")
Back to Journals » International Journal of General Medicine » Volume 15
Role of Hub Genes in the Occurrence and Development of Testicular Cancer Based on Bioinformatics
Authors Zhang C , Zhang W, Cui H, Zhang B, Miao P, Yang Q, Bai M, Jiao H, Chang D
Received 15 October 2021
Accepted for publication 23 December 2021
Published 18 January 2022 Volume 2022:15 Pages 645—660
DOI https://doi.org/10.2147/IJGM.S342611
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Chunlei Zhang,1,* Weijun Zhang,1,* Han Cui,2,* Bin Zhang,1,* Pengcheng Miao,1 Qi Yang,1 Mei Bai,1 Hongmei Jiao,1 Dehui Chang1
1Department of Urology, The 940 Hospital of Joint Logistics Support Force of Chinese PLA, Lanzhou, 730050, People’s Republic of China; 2Department of the First Clinic, The 940 Hospital of Joint Logistics Support Force of Chinese PLA, Lanzhou, 730050, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Dehui Chang; Hongmei Jiao Email [email protected]; [email protected]
Background: Testicular cancer severely affects male health, so finding effective diagnosis and prognostic indicators and exploring its pathogenesis are very important.
Purpose: This study aims to explore the hub genes that play important roles in the occurrence and development of testicular germ cell tumor (TGCT).
Methods: Data were obtained from Gene Expression Omnibus datasets (GSE3218 and GSE1818) and verified in The Cancer Genome Atlas database and the Genotype-Tissue Expression database and the Human Protein Atlas database. A protein–protein interaction network was constructed to obtain hub genes. GEO2R, R software and packages were used to analyze differentially expressed genes (DEGs), receiver operating characteristic curve assessment, Cox regression analysis, Kaplan–Meier survival curve assessment, Gene Ontology analysis, Kyoto Encyclopedia of Genes and Genomes analysis, the relationship with clinicopathological information, gene set enrichment analysis, the correlation with immune cells’ infiltration, and the expression in pan-cancers of the hub genes.
Results: PLK4, TRIP13, TPR, KIF18A, CDKN3, HMMR, PBK, PTTG1, CKS2, SYCP1, HSPA2, and MKI67 were selected as the hub genes. mRNA of PLK4, TRIP13, CDKN3, SYCP1, HSPA2, and MKI67 had high diagnostic values, and higher expression of CDKN3 and HSPA2 mRNA were poor prognostic factors for progression-free interval of TGCT. The hub genes involved organelle division and cell cycle, chromosome and centromeric region, heat shock protein binding, and more. Downregulated TPR and PLK4 were selected as research targets for continued study, and they may participate in multiple signaling pathways. The expression of TPR and PLK4 correlated with the infiltration of a variety of immune cells and differed in pan-cancers.
Conclusion: The mRNA levels of multiple hub genes have high diagnostic and prognostic values for TGCT. TPR and PLK4 may play a role in the occurrence and development of TGCT through cancer-related signaling pathways.
Keywords: testicular cancer, mRNA, hub gene, diagnosis, prognosis, biomarker, immune infiltration, signal pathway
Introduction
Testicular cancer accounts for 1% of newly diagnosed cancers in men worldwide. It is one of the most common cancers between the ages of 14 and 44 years for men in western countries, and its incidence has gradually increased in recent years.1,2 According to 2020 estimates by the American Cancer Society, there will be 1,806,590 new cancer cases and 606,520 cancer deaths in the United States (US), including 9,610 cases of testicular cancer and 440 deaths resulting from testicular cancer.3 Estimated cancer statistics for US adolescents and young adults in 2020 from Leading Sites of New Cancer Cases in Adolescents and Young Adults shows that the proportion of the number of cases with testicular seminoma ranks second among the 20–29-year-old male patients.4 Because testicular cancer is relatively rare, few clinical reports exist on the topic. Currently, alpha-fetoprotein (AFP) and human chorionic gonadotropin (HCG) are molecular diagnostic markers for testicular cancer; however, because of their low specificity and the insignificant increases in serum for most patients with seminoma, challenges still exist in the screening and diagnosis of testicular cancer, especially pure seminoma.5 In addition, no good prognostic indicators exist for testicular cancer. Germ cell tumors are particularly sensitive to cisplatin-based chemotherapy, which greatly improves the patient’s long-term survival rate. However, 40–60% of the patients with germ cell tumor fail to benefit from salvage chemotherapy and have a poor prognosis. The exploration of new targeted drug therapy is indispensable for these patients.6
At present, testicular cancer lacks ideal biomarkers with high sensitivity and specificity in diagnosis, treatment, and prognosis. Therefore, it is important to use bioinformatics methods to find clinically applicable biomarkers. Public databases, such as the Gene Expression Omnibus (GEO) database and The Cancer Genome Atlas (TCGA), offer methods to study the gene spectrum and pathogenesis of tumor diseases. By extracting data, we can obtain differentially expressed genes (DEGs) in disease, predict the genes related to the disease, and analyze the possible mechanism of these genes in the process of disease. Cancer is a genomic disease that is characterized by genome instability, and a large number of point mutations and structural changes appear during tumor progression. Genomic mutation may produce tumor antigens, which may be recognized by the immune system as non-self antigens and trigger cellular immune responses. The immune system plays a vital role in immune surveillance, because immune cells of the adaptive immune system and the innate immune system can penetrate into the tumor microenvironment and help regulate tumor progression.7,8 However, current research on testicular cancer is relatively one sided and is focused on the expression of a certain gene; it lacks comprehensive, systematic, and in-depth analysis of testicular cancer sequencing results in existing databases to explore molecular markers for the diagnosis and prognosis of testicular cancer and to explore hub genes that may play a role in the development. Current efforts also fail to effectively combine and analyze the only two datasets (GSE3218 and GSE1818) for mRNA sequencing of testicular cancer tissues and normal testicular tissues. Moreover, at present, few studies have explored the correlation between gene expression and immune infiltration in testicular cancer. To make up for these shortcomings, in this study, we selected hub genes that may be involved in the occurrence and development of testicular cancer through bioinformatics analysis combined with the two datasets. As the most common testicular cancer is testicular germ cell tumor (TGCT), we used data of TGCT obtained from TCGA and Genotype-Tissue Expression (GTEx) database to explore the function of hub genes. In addition to analyzing the relationship between these genes and patient clinicopathological information, the signal pathways that may be involved, and the diagnosis and prognostic value of corresponding mRNA, we also analyzed the relationship between these genes and immune cells' infiltration in TGCT, to explore genes as diagnostic and prognostic markers and hub molecules that may be selected as therapeutic targets.
Materials and Methods
Data Collection
The data on differentially expressed genes (DEGs) came from two datasets—GSE3218 and GSE1818—downloaded from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database.9 The gene expression profile of GSE3218, including 101 testicular cancer tissues and 5 normal tissues, was assessed by the GPL96 [HG-U133A] Affymetrix Human Genome U133A Array platform. The gene expression profile of GSE1818, including 20 testicular cancer tissues and 3 normal tissues, was assessed by the GPL885 Agilent-011521 Human 1A Microarray G4110A (Feature Number version) platform. For verification of DEGs, data in the UCSC XENA database (https://xenabrowser.net/datapages/)10 and RNA sequencing (RNAseq) in TPM format from TCGA and GTEx databases,11,12 processed uniformly by the Toil process,13 were extracted; data sources included 154 cases of testicular cancer tissue samples in TCGA and 165 cases of normal testicular tissue samples in the GTEx database. The pan-cancer analysis included 10,363 cancer tissue samples and 5,413 paracancerous and normal tissue samples in TCGA and the GTEx database. The RNAseq data in the TPM format were log2 transformed and compared between samples. The pan-cancer groups included adrenocortical carcinoma (ACC); bladder urothelial carcinoma (BLCA); breast invasive carcinoma (BRCA); cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC); cholangiocarcinoma (CHOL); colon adenocarcinoma (COAD); lymphoid neoplasm diffuse large b-cell lymphoma (DLBC); esophageal carcinoma (ESCA); glioblastoma multiforme (GBM); head and neck squamous cell carcinoma (HNSC); kidney chromophobe (KICH); kidney renal clear cell carcinoma (KIRC); kidney renal papillary cell carcinoma (KIRP); acute myeloid leukemia (LAML); brain lower grade glioma (LGG); liver hepatocellular carcinoma (LIHC); lung adenocarcinoma (LUAD); lung squamous cell carcinoma (LUSC); mesothelioma (MESO); ovarian serous cystadenocarcinoma (OV); pancreatic adenocarcinoma (PAAD); pheochromocytoma and paraganglioma (PCPG); prostate adenocarcinoma (PRAD); rectum adenocarcinoma (READ); sarcoma (SARC); skin cutaneous melanoma (SKCM); stomach adenocarcinoma (STAD); testicular germ cell tumors (TGCT); thyroid carcinoma (THCA); thymoma (THYM); uterine corpus endometrial carcinoma (UCEC); uterine carcinosarcoma (UCS); uveal melanoma (UVM).
Identification of DEGs
GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/)14 was used to identify DEGs between testicular cancer samples (including seminoma and nonseminoma) and normal samples using R 3.2.3, Biobase 2.30.0, GEOquery 2.40.0, and Limma 3.26.8. The cutoff criterion was an adjusted p<0.05.
Software and Packages for Analysis Between Gene Expression and Clinicopathological Information
R software (version 3.6.3, for statistical analysis and visualization) was used for differential expression analysis. The R packages were ggplot2 (version 3.3.3, for visualization) and the basic R package. The pROC package (version 1.17.0.1, for analysis) and ggplot2 package (version 3.3.3, for visualization) were used for ROC curve construction. The Survminer package version 0.4.9, for visualization) and the survival package (version 3.2–10, for statistical analysis of survival data) were used for prognostic analysis, and the survival package (version 3.2–10, for statistical analysis of survival data) was used for Cox regression analysis. Supplementary data were obtained from the study of Liu et al.15 The clusterProfiler package (version 3.14.3) was used for gene set enrichment analysis (GSEA); the reference gene set was c2.cp.v7. 2. symbols.gmt (curated), and the gene set database was MSigDB Collections.16,17 The false discovery rate (FDR) was <0.25, and adjusted p values <0.05 were considered significant. Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were based on clusterProfiler package [version 3.14.3] (for enrichment analysis) and org.Hs.eg.db package [version 3.10.0] (for ID conversion). The GSVA package [version 1.34.0] was used to analyze the correlation between gene expression and immune cells' infiltration.18 The immune infiltration algorithm was ssGSEA. Immune cells included aDC (activated DC); B cells; CD8 T cells; cytotoxic cells; DC; eosinophils; iDC (immature DC); macrophages; mast cells; neutrophils; NK CD56bright cells; NK CD56dim cells; NK cells; pDC (plasmacytoid DC); T cells; T helper cells; Tcm (T central memory); Tem (T effector memory); Tfh (T follicular helper); Tgd (T gamma delta); Th1 cells; Th17 cells; Th2 cells; Treg. Immune cell markers referred to the study of Bindea et al.19
Protein Interaction Network Construction and Screening of Hub Genes
The construction of a PPI network relied on the STRING database (version 11.5 https://www.string-db.org/).20 The minimum required interaction score was 0.4 and maximum number of interactors was 5. Hubba plug-in in Cytoscape software was used to select top 20 hub nodes ranked by degree. Functional gene clusters were constructed via MCODE software. Network scoring degree cutoff was 2, node score cutoff was 0.2, K-core was 2, and Max. depth was 100.
Protein Immunohistochemical Expression Analysis
The extraction of immunohistochemical results relied on the Human Protein Atlas database (https://www.proteinatlas.org/).21 The scoring method of immunohistochemistry results was intensity plus quantity. For intensity, negative, weak, medium, and strong corresponded to 0, 1, 2, and 3 points, respectively; for quantity, negative, <25%, 25–75%, and >75% corresponded to 0, 1, 2, and 3 points, respectively.
Statistical Analysis
The Mann–Whitney U-test or t test was used to analyze the difference of gene expression in samples according to whether the data were normally distributed based on the normality test. The chi-squared test and Fisher’s exact test were used to analyze the difference between clinicopathological information groups according to whether the data met the conditions of theoretical frequency >5 or total sample size >40. The Log rank test was used in the Kaplan-Meier prognostic analysis, and the prognostic factors of TGCT were analyzed by Cox regression analysis. The Spearman correlation coefficient or the Pearson correlation coefficient was calculated in correlation analysis according to whether the result of the Shapiro–Wilk normality test satisfied normal distribution. Nonsignificance (ns) was set as p≥0.05; p<0.05 was considered statistically significant.
Results
Screening for Differentially Expressed Genes
The two datasets, GSE3218 and GSE1818, in the GEO database were selected to analyze the DEGs between testicular cancer and normal tissues. Specimens represented 101 adult male germ cell cancers (including choriocarcinoma, embryonal carcinoma, seminoma, teratoma, and yolk sac tumor), and 5 normal testis specimens were obtained from the GSE3218 dataset; there was a relatively high degree of discrimination between the two groups of data (Figure 1A). A total of 15,518 genes were identified, and 6,765 genes were differentially expressed (adjusted p<0.05, Figure 1B), of which 3,027 genes were upregulated and 3,738 were downregulated in cancer tissues compared with normal tissues (Figure 1C). Twenty male germ cell cancer specimens (including choriocarcinoma, embryonal carcinoma, intratubular germ cell tumor, seminoma, teratoma, and yolk sac tumor) and 3 normal testis specimens were in the GSE1818 dataset. The distinction between the two groups of data was relatively high (Figure 1D), and 14,379 genes were identified; 944 genes were differentially expressed (adjusted p<0.05, Figure 1E), of which 81 were upregulated and 863 were downregulated in cancer tissues compared with normal tissues (Figure 1F).
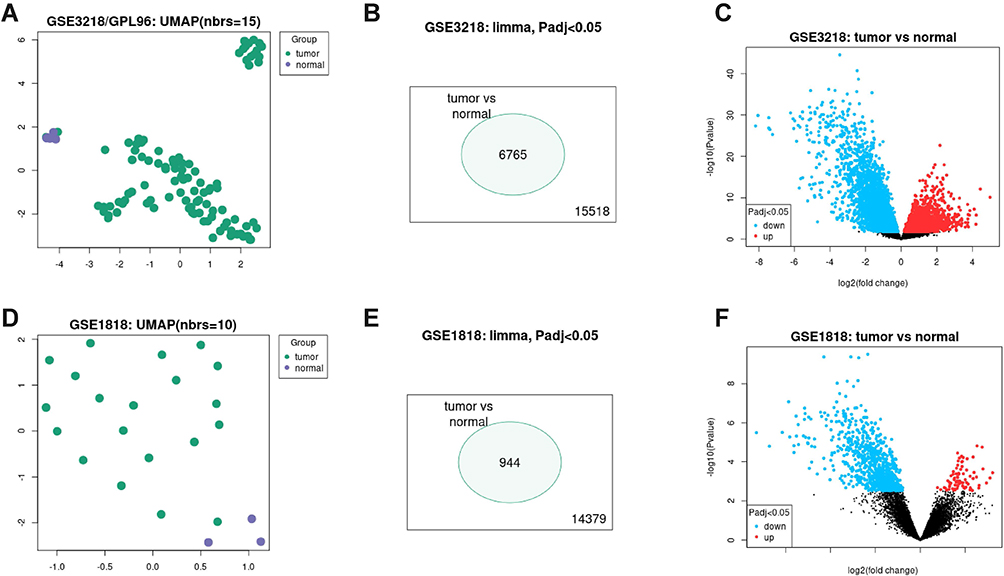 |
Figure 1 Identification of differentially expressed genes from GSE3218 and GSE1818. (A) UMAP of GSE3218; (B) a total of 15,518 genes were identified, and 6,765 genes were differentially expressed in GSE3218 (adjusted p<0.05); (C) 3,027 genes were upregulated, and 3,738 genes were downregulated in cancer tissues compared with control in GSE3218; (D) UMAP of GSE1818; (E) a total of 14,379 genes were identified, and 944 genes were differentially expressed in GSE1818 (adjusted p<0.05); (F) 81 were upregulated, and 863 were downregulated in cancer tissues compared with control in GSE1818. |
Screening for Hub Genes
The differentially expressed and co-expressed genes with log|fold change|>1 and adjusted p<0.05 were selected in the two datasets. Of the results, 32 genes were upregulated and 328 genes were downregulated in cancer tissues compared with normal tissues (Figure 2A and B); all DEGs are shown in Table 1. To screen for hub genes, we constructed a PPI network of these genes and selected the top 20 hub nodes ranked by degree with Hubba software (Figure 3A) and the top 2 functional gene clusters ranked by MCODE score with MCODE software (Figure 3B and C). Twelve hub genes were used as the target genes for the next analysis by taking the intersection; these genes were PLK4, TRIP13, TPR, KIF18A, CDKN3, HMMR, PBK, PTTG1, CKS2, SYCP1, HSPA2, and MKI67. Only the mRNA of MKI67 was upregulated in testicular cancer tissues. To verify the accuracy of the differential expression of these genes, we used data from 154 TGCT tissue samples in TCGA and from 165 normal testicular tissue samples in the GTEx database to analyze the mRNA expression level. The results of gene differential expression trends were consistent with the results from the GEO database (Figure 4). In addition, we analyzed the protein expression of these genes in normal testicular tissues and in cancer tissues using the Human Protein Atlas database, and immunohistochemical scores of these proteins in the two groups were compared. The results showed that PLK4, TRIP13, TPR, HMMR, PBK, PTTG1, SYCP1, and HSPA2 were downregulated in cancer tissues (all p<0.05), and there was no significant difference in the expression of KIF18A, CKS2 and MKI67, CDKN3 data could not be found in the database (Figure 5).
 |
Table 1 Differentially Expressed Genes |
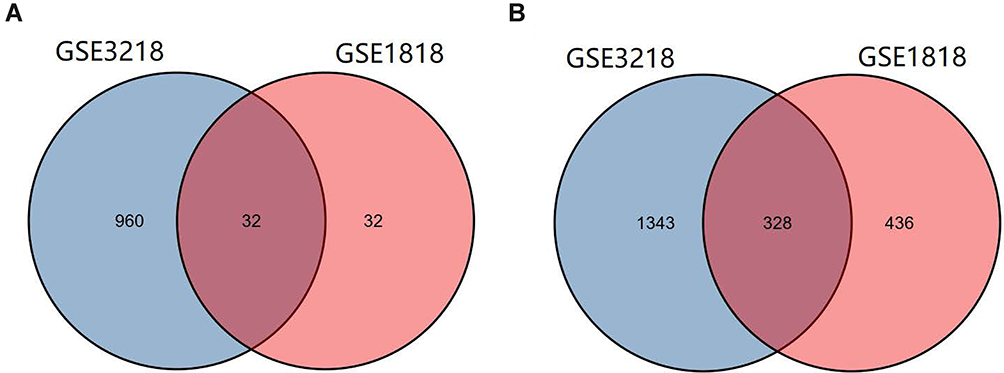 |
Figure 2 Identification of common genes from differentially expressed genes in GSE3218 and GSE1818. (A and B) 32 genes were upregulated, and 328 genes were downregulated (log |fold change|>1, adjusted p<0.05). |
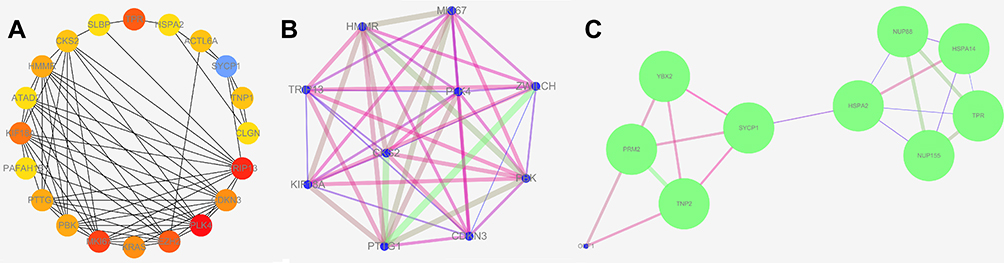 |
Figure 3 Screen for hub genes. (A) Top 20 hub nodes ranked by degree with Hubba software; (B and C) top 2 functional gene clusters ranked by MCODE score with MCODE software. |
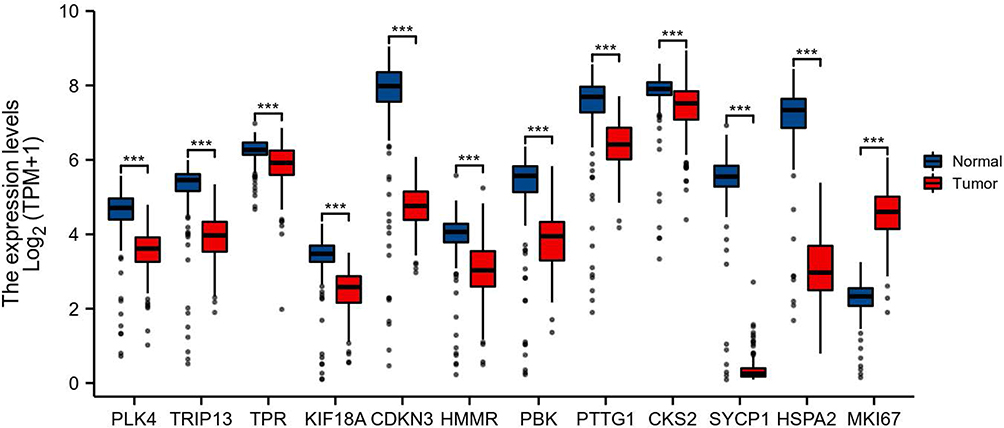 |
Figure 4 mRNA expression of PLK4, TRIP13, TPR, KIF18A, CDKN3, HMMR, PBK, PTTG1, CKS2, SYCP1, HSPA2, and MKI67 in 154 TGCT cancer tissue samples in TCGA database and 165 normal testicular tissue samples in the GTEx database. ***p<0.001. |
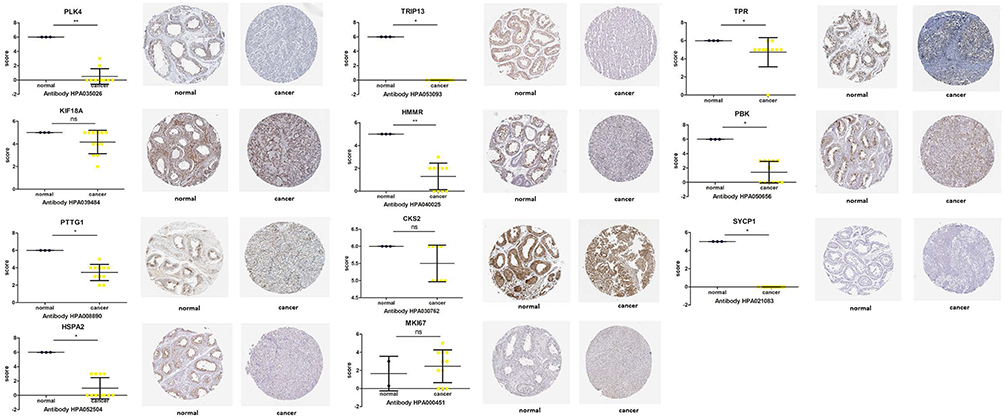 |
Figure 5 Protein immunohistochemical analysis of PLK4, TRIP13, TPR, KIF18A, HMMR, PBK, PTTG1, CKS2, SYCP1, HSPA2, and MKI67 in TGCT and normal tissues from the Human Protein Atlas database. Nonsignificant (ns): p≥0.05; *p<0.05; **p<0.01. |
Diagnostic and Prognostic Value of Hub Gene mRNA
The data of 139 TGCT samples with relatively complete clinicopathological information in TCGA were analyzed for the clinically related indicators (and clinicopathological information is shown in Table 2). The ROC curve was constructed for mRNA expression of these hub genes in TGCT and normal tissues. The results showed that the mRNA of PLK4, TRIP13, CDKN3, SYCP1, HSPA2, and MKI67 had high diagnostic value, with AUCs of 0.906, 0.926, 0.924, 0.986, 0.972, and 0.989, respectively (Figure 6). In the prognostic analysis, the progression-free interval was used as an endpoint because of the high censorship rate in patients with overall survival and disease-specific survival. Unfortunately, only high expression of CDKN3 (p=0.034, Figure 7) and HSPA2 (p=0.033, Figure 7) mRNA were factors for the poor prognosis of TGCT. We used univariate and multivariate Cox regression analyses to determine whether these hub gene mRNAs were independent risk factors for poor prognosis in terms of progression-free interval. The results showed that pathologic N stage and serum tumor markers were independent risk factors (all p<0.05). Although CDKN3 and HSPA2 mRNA also affected the prognosis of patients in univariate analysis (all p<0.05), the results of multivariate analysis did not support this conclusion (all p>0.05, Table 3).
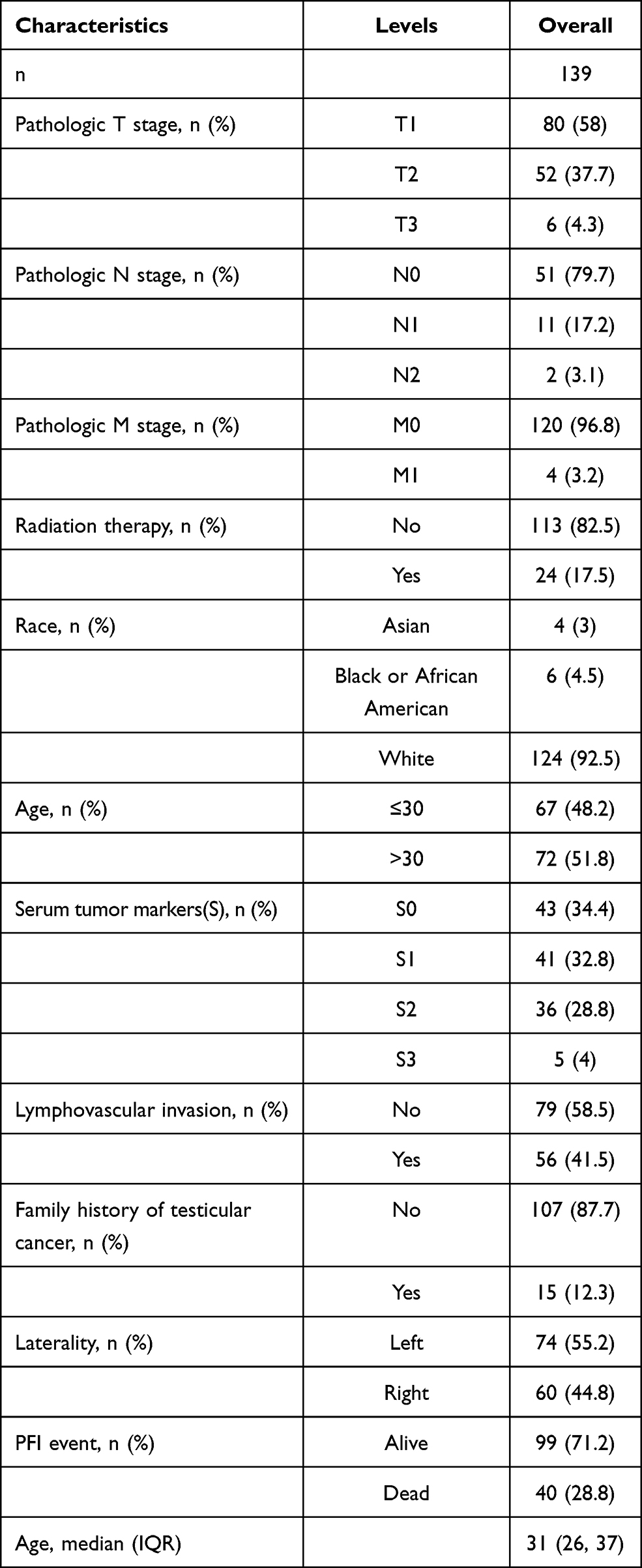 |
Table 2 Clinicopathological Information of TGCT Patients |
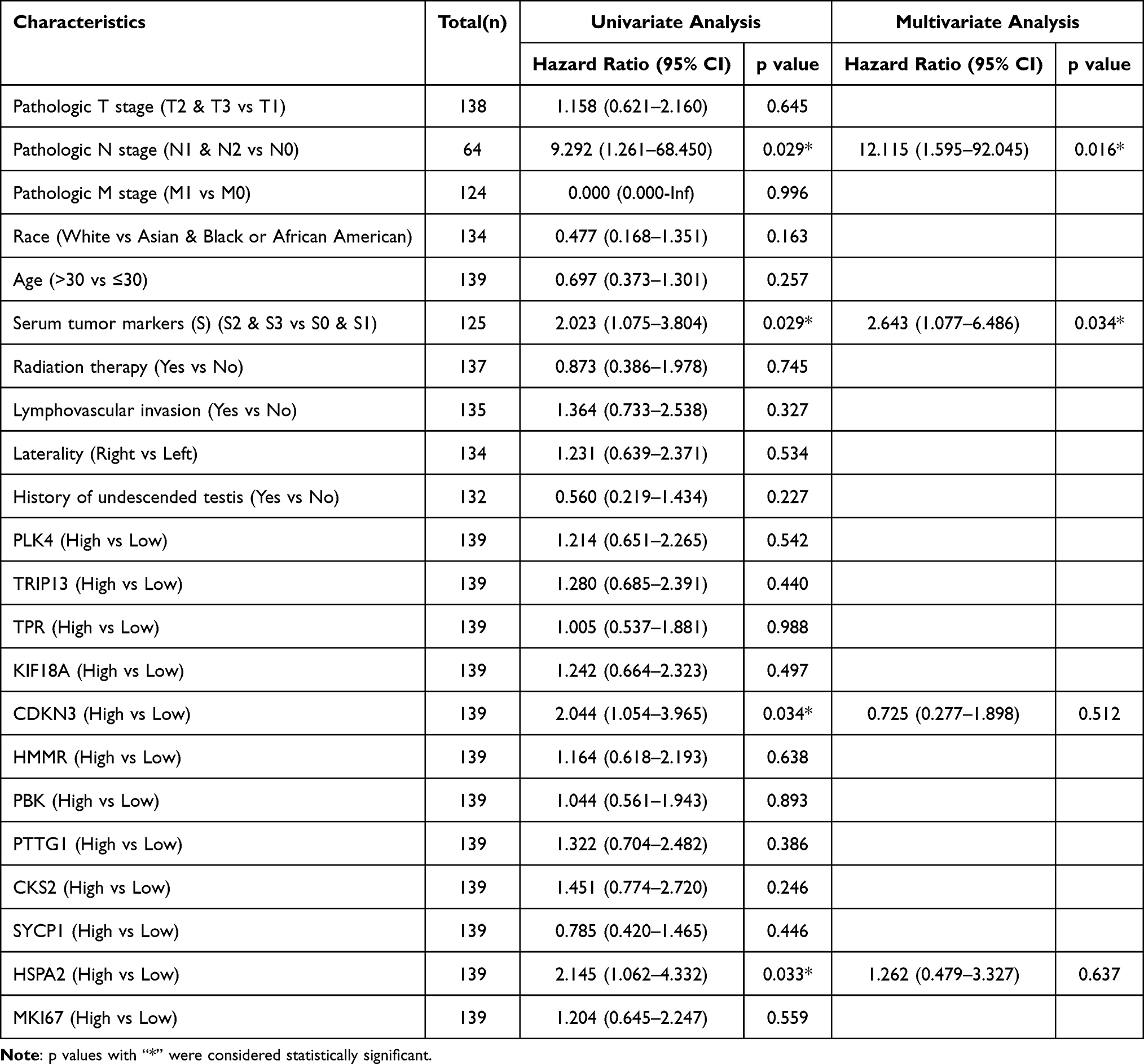 |
Table 3 Associations Between Clinicopathological Characteristics and PFI in TGCT Patients Using Cox Regression Analysis |
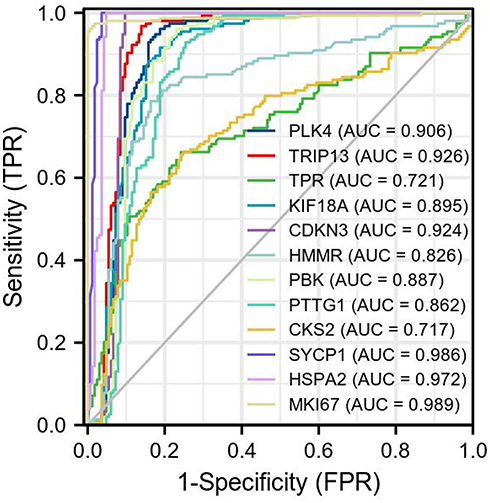 |
Figure 6 ROC curve of mRNA of PLK4, TRIP13, TPR, KIF18A, CDKN3, HMMR, PBK, PTTG1, CKS2, SYCP1, HSPA2, and MKI67 in TGCT and normal tissues. The abscissa is the false-positive rate (FPR), and the ordinate is the true-positive rate (TPR). |
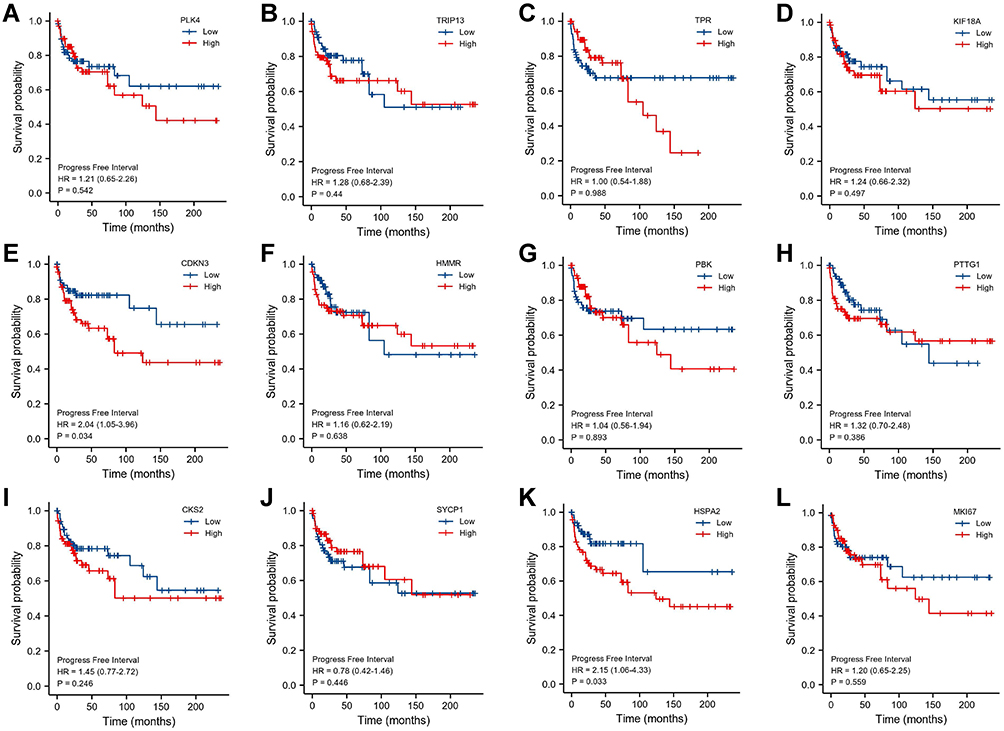 |
Figure 7 Kaplan–Meier curve of mRNA of (A) PLK4, (B) TRIP13, (C) TPR, (D) KIF18A, (E) CDKN3, (F) HMMR, (G) PBK, (H) PTTG1, (I) CKS2, (J) SYCP1, (K) HSPA2, and (L) MKI67 in patients with TGCT. |
Relationship Between Hub Genes and Clinicopathological Characteristics in Patients with TGCT
The results showed that, in samples with higher T stage, the mRNA of HMMR and PTTG1 was overexpressed, and the mRNA of TPR was downregulated (all p<0.05). In samples with lymphatic metastasis, the mRNA of TPR and PLK4 was downregulated (all p<0.05), and no differential gene mRNA was found between M0 and M1 samples (all p>0.05). In the group with higher serum tumor markers, the mRNA of CDKN3, HMMR, and PTTG1 was overexpressed, and no mRNA was downregulated (all p<0.05, Figure 8).
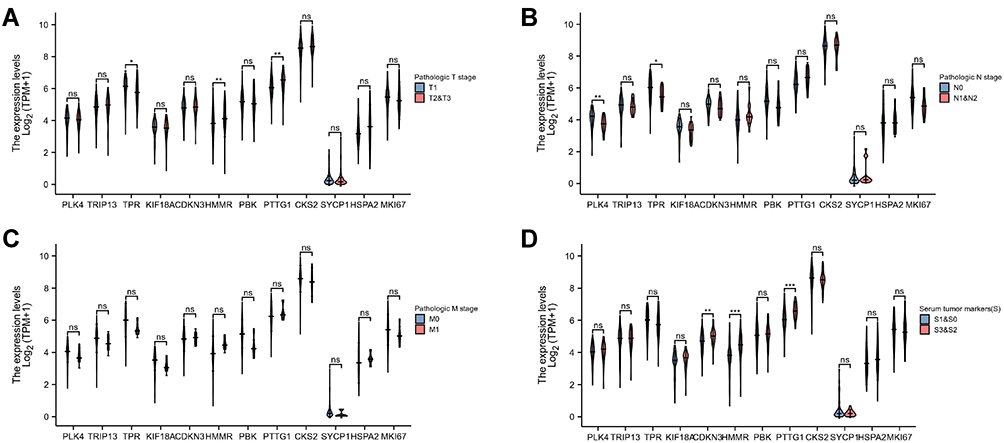 |
Figure 8 mRNA expression of PLK4, TRIP13, TPR, KIF18A, CDKN3, HMMR, PBK, PTTG1, CKS2, SYCP1, HSPA2, and MKI67 in different clinicopathological groups of patients with TGCT. (A) pathologic T stage; (B) pathologic N stage; (C) pathologic M stage; (D) serum tumor markers. Nonsignificant (ns): p≥0.05; *p<0.05; **p<0.01; ***p<0.001. |
Mechanism of Hub Genes in the Occurrence and Development of TGCT
Because the protein expression of KIF18A, CKS2 and MKI67 was not significantly different between cancer tissues and normal tissues, the remaining 9 genes were selected for functional analysis. Gene Ontology (GO) analysis results indicated that biological processes included meiotic nuclear division, nuclear division, meiotic cell cycle process, and organelle fission; the cellular components included male germ cell nucleus, germ cell nucleus, chromosome, centromeric region, and synaptonemal complex; and the molecular functions included heat shock protein binding, microtubule plus-end binding, ATPase activity, coupled, and hyaluronic acid binding (Figure 9). Unfortunately, no Kyoto Encyclopedia of Genes and Genomes (KEGG) signaling was matched to these genes. Among all downregulated DEGs in cancer tissues, only TPR was downregulated in the higher T-stage group and the lymphatic metastasis group, and PLK4 was downregulated in the lymphatic metastasis group. The mRNA expression trend was consistent with the protein expression. Therefore, TPR and PLK4 were chosen for additional functional mechanism analysis. GSEA analysis showed that the signal pathways that TPR may participate in were NABA_MATRISOME and REACTOME_METABOLISM_OF_LIPIDS (Figure 10A), and the signal pathways that PLK4 may participate in were NABA_MATRISOME and REACTOME_DEVELOPMENTAL_BIOLOGY (Figure 10B).
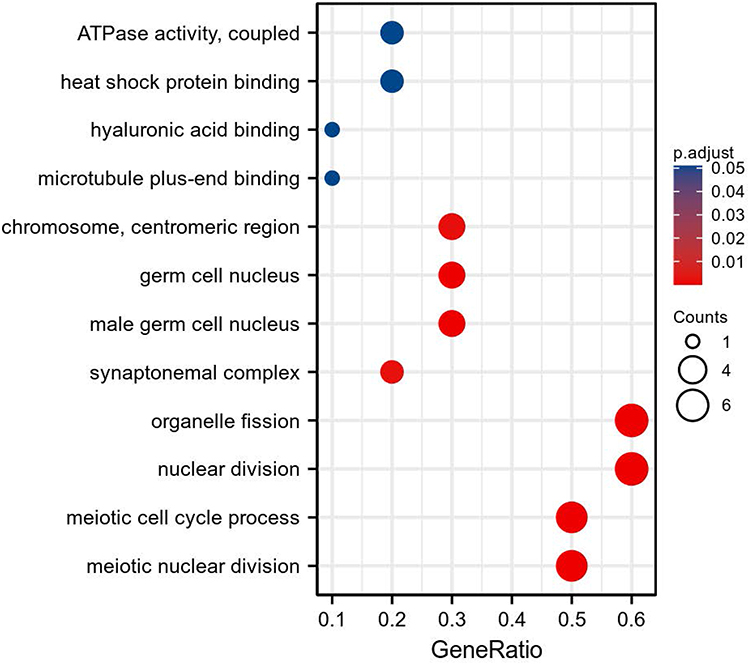 |
Figure 9 GO analysis of PLK4, TRIP13, TPR, CDKN3, HMMR, PBK, PTTG1, SYCP1, and HSPA2. |
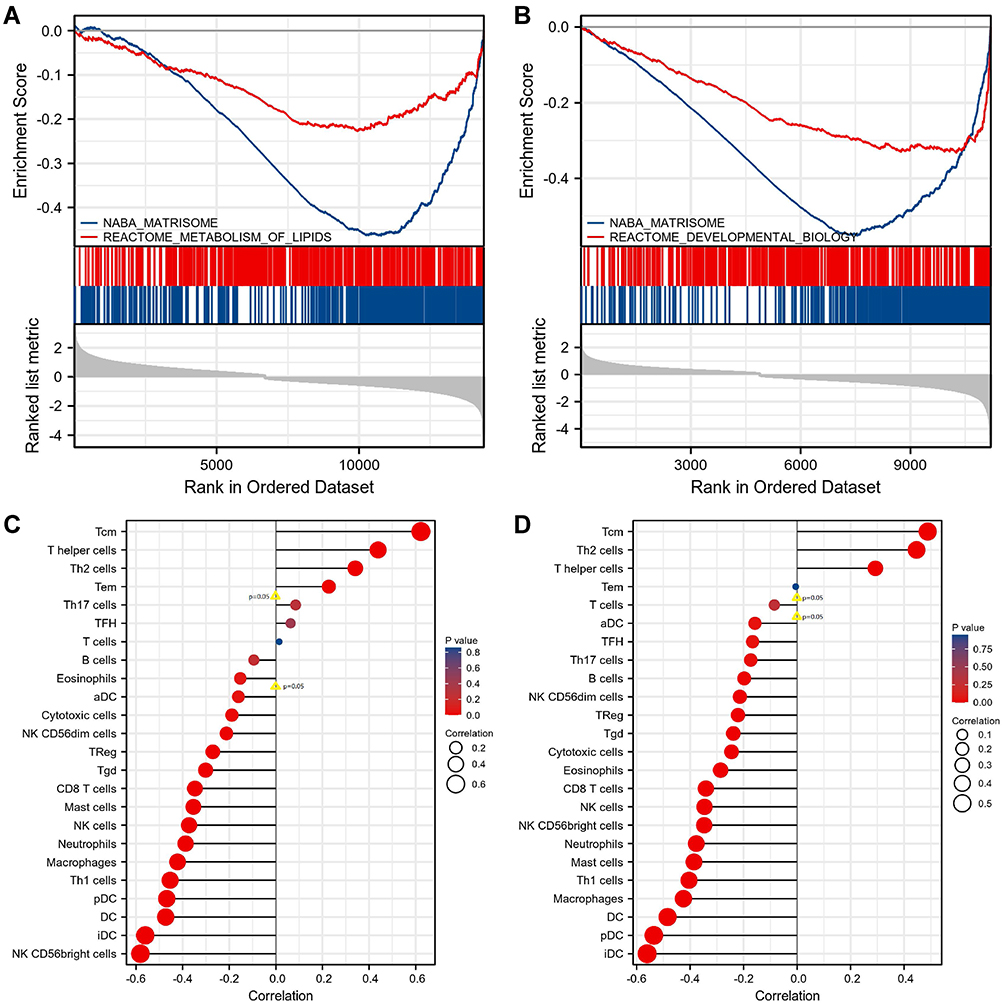 |
Figure 10 GSEA analysis and correlation with immune cell infiltration of mRNA of TPR/PLK4 in TGCT. (A) TPR may participate in NABA_MATRISOME and REACTOME_METABOLISM_OF_LIPIDS. (B) PLK4 may participate in NABA_MATRISOME and REACTOME_DEVELOPMENTAL_BIOLOGY. (C) mRNA of TPR and immune cells' infiltration. (D) mRNA of PLK4 and immune cells' infiltration. |
Analysis of Hub Genes Involved in Immune Cell Infiltration in TGCT
After analyzing the relationship between hub genes and immune cell infiltration in TGCT, we found that immune cells with infiltration that positively correlated with TPR expression were Tcm, T helper cells, Th2 cells, and Tem (all p<0.05, Figure 10C); those that negatively correlated with TPR expression were aDC, cytotoxic cells, NK CD56dim cells, TReg, Tgd, CD8 T cells, mast cells, NK cells, neutrophils, macrophages, Th1 cells, pDC, DC, iDC, and NK CD56bright cells (all p<0.05, Figure 10C). Immune cells with infiltration degrees that positively correlated with the expression of PLK4 were Tcm, Th2 cells, and T helper cells (all p<0.05, Figure 10D), and those with infiltration degrees that negatively correlated with the expression of PLK4 were aDC, TFH, Th17 cells, B cells, NK CD56dim cells, TReg, Tgd, cytotoxic cells, eosinophils, CD8 T cells, NK cells, NK CD56bright cells, neutrophils, mast cells, Th1 cells, macrophages, DC, pDC, and iDC (all p<0.05, Figure 10D).
Expression of Hub Genes in Pan-Cancer
Overall, 33 related samples of cancer tissues were included in the gene expression in pan-cancer analysis. The results showed that, compared with normal tissues, TPR was differentially expressed in 24 cancer tissues, of which it was upregulated in all cancers except ACC, KICH, OV, TGCT, and THCA (all p<0.05, Figure 11A). Compared with normal tissues, PLK4 was downregulated in TGCT and LAML, and was upregulated in almost all the other cancers (all p<0.05, Figure 11B).
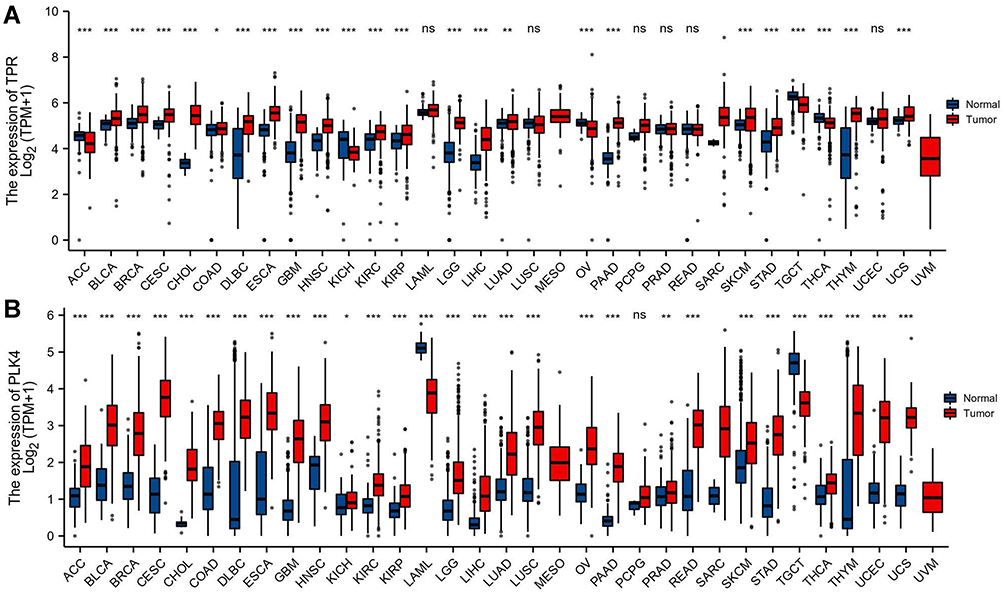 |
Figure 11 Expression of mRNA of TPR and PLK4 in pan-cancer. (A) Expression of mRNA of TPR in pan-cancer; (B) expression of mRNA of PLK4 in pan-cancer. Nonsignificant (ns): p≥0.05; *p<0.05; **p<0.01; ***p<0.001. |
Discussion
Although the incidence of testicular cancer is increasing year by year, its proportion is not high among all tumors; however, it seriously affects male health, including fertility, especially that of young men.1–4 Therefore, finding effective diagnosis and prognostic indicators and exploring its pathogenesis to seek treatment targets are important research goals. However, the current basic and translational research on testicular cancer is relatively scarce. Using bioinformatics analysis technology to assess the sequencing results of tissue samples in public databases, such as the GEO database, TCGA, and the GTEx database, provides a convenient way to obtain gene expression data and clinical information about patients. In this study, we used the tissue sequencing information of testicular cancer in the two datasets GSE3218 and GSE1818 from the GEO database to select 32 upregulated genes and 328 downregulated genes in testicular cancer tissues to compare with normal tissues through bioinformatics analysis. For additional study, we constructed a PPI network and screened out 12 hub genes as research targets: PLK4, TRIP13, TPR, KIF18A, CDKN3, HMMR, PBK, PTTG1, CKS2, SYCP1, HSPA2, and MKI67. The mRNA expression of these DEGs was verified in TGCT tissues from TCGA and in normal testis tissues from the GTEx database. We found via immunohistochemical analysis that mRNA expression was not completely consistent with the protein expression. For 3 genes, protein expression was not differential between cancer and normal tissues. One possible reason is that the process of mRNA translation into protein may be regulated by noncoding RNA, including circRNA, lncRNA, and miRNA,22–24 or may be subject to epigenetic modifications, such as m6A modification.25 Another reason may be the complex composition of normal testicular tissue, including spermatogonia, spermatocytes, and more. There was cellular heterogeneity in the expression of these genes in testicular tissues, but tumor cells are usually derived from the same type of cell. For example, CKS2 expresses in round or early spermatids in normal testicular tissues and not in other cell types, but it is uniformly expressed in cancer tissues.
mRNA carries genetic information and is a direct template for instructing protein biosynthesis. It links the genetic information in DNA with the translation and expression of proteins, and it plays a vital role in all cells, including tumor cells. The mRNA expression level can reflect the expression of hub genes to a certain extent and often can distinguish diseases and disease subgroups as a gene transcription manuscript.26–28 Therefore, mRNA has the potential to be used as a diagnostic and prognostic molecular of diseases, including cancer. The AUCs of mRNA of PLK4, TRIP13, CDKN3, SYCP1, HSPA2, and MKI67 in this study were all above 0.9, and this measure can distinguish cancer tissues from normal tissues well. Therefore, these genes have good potential to be diagnostic molecular markers. In the prognostic analysis, mRNA for CDKN3 and HSPA2 was a poor prognostic factor for the progression-free interval in TGCT; however, the Cox regression analysis did not indicate CDKN3 and HSPA2 mRNA expression but suggested that pathologic N stage and serum tumor markers were independent risk factors for the poor prognosis of TGCT. These results were consistent with the conclusions that lymphadenectomy can bring benefits to patients and that serum tumor markers can predict the prognosis of patients.29,30
In this study, we found that the mRNA expression of almost all hub genes, except MKI67, in cancer tissues was lower than that in normal tissues; but many of the genes were upregulated in higher tumor grades. For example, the mRNA of HMMR and PTTG1 was overexpressed in samples with higher T stages, and the mRNA of CDKN3, HMMR, and PTTG1 was overexpressed in the group with higher serum tumor markers. A possible explanation is that downregulation of genes is the initiating factor of TGCT, but the expression level gradually increases during the disease progression or as a result of other regulation of oncogene expression. Another possible explanation is that these genes may be related to the infiltration of immune cells, whereas the result of tissue sequencing referred to all the components, and the high expression of these genes in immune cells leads to genes higher expression.31 Immune cells also play an important role in the disease process of cancer. Innate immune cells are composed of natural killer (NK) cells, eosinophils, basophils, and phagocytes (including mast cells, neutrophils, monocytes, macrophages, and dendritic cells), and they participate in cancer suppression by directly killing tumor cells or triggering an adaptive immune response. The adaptive immune system works through lymphocytes (including B cells and T cells), among which B cells play a major role in humoral immune responses, whereas T cells participate in cell-mediated immune responses.32 The expression of genes may affect the immune cells’ infiltration in cancer. In this study, we found that the expression of the selected genes PLK4 and TPR was related to the content of specific immune cells, which may be used as interference targets for immunotherapy.
To analyze the role of hub genes in TGCT, we performed GO analysis and KEGG analysis on these genes. Although no signal pathway was matched to KEGG analysis, we obtained some important GO analysis results. For example, we found that biological processes involved organelle division and cell cycle, and molecular functions involved chromosome and centromeric region. The proper coordination of centromeric chromatin dynamics is essential in the cell cycle,33 and previous studies have shown that the level of centromeric transcripts is elevated in some tumors.34 Biological processes involve heat shock protein (HSP) binding. HSP is a large class of proteins involved in protein folding and maturation. Its expression is induced by heat shock or other stress factors,35 and it plays an important role in cell proliferation, differentiation, and carcinogenesis.36 HSP is the research target for cancer development and treatment.37 Therefore, these biological functions may be the mechanism of action of these hub genes in testicular cancer. To avoid ambiguity, we selected PLK4 and TPR as the target genes that may be involved in the mechanism of TGCT for additional research; the mRNA and protein of each gene were downregulated in cancer tissues and in higher-grade cancer tissues. GSEA was used to analyze the signal pathways in which a single gene may participate. The results showed that PLK4 may be involved in the signaling pathways NABA_MATRISOME and REACTOME_DEVELOPMENTAL_BIOLOGY and that TPR may be involved in the signaling pathways NABA_MATRISOME and REACTOME_METABOLISM_OF_LIPIDS to regulate tumor progression. Lipid metabolism reprogramming not only can be used as a molecular marker of tumor but also can affect the proliferation of tumor cells.38,39 Whether TPR functions through lipid metabolism is worthy of further study.
Polo-like kinase 4 (PLK4) is a serine threonine kinase that is located in the centrioles throughout the cell cycle and is essential for centriole replication.40 It promotes cell proliferation, invasion, and migration in cancers through its important role in the centrioles.41–44 Interestingly, PLK4 was specifically downregulated in TGCT and LAML, but its expression was relatively high in normal testicular tissue compared to other organ tissues, even higher than other malignancies. This phenomenon shows that PLK4 mRNA can be effectively used as a diagnostic indicator of TGCT but also indicates that PLK4 may function in TGCT through a unique mechanism. High PLK4 expression in testis tissue may result from the spermatogonia that can continue to duplicate as a kind of cell with active spermatogenesis function. If the balance of PLK4 expression is disrupted (eg, by a gene mutation), tumor development may occur. TPR encodes a large coiled-coil protein that forms inner nuclear filaments that attach to the inner surface of the nuclear pore complex (NPC).45 The TPR protein directly interacts with several components of NPC and is necessary for the nuclear export of mRNA and some proteins.46 There is no research on this protein in tumor diseases, so it is a valuable topic for study in TGCT. TPR is downregulated in several tumors with typical endocrine function, such as ACC, OV, TGCT, and THCA, so the relationship between TPR expression and hormone level in tumors is an interesting topic that is worthy of in-depth study.
Conclusion
PLK4, TRIP13, TPR, KIF18A, CDKN3, HMMR, PBK, PTTG1, CKS2, SYCP1, HSPA2, and MKI67 may be important genes for the development of TGCT. PLK4 and TPR in particular may affect the disease process by participating in a variety of signal pathways and regulating tumor-related genes. Influencing immune cell infiltration may be one way to regulate the disease. The hub genes in this article were screened out by bioinformatics technology; whether they can regulate the biological function of TGCT cells must be determined with additional experimental studies. The specific mechanism also must be verified and explored in more detail.
Abbreviations
TGCT, testicular germ cell tumor; DEGs, differentially expressed genes; GSEA, Gene Set Enrichment Analysis; FDR, false discovery rate; PPI, protein–protein interaction; AUC, area under the curve; PFI, progression-free interval; US, the United States; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; HSP, heat shock protein.
Acknowledgments
This study benefited from the Cancer Genome Atlas (TCGA) database, the Genotype-Tissue Expression (GTEx) database, and the Gene Expression Omnibus (GEO) database.
Funding
This work was supported by the Natural Science Foundation of Gansu Province (grant number 18JR3RA404); Natural Science Foundation of Gansu Province (grant number 20JR5RA601); and in-hospital projects of The 940 Hospital of Joint Logistics Support Force of Chinese PLA (grant numbers 2021yxky057 and 2021yxky034).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–386. doi:10.1002/ijc.29210
2. Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. doi:10.3322/caac.21590
4. Miller KD, Fidler-Benaoudia M, Keegan TH, et al. Cancer statistics for adolescents and young adults, 2020. CA Cancer J Clin. 2020;70(6):443–459. doi:10.3322/caac.21637
5. Leão R, Ahmad AE, Hamilton RJ. Testicular cancer biomarkers: a role for precision medicine in testicular cancer. Clin Genitourin Cancer. 2019;17(1):e176–e183. doi:10.1016/j.clgc.2018.10.007
6. Kondagunta GV, Bacik J, Donadio A, et al. Combination of paclitaxel, ifosfamide, and cisplatin is an effective second-line therapy for patients with relapsed testicular germ cell tumors. J Clin Oncol. 2005;23(27):6549–6555. doi:10.1200/JCO.2005.19.638
7. Pulendran B, Davis MM. The science and medicine of human immunology. Science. 2020;369(6511). doi:10.1126/science.aay4014
8. Chovanec M, Mardiak J, Mego M. Immune mechanisms and possible immune therapy in testicular germ cell tumours. Andrology. 2019;7(4):479–486. doi:10.1111/andr.12656
9. Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013;41(Database issue):D991–995. doi:10.1093/nar/gks1193
10. Goldman MJ, Craft B, Hastie M, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38(6):675–678. doi:10.1038/s41587-020-0546-8
11. Weinstein JN, Collisson EA, Mills GB, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–1120. doi:10.1038/ng.2764
12. Ardlie KG, Deluca DS, Segrè AV. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–660. doi:10.1126/science.1262110
13. Vivian J, Rao AA, Nothaft FA, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol. 2017;35(4):314–316. doi:10.1038/nbt.3772
14. Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–1847. doi:10.1093/bioinformatics/btm254
15. Liu J, Lichtenberg T, Hoadley KA, et al. An Integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400–416.e411. doi:10.1016/j.cell.2018.02.052
16. Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
17. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
18. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7. doi:10.1186/1471-2105-14-7
19. Bindea G, Mlecnik B, Tosolini M, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39(4):782–795. doi:10.1016/j.immuni.2013.10.003
20. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–452. doi:10.1093/nar/gku1003
21. Thul PJ, Lindskog C. The human protein atlas: a spatial map of the human proteome. Protein Sci. 2018;27(1):233–244. doi:10.1002/pro.3307
22. Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–352. doi:10.1038/nature12986
23. Panni S, Lovering RC, Porras P, et al. Non-coding RNA regulatory networks. Biochim Biophys Acta - Gene Regul Mech. 2020;1863(6):194417. doi:10.1016/j.bbagrm.2019.194417
24. Noh JH, Kim KM, McClusky WG, et al. Cytoplasmic functions of long noncoding RNAs. Wiley Interdiscip Rev RNA. 2018;9(3):e1471. doi:10.1002/wrna.1471
25. Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015;29(13):1343–1355. doi:10.1101/gad.262766.115
26. Zheng Y, Luo Y, Chen X, et al. The role of mRNA in the development, diagnosis, treatment and prognosis of neural tumors. Mol Cancer. 2021;20(1):49. doi:10.1186/s12943-021-01341-7
27. Wieczorek E, Reszka E. mRNA, microRNA and lncRNA as novel bladder tumor markers. Clin Chim Acta. 2018;477:141–153. doi:10.1016/j.cca.2017.12.009
28. Chen Q, Yu D, Zhao Y, et al. Screening and identification of hub genes in pancreatic cancer by integrated bioinformatics analysis. J Cell Biochem. 2019;120(12):19496–19508. doi:10.1002/jcb.29253
29. Mano R, Di Natale R, Sheinfeld J. Current controversies on the role of retroperitoneal lymphadenectomy for testicular cancer. Urol Oncol. 2019;37(3):209–218. doi:10.1016/j.urolonc.2018.09.009
30. Murray MJ, Huddart RA, Coleman N. The present and future of serum diagnostic tests for testicular germ cell tumours. Nat Rev Urol. 2016;13(12):715–725. doi:10.1038/nrurol.2016.170
31. Cao Y, Jiao N, Sun T, et al. CXCL11 correlates with antitumor immunity and an improved prognosis in colon cancer. Front Cell Dev Biol. 2021;9:646252. doi:10.3389/fcell.2021.646252
32. Baxter D. Active and passive immunization for cancer. Hum Vaccin Immunother. 2014;10(7):2123–2129. doi:10.4161/hv.29604
33. Müller S, Almouzni G. Chromatin dynamics during the cell cycle at centromeres. Nat Rev Genet. 2017;18(3):192–208. doi:10.1038/nrg.2016.157
34. Chan FL, Wong LH. Transcription in the maintenance of centromere chromatin identity. Nucleic Acids Res. 2012;40(22):11178–11188. doi:10.1093/nar/gks921
35. Jacob P, Hirt H, Bendahmane A. The heat-shock protein/chaperone network and multiple stress resistance. Plant Biotechnol J. 2017;15(4):405–414. doi:10.1111/pbi.12659
36. Wu J, Liu T, Rios Z, et al. Heat Shock Proteins and Cancer. Trends Pharmacol Sci. 2017;38(3):226–256. doi:10.1016/j.tips.2016.11.009
37. Yun CW, Kim HJ, Lim JH, et al. Heat shock proteins: agents of cancer development and therapeutic targets in Anti-Cancer Therapy. Cells. 2019;9(1):60. doi:10.3390/cells9010060
38. Cheng C, Geng F, Cheng X, et al. Lipid metabolism reprogramming and its potential targets in cancer. Cancer commun. 2018;38(1):27. doi:10.1186/s40880-018-0301-4
39. Currie E, Schulze A, Zechner R, et al. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18(2):153–161. doi:10.1016/j.cmet.2013.05.017
40. Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, et al. SAK/PLK4 is required for centriole duplication and flagella development. Current Biol. 2005;15(24):2199–2207. doi:10.1016/j.cub.2005.11.042
41. Kazazian K, Go C, Wu H, et al. Plk4 promotes cancer invasion and metastasis through Arp2/3 complex regulation of the actin cytoskeleton. Cancer Res. 2017;77(2):434–447. doi:10.1158/0008-5472.CAN-16-2060
42. Maniswami RR, Prashanth S, Karanth AV, et al. PLK4: a link between centriole biogenesis and cancer. Expert Opin Ther Targets. 2018;22(1):59–73. doi:10.1080/14728222.2018.1410140
43. Liao Z, Zhang H, Fan P, et al. High PLK4 expression promotes tumor progression and induces epithelial‑mesenchymal transition by regulating the Wnt/β‑catenin signaling pathway in colorectal cancer. Int J Oncol. 2019;54(2):479–490. doi:10.3892/ijo.2018.4659
44. Garvey DR, Chhabra G, Ndiaye MA, et al. Role of polo-like kinase 4 (PLK4) in epithelial cancers and recent progress in its small molecule targeting for cancer management. Mol Cancer Ther. 2021;20(4):632–640. doi:10.1158/1535-7163.MCT-20-0741
45. Gallardo P, Salas-Pino S, Daga RR. A new role for the nuclear basket network. Microbial Cell. 2017;4(12):423–425. doi:10.15698/mic2017.12.604
46. Krull S, Thyberg J, Björkroth B, et al. Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol Biol Cell. 2004;15(9):4261–4277. doi:10.1091/mbc.e04-03-0165
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.