ILC3) according to their phenotype and function. ILCs are widely expressed in many tissues such as skin, mucosal membranes, and lung tissues.80
Most studies examining ILC expression and function in airway disease have focused on asthma. ILC3s play a role in driving neutrophilic inflammation, and the number of natural cytotoxicity receptor (NCR−) expressing ILC3 cells was increased in COPD lung.81 ILC3s can activate TGF-β, which is a key mediator for tissue and mucosal repair.82 More recent evidence indicates the enhanced presence of primed NRP1+ ILC3s, which produce high amounts of cytokines in the lungs of smokers with and without COPD where they may play a role in angiogenesis and/or the initiation of lymphoid follicles.83 This result implies that ILC3s may participate in the process of airway remodeling in COPD.
ILC1 frequency has also been reported to be increased in COPD and to correlate with disease severity and susceptibility to exacerbations. This may reflect the functional plasticity of ILC2 cells and an attenuation of antiviral immunity.84 Most recently, the combination of microCT analysis, histology, and gene expression profiling indicated that signatures for ILC1s, but not ILC2s or ILC3s, were associated with centrilobular emphysema. This suggests that the alveolar destruction observed in COPD is driven by a Th1 response activated by infiltrating ILC1s.85
Overall, there are few studies that focus on the relationship between ILCs and airway remodeling of COPD. Considering the role of the immune response in COPD pathogenesis, further elucidation of the functional role of ILCs subsets in COPD and its correlation with other inflammatory cells is essential.
Conclusions
COPD is a chronic inflammatory disease involving the infiltration of various inflammatory cells including neutrophils, macrophages, lymphocytes, mast cells, and ILCs.3 The infiltration of inflammatory cells can contribute to the detrimental changes observed in structural cells such as airway epithelial cells, stromal cells, and parenchyma cells. The effects of these inflammatory cells on remodeling are attributed to direct or indirect release of factors such as inflammatory cytokines, proteases, and growth factors (Figure 1). However, definitive proof of their role will require controlled clinical studies targeting specific cell types and/or remodeling factors with airway remodeling as a defined outcome. Currently, we do not have good biomarkers of remodeling, and imaging techniques are not yet sensitive to directly visualize airway remodeling changes.
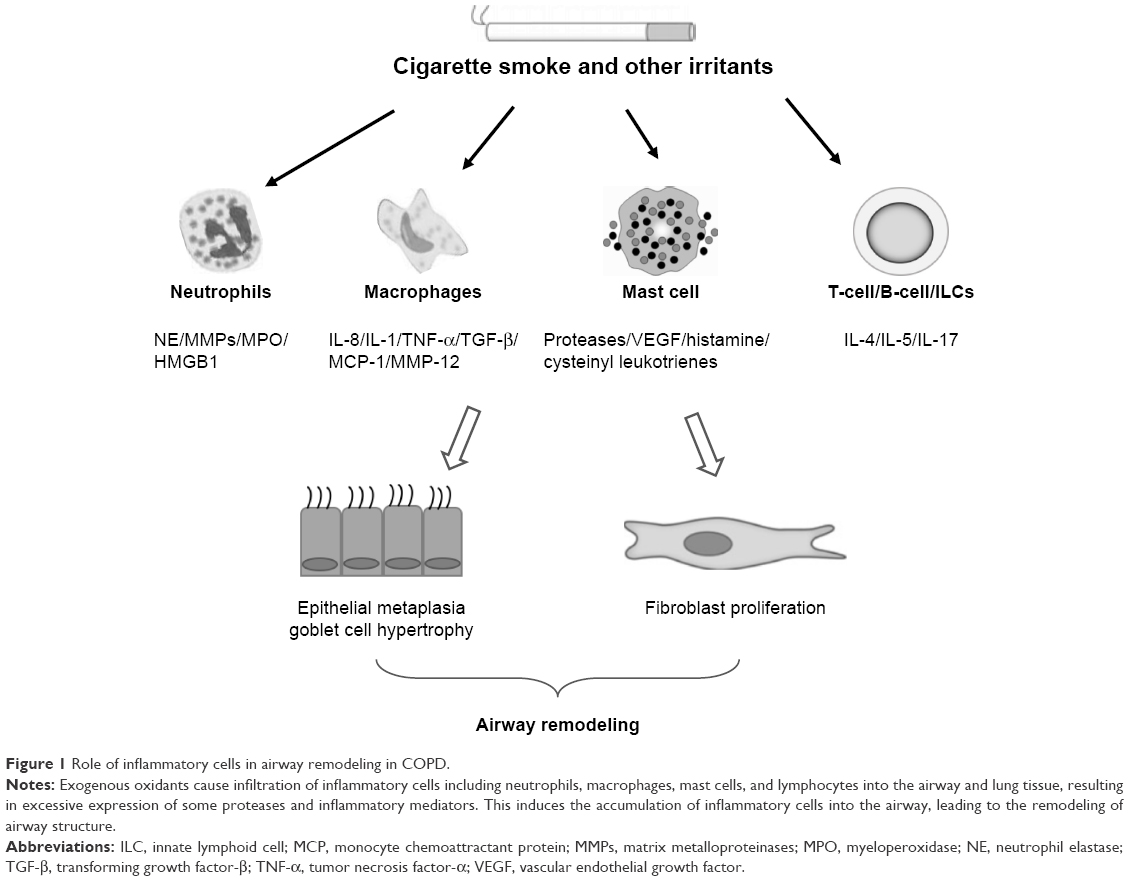 | Figure 1 Role of inflammatory cells in airway remodeling in COPD. |
The mechanisms of airway remodeling are poorly studied in COPD compared with asthma. Airway remodeling is characterized by the changes in tissue, cellular, and molecular components, thereby contributing to pathological changes to the epithelium, airway smooth muscle, vessels, and ECM.86 Inflammation in the airways of COPD is largely attributed to smoking and usually enhanced by bacterial and viral infection although this may also be present in ex-smokers.87 Experimental studies show that CS exposure can directly lead to the changes in structural cells seen in the lung tissue and small airways as a result of inflammatory response and oxidative stress.88 It is notable that smoking cessation does not prevent the progression of chronic inflammation and oxidative stress and that these are associated with persistent tissue destruction and remodeling.89
We posit that in COPD lungs, inflammatory cells infiltrate into the bronchial mucosa and lung parenchyma. They affect airway destruction and remodeling by directly secreting enzymes and inflammatory cytokines or by indirectly regulating other cellular functions. Some of the above factors can promote airway destruction and remodeling, whereas other factors may protect from tissue damage and reconstruction. Overall, inflammatory cells influence the structural cell destruction, hyperplasia of smooth muscle cells, metaplasia of goblet cells, and subepithelial fibrosis seen in COPD.
It is not completely known how different inflammatory mediators function in the process of airway remodeling and how remodeling contributes to the decrease in lung function. Present treatments can at best only partially reduce the inflammatory response and barely prevent or reverse airway remodeling. Given the fact that inflammatory cells induce such significant effects on airway remodeling in COPD pathogenesis, it is imperative to explore the mechanisms of airway remodeling in COPD and delineate new therapeutic avenues.
Acknowledgments
This work was supported by National Natural Science Foundation of China (Grants 81070025 and 81470237), the Jiangsu Health Promotion Project, and the Priority Academic Program Development of Jiangsu Higher Education Institutions (Grant JX10231802). Research by IMA is also supported by the NIHR Biomedical Research Center at Imperial College London.
Disclosure
IMA is supported by the MRC (G1001367/1) and the Wellcome Trust (093080/Z/10/Z). The authors report no conflicts of interest in this work.
References
Vestbo J, Hurd SS, Agustí AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. | ||
Polosukhin VV, Richmond BW, Du R-H, et al. Secretory IgA deficiency in individual small airways is associated with persistent inflammation and remodeling. Am J Respir Crit Care Med. 2017;195(8):1010–1021. | ||
Richmond BW, Du RH, Han W, et al. Bacterial-derived neutrophilic inflammation drives lung remodeling in a mouse model of COPD. Am J Respir Cell Mol Biol. 2018;58(6):736–744. | ||
Hirota N, Martin JG. Mechanisms of airway remodeling. Chest. 2013;144(3):1026–1032. | ||
Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol. 2009;4:435–459. | ||
Sohal SS, Ward C, Danial W, Wood-Baker R, Walters EH. Recent advances in understanding inflammation and remodeling in the airways in chronic obstructive pulmonary disease. Expert Rev Respir Med. 2013;7(3):275–288. | ||
Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161(5):1720–1745. | ||
Singh D, Edwards L, Tal-Singer R, Rennard S. Sputum neutrophils as a biomarker in COPD: findings from the ECLIPSE study. Respir Res. 2010;11(1):77. | ||
Mendy A, Forno E, Niyonsenga T, Gasana J. Blood biomarkers as predictors of long-term mortality in COPD. Clin Respir J. 2018;12(5):1891–1899. | ||
Bartoli ML, Costa F, Malagrinò L, et al. Sputum inflammatory cells in COPD patients classified according to GOLD 2011 guidelines. Eur Respir J. 2016;47(3):978–980. | ||
Bardoel BW, Kenny EF, Sollberger G, Zychlinsky A. The balancing act of neutrophils. Cell Host Microbe. 2014;15(5):526–536. | ||
Wang Y, Jia M, Yan X, et al. Increased neutrophil gelatinase-associated lipocalin (NGAL) promotes airway remodelling in chronic obstructive pulmonary disease. Clin Sci. 2017;131(11):1147–1159. | ||
Baek KJ, Cho JY, Rosenthal P, Alexander LE, Nizet V, Broide DH. Hypoxia potentiates allergen induction of HIF-1α, chemokines, airway inflammation, TGF-β1, and airway remodeling in a mouse model. Clin Immunol. 2013;147(1):27–37. | ||
Hendrix AY, Kheradmand F. The role of matrix metalloproteinases in development, repair, and destruction of the lungs. Prog Mol Biol Transl Sci. 2017;148:1–29. | ||
Meijer M, Rijkers GT, van Overveld FJ. Neutrophils and emerging targets for treatment in chronic obstructive pulmonary disease. Expert Rev Clin Immunol. 2013;9(11):1055–1068. | ||
D’Armiento JM, Goldklang MP, Hardigan AA, et al. Increased matrix metalloproteinase (MMPs) levels do not predict disease severity or progression in emphysema. PLoS One. 2013;8(2):e56352. | ||
Ostridge K, Williams N, Kim V, et al. Relationship between pulmonary matrix metalloproteinases and quantitative CT markers of small airways disease and emphysema in COPD. Thorax. 2016;71(2):126–132. | ||
Royer P-J, Henrio K, Pain M, et al. TLR3 promotes MMP-9 production in primary human airway epithelial cells through Wnt/β-catenin signaling. Respir Res. 2017;18(1):208. | ||
Aida Y, Shibata Y, Abe S, et al. Inhibition of elastase-pulmonary emphysema in dominant-negative MafB transgenic mice. Int J Biol Sci. 2014;10(8):882–894. | ||
Polverino E, Rosales-Mayor E, Dale GE, Dembowsky K, Torres A. The role of neutrophil elastase inhibitors in lung diseases. Chest. 2017;152(2):249–262. | ||
Guyot N, Wartelle J, Malleret L, et al. Unopposed cathepsin G, neutrophil elastase, and proteinase 3 cause severe lung damage and emphysema. Am J Pathol. 2014;184(8):2197–2210. | ||
Lerman I, Hammes SR. Neutrophil elastase in the tumor microenvironment. Steroids. 2018;133:96–101. | ||
Guo Y, Ma L, Zhang F, Sun R, Li T. Neutrophil elastase ameliorates matrix metalloproteinase-9 to promote lipopolysaccharide-induced acute lung injury in mice 1. Acta Cirurgica Brasileira. 2016;31(6):382–388. | ||
Gregory AD, Kliment CR, Metz HE, et al. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J Leukoc Biol. 2015;98(2):143–152. | ||
Arai N, Kondo M, Izumo T, Tamaoki J, Nagai A. Inhibition of neutrophil elastase-induced goblet cell metaplasia by tiotropium in mice. Eur Respir J. 2010;35(5):1164–1171. | ||
Nauseef WM. Biosynthesis of human myeloperoxidase. Arch Biochem Biophys. 2018;642:1–9. | ||
O’Donnell C, Newbold P, White P, Thong B, Stone H, Stockley RA. 3-Chlorotyrosine in sputum of COPD patients: relationship with airway inflammation. COPD. 2010;7(6):411–417. | ||
Churg A, Marshall CV, Sin DD, et al. Late intervention with a myeloperoxidase inhibitor stops progression of experimental chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;185(1):34–43. | ||
Askari AT, Brennan ML, Zhou X, et al. Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J Exp Med. 2003;197(5):615–624. | ||
Qiu S-L, Zhang H, Tang Q-Ya, et al. Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. Thorax. 2017;72(12):1084–1093. | ||
Dicker AJ, Crichton ML, Pumphrey EG, et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2018;141(1):117–127. | ||
Gangemi S, Casciaro M, Trapani G, et al. Association between HMGB1 and COPD: a systematic review. Mediators Inflamm. 2015;2015(8):1–8. | ||
Ojo OO, Ryu MH, Jha A, Unruh H, Halayko AJ. High-mobility group box 1 promotes extracellular matrix synthesis and wound repair in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2015;309(11):L1354–L1366. | ||
Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4–5):383–396. | ||
Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):71–86. | ||
Jundi K, Greene CM. Transcription of interleukin-8: how altered regulation can affect cystic fibrosis lung disease. Biomolecules. 2015;5(3):1386–1398. | ||
Simpson JL, Powell H, Baines KJ, et al. The effect of azithromycin in adults with stable neutrophilic COPD: a double blind randomised, placebo controlled trial. PLoS One. 2014;9(8):e105609. | ||
Lerner CA, Lei W, Sundar IK, Rahman I. Genetic ablation of CXCR2 protects against cigarette smoke-induced lung inflammation and injury. Front Pharmacol. 2016;7:391. | ||
Churg A, Zhou S, Wang X, Wang R, Wright JL. The role of interleukin-1beta in murine cigarette smoke-induced emphysema and small airway remodeling. Am J Respir Cell Mol Biol. 2009;40(4):482–490. | ||
Calverley PMA, Sethi S, Dawson M, et al. A randomised, placebo-controlled trial of anti–interleukin-1 receptor 1 monoclonal antibody MEDI8968 in chronic obstructive pulmonary disease. Respir Res. 2017;18(1):153. | ||
Lappalainen U, Whitsett JA, Wert SE, Tichelaar JW, Bry K. Interleukin-1beta causes pulmonary inflammation, emphysema, and airway remodeling in the adult murine lung. Am J Respir Cell Mol Biol. 2005;32(4):311–318. | ||
Ji M, Wang Y, Li X, Qian Z. Up-regulation of ICAM-1mRNA and IL-1βmRNA in lung tissues of a rat model of COPD. Int J Clin Exp Med. 2015;8(11):21956–21963. | ||
Reynaert N, Eurlings I, Mercken E, et al. Involvement of JNK in TNFα driven remodelling. Eur Respir J. 2015;46:A5058. | ||
di Stefano A, Sangiorgi C, Gnemmi I, et al. TGF-beta signaling pathways in different compartments of the lower airways of patients with stable COPD. Cheast. 2018;153(4):851–862. | ||
Michaeloudes C, Kuo C-H, Haji G, et al. Metabolic re-patterning in COPD airway smooth muscle cells. Eur Respir J. 2017;50(5):1700202. | ||
Gohy ST, Hupin C, Fregimilicka C, et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J. 2015;45(5):1258–1272. | ||
Hoang LL, Nguyen YP, Aspeé R, et al. Temporal and spatial expression of TGF-β following airway remodeling to tobacco smoke in rats. Am J Respir Cell Mol Biol. 2016;54(6):872–881. | ||
Xu J, Tao B, Guo X, et al. Macrophage-restricted Shp2 tyrosine phosphatase acts as a Rheostat for MMP12 through TGF-β activation in the prevention of age-related emphysema in mice. J Immunol. 2017;199(7):2323–2332. | ||
Trojanek JB, Cobos-Correa A, Diemer S, et al. Airway mucus obstruction triggers macrophage activation and matrix metalloproteinase 12-dependent emphysema. Am J Respir Cell Mol Biol. 2014;51(5):709–720. | ||
Yoshimura T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol Immunol. 2018;15(4):335–345. | ||
Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277(5334):2002–2004. | ||
Qian M, Fang X, Wang X. Autophagy and inflammation. Clin Transl Med. 2017;6(1):24. | ||
Yin H, Zhang S, Sun Y, et al. MicroRNA-34/449 targets IGFBP-3 and attenuates airway remodeling by suppressing Nur77-mediated autophagy. Cell Death Dis. 2017;8(8):e2998. | ||
Erjefält JS. Mast cells in human airways: the culprit? Eur Respir Rev. 2014;23(133):299–307. | ||
Cruse G, Bradding P. Mast cells in airway diseases and interstitial lung disease. Eur J Pharmacol. 2016;778:125–138. | ||
Virk H, Arthur G, Bradding P. Mast cells and their activation in lung disease. Transl Res. 2016;174:60–76. | ||
Andersson CK, Mori M, Bjermer L, Löfdahl CG, Erjefält JS. Alterations in lung mast cell populations in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(3):206–217. | ||
Ballarin A, Bazzan E, Zenteno RH, et al. Mast cell infiltration discriminates between histopathological phenotypes of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2012;186(3):233–239. | ||
Soltani A, Ewe YP, Lim ZS, et al. Mast cells in COPD airways: relationship to bronchodilator responsiveness and angiogenesis. Eur Respir J. 2012;39(6):1361–1367. | ||
Li H, Yang T, Ning Q, et al. Cigarette smoke extract–treated mast cells promote alveolar macrophage infiltration and polarization in experimental chronic obstructive pulmonary disease. Inhal Toxicol. 2015;27(14):822–831. | ||
Roos AB, Sandén C, Mori M, Bjermer L, Stampfli MR, Erjefält JS. IL-17A is elevated in end-stage chronic obstructive pulmonary disease and contributes to cigarette smoke–induced lymphoid neogenesis. Am J Respir Crit Care Med. 2015;191(11):1232–1241. | ||
Roos AB, Mori M, Gura HK, et al. Increased IL-17RA and IL-17RC in end-stage COPD and the contribution to mast cell secretion of FGF-2 and VEGF. Respir Res. 2017;18(1):48. | ||
Gosman MM, Postma DS, Vonk JM, et al. Association of mast cells with lung function in chronic obstructive pulmonary disease. Respir Res. 2008;9(1):64. | ||
Kosanovic D, Dahal BK, Peters DM, et al. Histological characterization of mast cell chymase in patients with pulmonary hypertension and chronic obstructive pulmonary disease. Pulm Circ. 2014;4(1):128–136. | ||
Mortaz E, Folkerts G, Redegeld F. Mast cells and COPD. Pulm Pharmacol Ther. 2011;24(4):367–372. | ||
Shaykhiev R, Crystal RG. Innate immunity and chronic obstructive pulmonary disease: a mini-review. Gerontology. 2013;59(6):481–489. | ||
Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. The Lancet. 2011;378(9795):1015–1026. | ||
Aurora AB, Baluk P, Zhang D, et al. Immune complex-dependent remodeling of the airway vasculature in response to a chronic bacterial infection. J Immunol. 2005;175(10):6319–6326. | ||
Mikko M, Forsslund H, Cui L, et al. Increased intraepithelial (CD103+) CD8+ T cells in the airways of smokers with and without chronic obstructive pulmonary disease. Immunobiology. 2013;218(2):225–231. | ||
Ravensberg AJ, Slats AM, van Wetering S, et al. CD8+ T cells characterize early smoking-related airway pathology in patients with asthma. Respir Med. 2013;107(7):959–966. | ||
Kim W-D, Chi H-S, Choe K-H, et al. A possible role for CD8+ and non-CD8+ cell granzyme B in early small airway wall remodelling in centrilobular emphysema. Respirology. 2013;18(4):688–696. | ||
Gadgil A, Duncan SR. Role of T-lymphocytes and pro-inflammatory mediators in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3(4):531–541. | ||
Majo J, Ghezzo H, Cosio MG. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J. 2001;17(5):946–953. | ||
Barczyk A, Pierzchała W, Kon OM, Cosio B, Adcock IM, Barnes PJ. Cytokine production by bronchoalveolar lavage T lymphocytes in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2006;117(6):1484–1492. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Vuga LJ, Tedrow JR, Pandit KV, et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;189(8):966–974. | ||
Bracke KR, Verhamme FM, Seys LJ, et al. Role of CXCL13 in cigarette smoke-induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(3):343–355. | ||
Duan MC, Zhang JQ, Liang Y, et al. Infiltration of IL-17-producing T cells and Treg cells in a mouse model of smoke-induced emphysema. Inflammation. 2016;39(4):1334–1344. | ||
Tan HL, Rosenthal M. IL-17 in lung disease: friend or foe? Thorax. 2013;68(8):788–790. | ||
Robinette ML, Colonna M. Innate lymphoid cells and the MHC. HLA. 2016;87(1):5–11. | ||
de Grove KC, Provoost S, Verhamme FM, et al. Characterization and quantification of innate lymphoid cell subsets in human lung. PLoS One. 2016;11(1):e0145961. | ||
Marashian SM, Mortaz E, Jamaati HR, et al. Role of innate lymphoid cells in lung disease. Iran J Allergy Asthma Immunol. 2015;14(4):346–360. | ||
Shikhagaie MM, Björklund Åsa K, Mjösberg J, et al. Neuropilin-1 is expressed on lymphoid tissue residing LTi-like group 3 innate lymphoid cells and associated with ectopic lymphoid aggregates. Cell Rep. 2017;18(7):1761–1773. | ||
Silver JS, Kearley J, Copenhaver AM, et al. Inflammatory triggers associated with exacerbations of COPD orchestrate plasticity of group 2 innate lymphoid cells in the lungs. Nat Immunol. 2016;17(6):626–635. | ||
Suzuki M, Sze MA, Campbell JD, et al. The cellular and molecular determinants of emphysematous destruction in COPD. Sci Rep. 2017;7(1):9562. | ||
Jeffery PK. Remodeling in asthma and chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(10 Pt 2):S28–S38. | ||
Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31(6):1334–1356. | ||
Reidel B, Radicioni G, Clapp P, et al. E-Cigarette use causes a unique innate immune response in the lung involving increased neutrophilic activation and altered mucin secretion. Am J Respir Crit Care Med. 2018;197(4):492–501. | ||
Rutgers SR, Postma DS, Ten Hacken NH, et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Chest. 2000;117(5):p. 262S. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.