")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Role of heat shock protein 22 in the protective effect of geranylgeranylacetone in response to oxidized-LDL
Authors Gong R, Li XY, Chen HJ, Xu CC, Fang HY, Xiang J, Wu YQ
Received 21 March 2019
Accepted for publication 4 June 2019
Published 30 July 2019 Volume 2019:13 Pages 2619—2632
DOI https://doi.org/10.2147/DDDT.S209598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jianbo Sun
Ren Gong,1 Xi-Yong Li,1 Huai-Jing Chen,2 Cong-Cong Xu,1 Hai-Yang Fang,1 Jian Xiang,1 Yan-Qing Wu1
1Department of Cardiology, The Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi, People’s Republic of China; 2Department of Cardiology, Baoan Central Hospital of Shenzhen, Shenzhen, People’s Republic of China
Objective: The aim was to investigate the role and potential mechanism of geranylgeranylacetone (GGA) in the development of atherosclerosis, and to explore the role of heat shock protein 22 (HSP22) in mediating GGA effect.
Methods: Human coronary artery endothelial cell (HCAEC) was used for in vitro study. RNA interference was applied to suppress HSP22 in the cells. Cellular apoptosis and intracellular level of reactive oxygen species (ROS) were detected by flow cytometer, and proteins of HSP22, NF-κB, eNOS, and ICAM-1 were assessed by immunoblotting. HSP22-/-//ApoE,-/- and HSP22+/+//ApoE-/- mice were used to investigate the effect of GGA in the animal model of atherosclerosis. Atherosclerotic lesion of the mice aortas was evaluated by Oil Red O staining and H&E staining.
Results: GGA significantly inhibited HCAEC apoptosis in response to oxidized-LDL (ox-LDL), but stimulated HSP22 synthesis in the cells. Transfection of HSP22-siRNA in the cells resulted in complete blockage of the GGA effect on apoptosis. GGA also significantly inhibited ROS, NF-κB, and ICAM-1 in the cells transfected control siRNA, but not in the cells transfected with HSP22-siRNA. Atherosclerotic plaque in the aorta was significantly less in the wild type (WT) animals treated with GGA as stained either by Oil Red O or by H&E staining, but not in the HSP22-KO mice. GGA significantly inhibited expression of NF-κB and ICAM-1 in the WT mice, but not in the HSP22-KO mice.
Conclusion: GGA-induced HSP22, and inhibited ox-LDL-induced apoptosis as well as expression of NF-κB and ICAM-1 in the HCAECs. GGA also attenuated formation of atherosclerotic plaques in mice aorta. Suppression of HSP22 by siRNA resulted in blockage of the GGA inhibition on apoptosis or stimulation on NF-κB and ICAM-1. These findings suggested that GGA protects endothelial cells from injury in response to ox-LDL and block atherosclerotic development in mice aorta through induction of HSP22.
Keywords: geranylgeranylacetone (GGA), heat shock protein 22 (HSP22), endothelial cells, inflammatory response, atherosclerosis
Introduction
Geranylgeranylacetone, GGA, also known as Teprenone, is an anti-ulcer drug that can protect gastric mucosa from variety kinds of insults. GGA is rapidly absorbed and widely distributed in variety kinds of organs including gastrointestinal tract, liver, and kidney.1 GGA could antagonize gastric ulcers through the following mechanisms: 1) Regulating the cellular metabolism of gastric mucosa; 2) Improving micro-circulation through augmenting the synthesis of prostaglandin (PG), nitric oxide (NO), but inhibiting endothelin-1 (ET-1) synthesis; 3) Eliminating oxygen-free radicals, and suppressing infiltration of inflammatory cells and inflammatory reaction.2,3 Studies also demonstrated that GGA could protect variety types of tissues from injury through induction of heat shock proteins (HSPs) in the milieu of oxidative stress, hypoxia, or ischemia.3–8 Vitamin A-like structure of GGA may contribute to its protective effect in response to variety insult.3
In addition to its anti-ulcer9 and anti-depressant effect,10 recently, it has been reported that GGA may have preventive and therapeutic effect in the cardiovascular system due to its induction of HSPs including HSP70/HSP72, HSP90, HSP27, and HSP22. In this regard, Ooie et al,8 reported that GGA protects myocardium from ischemia-reperfusion injury through HSP72 induction, and Shinohara et al,11 further demonstrated that protective effect of GGA in the ischemia-reperfusion injury was through improving the function of the mitochondria. GGA-induced HSPs are also involved in preventing arrhythmia or atrial fibrillation by prolonging action potential duration.12–14 Role of GGA in the development of atherosclerosis, however, has not been extensively studied although Yamamoto et al,15 have reported that GGA inhibited IL-1ß-induced iNOS expression in vascular smooth muscle cells through HSP70 induction and its regulation on NF-κB activation.
HSPs are highly conservative proteins. Currently, several members of HSP family proteins including HSP60, HSP70, HSP90, and small HSP (HSP27 and HSP22) have been found to be associated with cardiovascular diseases including atherosclerosis. In this regard, we have previously reported that HSP22 prevent atherosclerosis.16 HSP22 is a serine/threonine kinase with molecular weight of 22 kDa, a small heat shock protein (sHSP).3,17 As a molecular chaperone, HSP22 interacts with a broad range of unfolded proteins and regulates cellular apoptosis,18,19 and by which mechanism, it has cytoprotective effect on myocardial cells.20,21
Chronic inflammation plays an important role in the pathogenesis of atherosclerosis. Oxidized-LDL (ox-LDL) initiates endothelial injury and inflammation, which leads to the development of atherosclerosis. It has been reported that HSP22 could partially block ox-LDL-induced endothelial injury and apoptosis through activating ERK but inhibiting p38 MAPK.22,23 Therefore, we hypothesized that GGA may protect endothelial cells from ox-LDL-induced injury through HSP22 induction. To test this, treatment on the in vitro cell culture and clinically relative animal model of atherosclerosis were used to investigate the effect of GGA on HSP22 induction and its role in the intervention of atherosclerosis.
Materials and methods
This research was approved by the Ethics Committee of The Second Affiliated Hospital of Nanchang University. All methods were carried out in accordance with relevant guidelines and regulations. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. Cell culture and treatment human coronary artery endothelial cells (HCAECs) were purchased from the Shanghai Yu Bo Biological Technology Co. Ltd. (Shanghai, China). Cells were cultured with human endothelial cell growth medium (RPMI 1640 supplemented with 10% fetal calf serum), passages 4 and 5 were used for the experiments.
GGA solution was purchased from Eisai, Japan (Tokyo, Japan). GGA capsule was purchased from Eisai China Inc. (Shanghai, China).
The cells were treated as followings. Group 1: vehicle control; Group 2: GGA treatment: HCAECs were treated with GGA at varying concentrations (0, 0.01, 0.1, 1.0, 10, and 100 µmol/L) in 4 mL culture medium for 12 hrs; Group 3: HSP22 intervention group: cells were transfected with siRNA specifically targeting HSP22 (see detail below); Group 4: HSP22 intervention + GGA treatment.
Cells were also treated with 80 µg/mL ox-LDL for 24 hrs.
Transfection of siRNA: HCAECs were grown in six-well plate to 30–50% confluent. Lipofectamine 2000 (4 µL in 200 µL serum-free medium) was mixed with FAM-siRNA (8 µL in 200 µL serum-free medium) and sit in room temperature for 20 mins. The mixture of lipofectamine and FAM-siRNA were then added into the six-well plate, in which additional 1,600 µL/well serum-free medium was applied so that the final volume was 2 mL/well. Cells were transfected for 18–24 hrs.
Animals and treatment: ApoE knockout (ApoE-/-) male C57BL/6 mice, aged 8–10 weeks, weighed 20–24 g, were purchased from the Suzhou Aiermet Technology Co. Ltd. (Suzhou, China). HSP22//ApoE knockout (HSP22-/-//ApoE-/-) male C57BL/6 mice, aged 8–10 weeks, weighed 20–24 g, were established by Beijing Vitalstar Biotechnology Co. Ltd. (Beijing, China). Sixteen mice in each phenotype were used. All animals were housed in the SPF grade animal facility of Nanchang University, Institution of Translational Medicine. Animals were fed with either ordinary diet or high fat diet (0.15% cholesterol, 21.0% fat, 20.0% protein, and 50.0% carbohydrate) for 12 weeks and treated with or without GGA (400 mg/(kg.d)) through gavage for the first 8 weeks.
Before the GGA treatment, blood samples were collected from the venous sinus. At the end of the 12-week experiments, mice were fastened for 12 hrs before scarification. Animals were anesthetized by intraperitoneal injection of 10% chloral hydrate (0.07–0.09 mL). Blood was collected from the venous sinus followed by heart perfusion with clean PBS. Arteries from aorta down to common iliac bifurcation were harvested and fixed with formaldehyde or frozen with liquid nitrogen.
Apoptosis analysis by flow cytometry: HCAECs were harvested by trypsinization. Cell pellets were washed once with cold PBS, and suspended with 300 µL binding buffer. Cells were then allowed to react with annexin V-FITC (5 µL) at dark for 15 mins. Propidium iodide (5 µL) was applied for 5 mins before the assay, and 200 µL binding buffer was added right before the cytometry assay.
Assay of the reactive oxygen species (ROS): HCAECs were washed three times with PBS. DCFH-DA probe at 10 µM in culture medium was added and incubated for 30–60 mins. Cells were then harvested by trypsinization followed by washing with PBS two times. ROS was assessed by flow cytometry.
Oil Red O staining of the arteries: The arteries were harvested and fixed with formaldehyde as aforementioned. Tissues were dehydrated with isopropanol for 3 mins followed by staining with Oil Red O (100 mg Oil Red O powder was dissolved in 10 mL isopropanol at 60°C water bath for 40 mins, 10 mL ddH2O was then added and filtered through paper filter) for 3 hrs in the dark. Tissues were then washed with 85% isopropanol three times, 3 mins each time, followed by washing with PBS. Lipid staining by the Oil Red O in the arteries was observed and photographed.
Immunoblotting: The arteries were harvested, frozen with liquid nitrogen, and stored at −80°C as aforementioned. After weighing, the tissues were cut in small pieces and minced in the buffer (100 µL per 10 mg tissue) plus cocktail of proteinase inhibitor (1:100). Protein extraction buffer was then added (one kit per 500 µL minced tissue solution, ApplyGen, Beijing, China), mixed and sat at room temperature for 10 mins. After centrifuging at 4°C, 10,000 g for 10 mins, supernatant was discarded, and proteins at the middle layer were harvested. Samples were air dried at room temperature, and 3% SDS (2–5 µL/mg tissue) was added and shaking overnight at 4°C. After centrifuging at 4°C, 12,000 g for 5 mins, supernatant was harvested and diluted with 5 x loading buffer (Solarbio, Beijing, China) so that the final concentration was 1 x loading buffer. After boiling for 10 mins, the samples were aliquoted and stored at −20°C. Protein concentration was determined by BCA methods following the manufacturer's instruction (Bio-Rad, Beijing, China). Proteins were separated by 10% SDS-PAGE gel and transferred to PVDF membrane. After blocking with 5% milk in TBST buffer, primary antibodies were added and incubated at 4°C overnight. After washing, 2nd antibodies were applied for 1 hr at room temperature. Blotting bands were visualized with ECL reagent and photographed (Beyotime, Beijing, China). Density of the blotting bands was analyzed by Image Lab software (GelDoc, Bio-Rad, Beijing, China).
Statistical analysis: GraphPad Prism 5.0 software was used to perform statistical analysis. Normal distribution was examined by Kolmogorov–Smirnov method. Data were expressed by mean ± SD. Paired Student's t–test was used to analyze the data with equal variance, and Wilcoxon rank sum test was used to analyze the data with unequal variance. One-way ANOVA was used for comparison of multiple groups. P-value <0.05 was considered as significant.
Results
Effect of GGA on HCAEC viability in response to ox-LDL
o x-LDL-induced apoptosis of HCAECs as evidenced by positive Annexin-V (31.78±3.27%, Figure 1A and B). While GGA at lower concentrations (0.01 and 0.01 µmol/L) had no effect on ox-LDL-induced HCAEC apoptosis, 1.0 µmol/L GGA significantly blocked ox-LDL-induced HCAEC apoptosis (23.99±2.03%, Figure 1A and B, P<0.05 compared to ox-LDL only). At higher concentration of GGA (100 µmol/L), however, apoptosis of HCAEC was significantly enhanced (Figure 1A and 1B, P<0.01 compared to ox-LDL only).
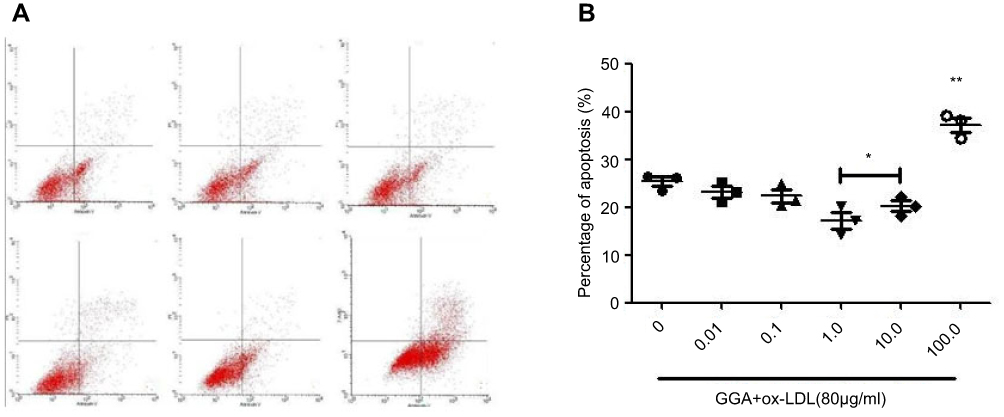 |
Figure 1 Effect of GGA on the ox-LDL-induced apoptosis of HCAEC. (A) Representative histogram of flow cytometry. HCAECs were treated with varying concentrations of GGA in the presence of ox-LDL (80 µg/mL). Cells were then harvested and Annexin-V was assessed by flow cytometry as described in the Materials and methods section. (B) Concentration-dependent effect of GGA on HCAEC apoptosis. Vertical axis: percentage of apoptosis (%); horizontal axis: concentration of GGA (µmol/L). Data presented were an average of three separate experiments. *P<0.05, **P<0.01 compared to control. |
Effect of GGA on HSP22 and NF-B expression
As shown in Figure 2, in the presence of ox-LDL (80 µg/mL), GGA stimulated HSP22 proteins in a concentration-dependent manner (0.01–100 µmol/L, Figure 2A and C). By semi-quantification of immunoblotting, it was found that HSP22 level in response to ox-LDL plus GGA was significantly increased: 0.231±0.025 of ox-LDL alone vs 0.364±0.058 of 1.0 µmoL/L GGA plus ox-LDL (P<0.05, Figure 2B and D).
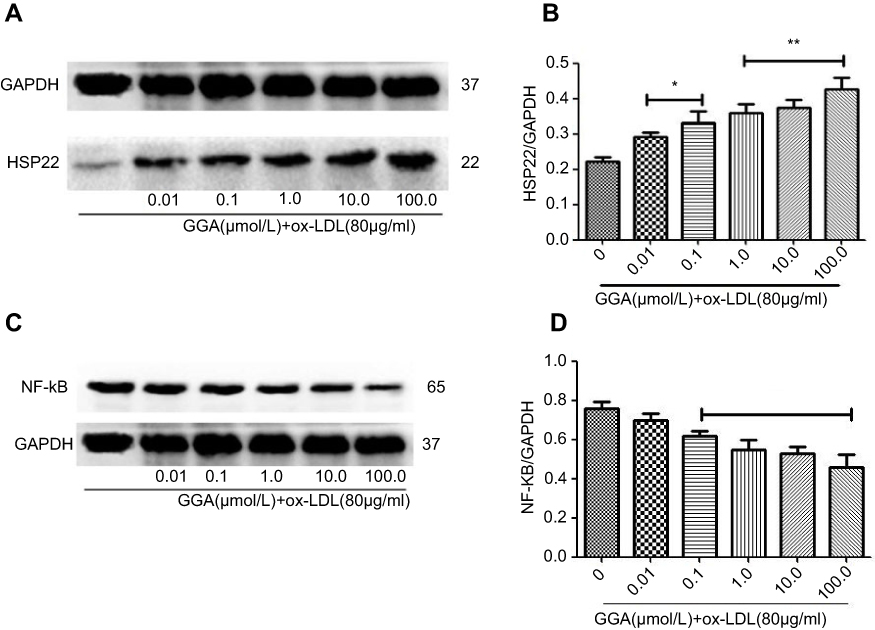 |
Figure 2 Concentration-dependent effect of GGA on HSP22 expression in response to ox-LDL exposure. (A) Representative data of immunoblotting to HSP22. HCAECs were treated and immunoblotting was performed as described in the Materials and methods section. Data presented were one representative data from at least three separate experiments. (B) Semi-quantitative comparison of the GGA effect on HSP22 expression. Vertical axis: relative level of HSP22 over GAPDH; horizontal axis: varying concentrations of GGA in the presence of ox-LDL. *P<0.05; **P<0.01. |
In contrast to the effect on HSP22, in the presence of ox-LDL (80 µg/mL), GGA significantly inhibited NF-kB (p65) levels in HCAECs (Figure 3A and B). Semi-quantification of immunoblotting revealed that p65 NF-kB level in response to ox-LDL plus GGA was significantly decreased: 0.781±0.092 of ox-LDL alone vs 0.595±0.068 of 1.0 µmol/L GGA plus ox-LDL (P<0.01, Figure 3B).
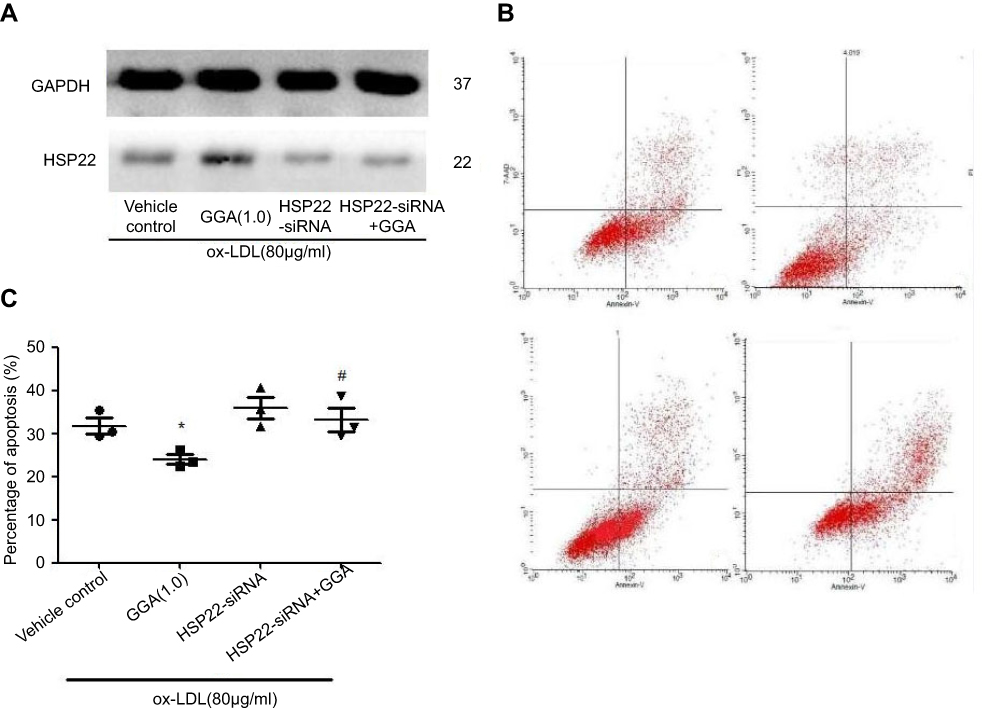 |
Figure 3 Effect of HSP22 suppression on the GGA protective effect on HCAEC apoptosis. (A) Representative data of immunoblotting. Cells were transfected with HSP22-siRNA, and immunoblotting was performed as described in the methods. Data presented were one representative of three separate experiments. (B) Representative histogram of flow cytometry. Cells were transfected with HSP22-siRNA, and Annexin-V was assessed as described in the Materials and methods section. Data presented were one representative of three separate experiments. (C) Pooled data of the effect of GGA on apoptosis following HSP22 suppression. Vertical axis: eo mean percentage of positive Annexin-V staining (%) (percentage of HCAEC apoptosis), horizontal axis: cell treatment. *P<0.05 compared to control, #P means HSP22-siRNA +GGA compared to HSP22-siRNA. |
Effect of HSP22 suppression on HCAEC viability in response to ox-LDL and GGA
HCAECs were transfected with siRNA targeting HSP22 or non-specific control siRNA 24 hrs prior to exposure to 80 µg/mL ox-LDL. Following the transfection of HSP22-siRNA (Figure S1), HSP22 expression was significantly suppressed in the presence or absence of GGA (Figure 3A), and inhibitory effect of GGA on ox-LDL-induced apoptosis of HCAECs was significantly blocked (23.99±2.03% of control-siRNA vs 33.24±4.85% of HSP22-siRNA, P<0.05, Figure 3B and C).
Effect of HSP22 suppression on ROS levels in the HCAECs
ROS level in response to ox-LDL and GGA in the cells transfected with HSP22-siRNA or control-siRNA was assessed by flow cytometry. As shown in Figure 4, ROS level was significantly reduced in response to GGA in the cells transfected with control-siRNA (308.0±23.52 of ox-LDL alone vs 218.0±26.21 of GGA + ox-LDL, P<0.05, Figure 4A). In the cells lacking HSP22, however, ROS level was significantly increased in response to ox-LDL exposure (381.7±56.22 of the cells transfected with control-siRNA vs 422.7±41.26 of the cells transfected with HSP22-siRNA, P<0.05, Figure 4B).
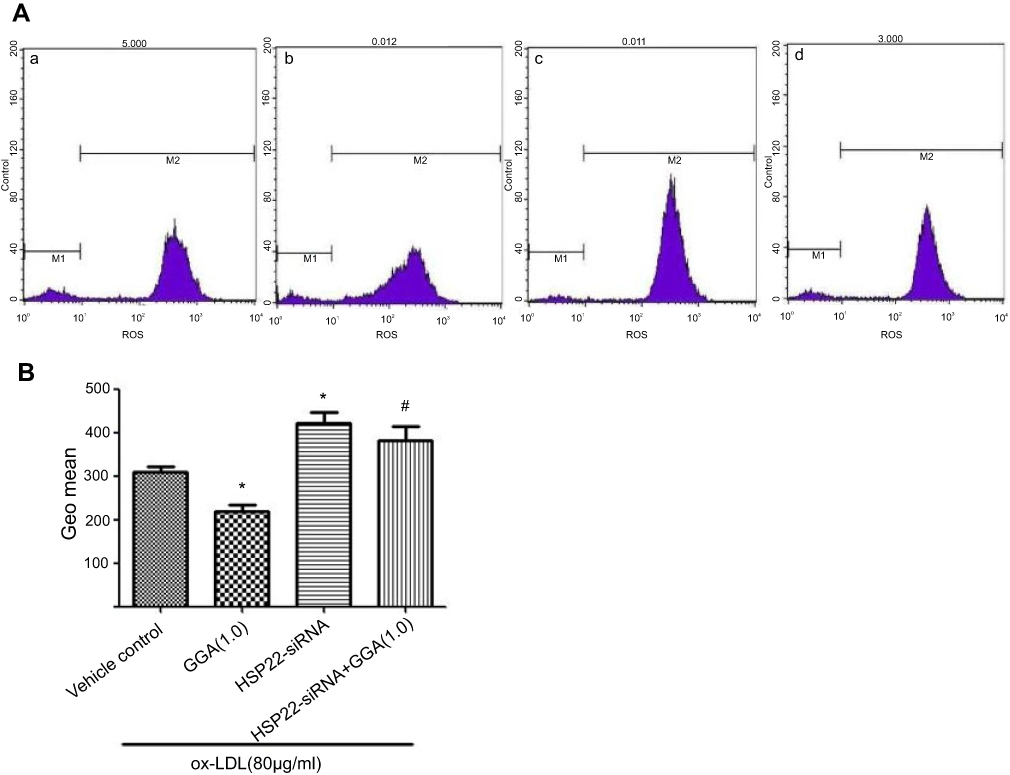 |
Figure 4 Effect of HSP22 suppression on ROS level in HCAECs. (A) Representative histogram of flow cytometry. HCAECs were transfected with HSP22-siRNA followed by treatment with or without GGA (1.0 µmol/L) in the presence of ox-LDL. Cells were then harvested and ROS content was assessed by flow cytometry as described in the Materials and methods section. a: vehicle control, b: GGA (1.0 µmol/L), c: cells transfected with HSP22-siRNA, d: cells transfected with HSP22-siRNA +GGA. Data presented were one representative of at least three separate experiments. (B) Pooled data of the effect of GGA on ROS level in HCAEC following HSP22 suppression. Vertical axis: Geo mean ROS level; horizontal axis: cell treatment. *P<0.05 compared to control, #P means HSP22-siRNA +GGA compared to HSP22-siRNA. |
Effect of HSP22 suppression on the expression of NF-kB, endothelial NOS (eNOS), and ICAM-1
Immunoblotting revealed that NF-kB level was significantly reduced in the cells treated with GGA (0.595±0.068) compared to that in the control cells (0.781±0.092, P<0.05, Figure 5A and B). Suppression of HSP22 resulted in a significant increase of NF-kB expression not only in the absence of GGA (1.042±0.102, P<0.05 compared to control, Figure 5A and B), but also in the presence of GGA (0.805±0.090, P<0.05 compared to the cells transfected with HSP22-siRNA, Figure 5A and B).
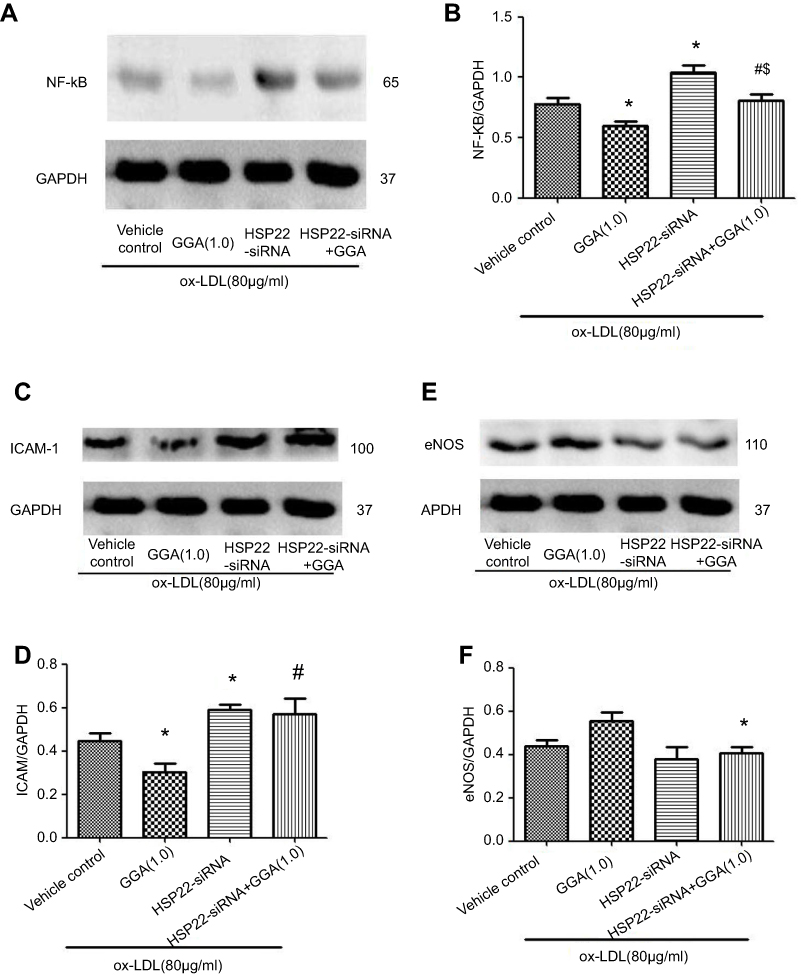 |
Figure 5 Effect of HSP22 suppression on expression of NF-kB (p65), ICAM-1, and eNOS. HCAECs were transfected with HSP22-siRNA and treated with or without GGA in the presence of ox-LDL. Total protein was extracted and immunoblotted as described in the Materials and methods section. (A) Representative image of immunoblotting to p65. (B) Semi-quantification of NF-kB (p65) expression in HCAEC following HSP22 suppression. (C) Representative image of immunoblotting to ICAM-1. (D) Semi-quantification of ICAM-1 expression in HCAEC following HSP22 suppression. (E) Representative image of immunoblotting to eNOS. (F) Semi-quantification of eNOS expression in HCAEC following HSP22 suppression. *P<0.05 compared to control, #P means HSP22-siRNA +GGA compared to HSP22-siRNA. $P < 0.05 compared to GGA only. |
Similarly, expression of ICAM-1 was significantly inhibited by GGA (0.447±0.061 of control vs 0.305±0.064 of GGA treated cells, P<0.05, Figure 5C and D). Suppression of HSP22 by siRNA could significantly block GGA inhibition on ICAM-1 expression (0.589±0.045 of HSP22-siRNA vs 0.571±0.127 of HSP22-siRNA + GGA, P<0.05, Figure 5C and D).
In contrast, while GGA could slightly but not significantly increased expression of eNOS in the cells transfected with control-siRNA (0.438±0.053 of control vs 0.556±0.065 of GGA treated cells, P>0.05, Figure 5E and F), in the cells transfected with HSP22-siRNA, however, effect of GGA on eNOS expression was significantly blocked (0.381±0.092 of GGA treated cells transfected with control-siRNA vs 0.406±0.048 of GGA treated cells transfected with HSP22-siRNA, P<0.05, Figure 5E and F).
In order to further explore the effect of GGA in the formation of atherosclerosis, effect of GGA on HSP22, NF-kB, ICAM-1, and eNOS expression was also investigated in the HSP22-knockout (as shown in Figure S2) and wild type (WT) mice. Similar to the findings of in vitro study, GGA significantly stimulated HSP22 expression in the aorta WT animals (0.297±0.090 of control vs 0.593±0.080 of GGA, P<0.05, data not shown), while expression of NF-kB in the animal aorta was significantly reduced in the WT animals treated with GGA (0.630±0.070 of control vs 0.357±0.090 of GGA treated WT mice, P<0.05, Figure 6A and B), which was slightly increased in the HSP22-KO mice treated with GGA (0.703±0.133, Figure 6A and B). Expression of ICAM-1 was significantly inhibited by GGA in the WT animals (0.183±0.031 of control vs 0.113±0.031 of GGA treated WT mice, P<0.05, Figure 6C and D), which was also slightly increased in the HSP22-KO mice treated with GGA (0.297±0.090 of HSP22-KO with GGA treatment, Figure 6C and D). Expression of eNOS, however, was not affected by GGA in either WT mice (0.263±0.040 of control vs 0.277±0.040 of GGA treated WT mice, P>0.05, Figure 6E and F) or HSP22-KO mice (0.263±0.04 of control vs 0.160±0.046 of GGA treated WT mice, P>0.05, Figure 6E and F) although eNOS expression was significantly reduced in the HSP22-KO mice compared with WT mice (P<0.05, Figure 6E and F).
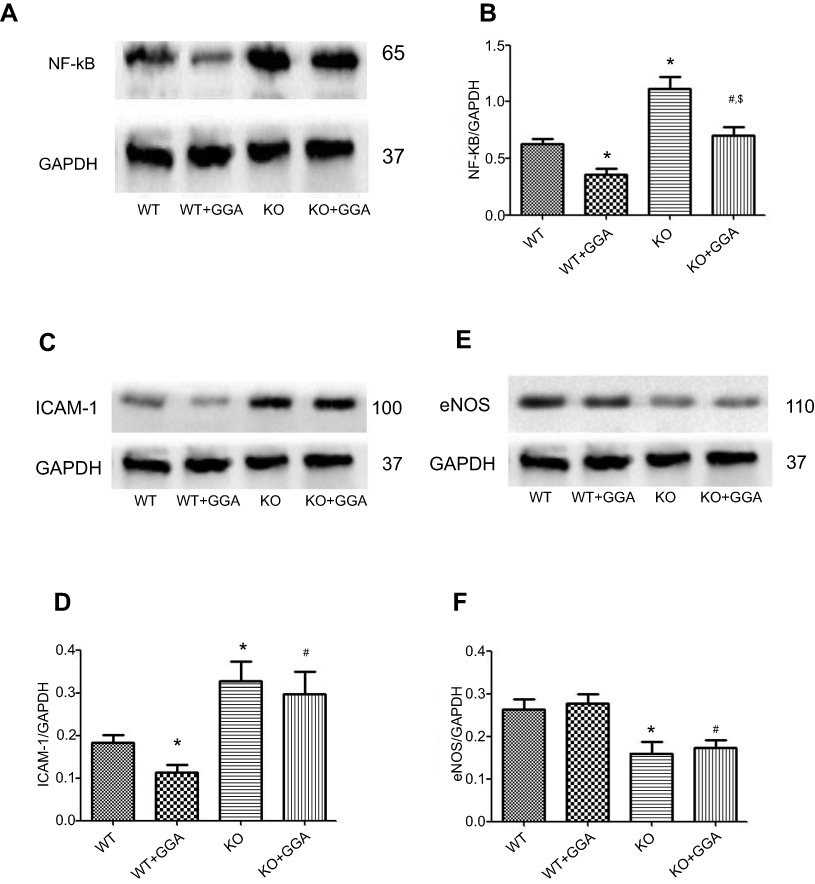 |
Figure 6 Effect of GGA on the expression of NF-kB (p65), ICAM-1, and eNOS in the wild type and HSP22-knockout mice. Artery tissues were harvested and total protein was extracted from the tissues. Immunoblotting was then performed as described in the Materials and methods section. (A) Representative image of immunoblotting to p65. (B) Semi-quantification of NF-kB (p65) expression. (C) Representative image of immunoblotting to ICAM-1. (D) Semi-quantification of ICAM-1 expression. (E) Representative image of immunoblotting to eNOS. (F) Semi-quantification of eNOS expression. *P<0.05 compared to control, #P means HSP22-siRNA +GGA compared to HSP22-siRNA. *P<0.05 compared to WT, #P means KO +GGA compared to KO animal; $P < 0.05 compared to WT+GGA. |
Pathologic findings in animal aorta
Animal models were used to further explore the role HSP22 in mediating GGA effect. Plasma lipoprotein levels at baseline as well as after high-lipid diet were measured in the animal models. While total cholesterol, triglycerides, LDL, and HDL were increased in the animals after high-lipid diet (Table 1) compared to that of baseline (Table 2), there was no significant difference between the WT animals and HSP22-KO animals regardless of GGA treatment.
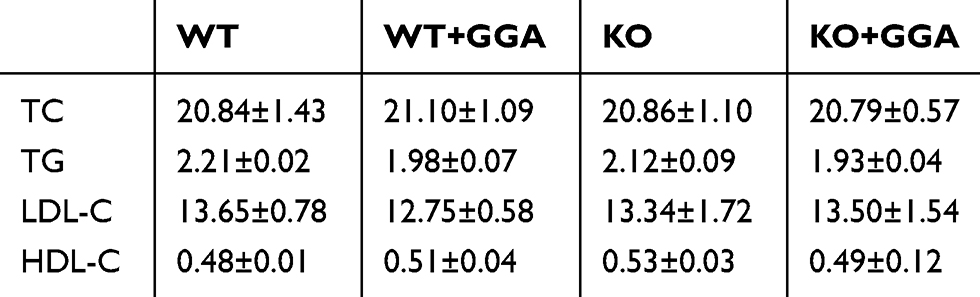 |
Table 1 Lipoprotein level after high lipid diet (mmol/L, n=9) |
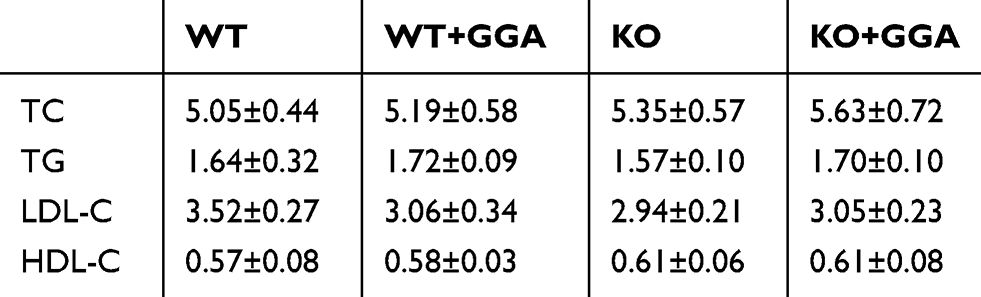 |
Table 2 Baseline of lipoproteins in the animal plasma (mmol/L, n=8) |
Effect of GGA on the WT and HSP22-KO mice was further examined by Oil Red O staining and H&E staining of the aorta tissues. As shown in Figure 7, Oil Red O staining was observed in WT control and GGA treated as well as HSP22-KO control and GGA treated animal arteries, especially, in the aorta arch, abdominal aorta, and bifurcation of aorta arch (Figure 7A). Relative area of positive Oil Red O staining vs whole artery area (%) comparison indicated that it was significantly less in the animals treated with GGA (WT: 6.82±0.83%, HSP22-KO: 7.16±2.29%, respectively) compared to the animals without GGA treatment (WT: 12.42±1.49%, HSP22-KO: 13.70±0.86%, respectively, P<0.01 in both comparison, Figure 7B).
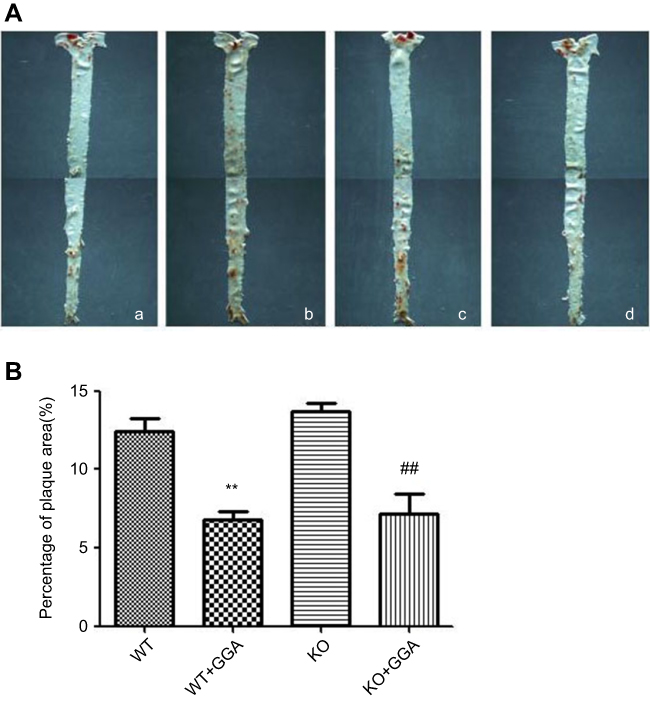 |
Figure 7 Comparison of lipid deposition in the arteries of wild type and HSP22-knockout mice. Animals were treated with GGA and arteries were harvested. Lipid deposition on the arterial walls was then evaluated by Oil Red O staining as described in the Materials and methods section. (A) Representative image of the Oil Red O positive staining arteries. a: Wild type animal; b: wild type animal treated with GGA; c: HSP22-knockoout mice; d: HSP22-knockout mice treated with GGA. (B) Semi-quantitative evaluation on the effect of GGA on lipid deposition in the arteries of wild type and HSP22-knockout mice. Vertical axis: percentage of plaque area, horizontal axis: wild or HSP22-KO mice. **P<0.05 compared to WT, ##P means KO +GGA compared to KO animals. |
H&E staining of the aortic arches indicated that atherosclerotic plaques were found in all animals, which were composed of fibrous cap and inflammatory cells or foam cells (Figure 8). Under higher magnification, rougher surface of the fibrous cap, accumulated inflammatory cells or foam cells under the cap, accumulated cholesterol crystal and necrotic tissues, damaged inner elastic membrane, and disrupted intimal smooth muscle cells and tissue were observed in the animals of WT (Figure 8(A), A/a) or HSP22-KO mice (Figure 8(A), C/c). In contrast, animals treated with GGA had a smoother surface of the fibrous cap, less cholesterol crystal or necrotic tissues, mild damage to inner elastic membrane, and fairly normal intima and smooth muscle cell distribution (Figure 8(A), B/b and Figure 8(A), D/d). Compared to the WT animals, the area of atherosclerotic plaque was significantly less in the WT animals treated with GGA (WT + GGA) 41.80±3.05 of WT vs 29.21±5.46 of WT + GGA, P<0.05, Figure 8(B)). In contrast, area of atherosclerotic plaque was significantly increased in the HSP22-knockout (KO) animals (48.24±2.40, P<0.05, Figure 8(B)) compared with that of WT animals. Treatment with GGA in the HSP22-knockout mice (KO + GGA) resulted in a significant reduction in the area of atherosclerotic plaque (35.82±3.67) compared to that of HSP-22-KO animals (48.24±2.40, P<0.05, Figure 8(B)). However, GGA treatment did not make difference in the area of plaque between the wild type (WT + GGA) and HSP22-knockout (KO + GGA) mice (P>0.05, Figure 8(B)).
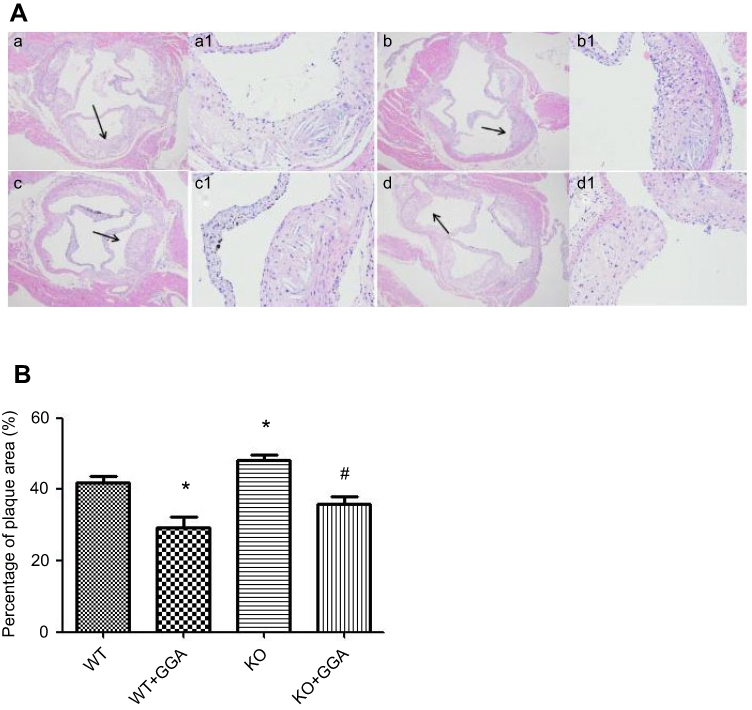 |
Figure 8 Comparison of plaque size in the arteries of wild type and HSP22-knockout mice. Animals were treated with GGA and arteries were harvested, fixed, and H&E stained as described in the Materials and methods section. (A) Representative image of atherosclerotic plaque stained with H&E. (B) Semi-quantitative evaluation on the effect of GGA on atherosclerotic plaque size in the arteries of wild type and HSP22-knockout mice. Vertical axis: percentage of plaque area, horizontal axis: wild or HSP22-KO mice. P<0.05 compared to control, #P means HSP22-siRNA +GGA compared to HSP22-siRNA. *P<0.05 compared to WT, #P means KO +GGA compared to KO animals. |
Discussion
In the current study, effect of GGA in the development of atherosclerotic plaque in animals as well as endothelial cell injury in response to ox-LDL in the cell culture was invested. It was found that GGA-induced HSP22 in both in vitro cell culture and in vivo animal models, and that GGA inhibited ox-LDL-induced apoptosis as well as expression of NF-κB and ICAM-1 in the HCAECs. GGA also attenuated formation of atherosclerotic plaques in mice aorta. Suppression of HSP22 by siRNA resulted in blockage of the GGA inhibition on apoptosis or stimulation on NF-κB and ICAM-1.
GGA or Teprenone is a pharmaceutical drug used for the treatment of gastric ulcers.24,25 GGA is rapidly absorbed without metabolizing by liver or kidney, and widely distributed in variety kinds of organs including gastrointestinal tract, liver, and kidney.1 Mechanisms of GGA in the treatment of gastric ulcers included: 1) Regulation on cellular metabolism of the gastric mucosa; 2) enhancement on the synthesis of PG, NO, but inhibited ET-1 synthesis, and thus improved micro-circulation; 3)elimination of oxygen free radicals, and suppression of inflammatory cell infiltration and inflammatory reaction.2,3
Recently, accumulating evidence indicated that, through inducing HSPs, GGA may have protective or therapeutic effect in the cardiovascular diseases as well.12–14,26 In this regard, it has been demonstrated that GGA could protect myocardium from ischemia-reperfusion-induced injury, prevent atrial fibrillation, and protect heart from interstitial fibrosis and enlargement.8,11–14
Study of GGA on atherosclerosis, however, was limited.26 In this content, it has been reported that GGA could inhibit iNOS activation in the smooth muscle cells through inhibiting IL-1ß but stimulating HSP70 in the cells,21,27 and thus GGA could protect the cells from injury. Consistent with these studies, in vitro cell culture of the current study demonstrated that GGA significantly inhibited ox-LDL-induced apoptosis of HCAECs, suggesting GGA could protect the endothelial cells from ox-LDL-induced injury. Furthermore, the current study of animal models demonstrated that GGA reduced lipid deposition as evidenced by Oil Red O staining, and that GGA could not only inhibit formation of the atherosclerotic plaque in the aorta and abdominal arteries, but also stabilize the atherosclerotic plaques as evidenced by H&E staining of the aorta and abdominal arteries.
The current study further invested potential mechanisms of GGA protection of arteries from ox-LDL-induced atherosclerosis. Studies have indicated that GGA is an inducer of several isoforms of HSP proteins,5–7 which is associated with the vitamin A-like structure of the GGA. Therefore, we anticipated that GGA might protect arteries from the development of atherosclerosis through inducing HSP proteins including HSP70/HSP72, HSP90, HSP27, and HSP22.
HSP proteins are highly conservative proteins. Members of HSP family include HSP110, HSP90, HSP70, HSP60, HSP40, sHSPs, and ubiquitin.2,22,23,28 Many of HSP family proteins perform chaperone function by stabilizing new proteins to ensure correct folding or by helping to refold proteins that are damaged by the cell stress.29 It has been known that HSP60, HSP70, HSP90, and sHSPs may be associated with the development of atherosclerosis, and that HSP70, HSP90 and sHSPs (HSP22 and HSP 27) may have a protective role in the development of atherosclerosis. In this regard, HSP22 has been reported to have protective effect in atherosclerosis.26 Consistently, we found that GGA-induced HSP22 expression in the HCAECs in a concentration-dependent manner, and that GGA could significantly block ox-LDL-induced apoptosis of HCAECs. Suppression of HSP22 by siRNA resulted in up-regulation of NF-kB, ICAM-1, and ROS level, and significantly blocked protective effect of GGA on ox-LDL-induced endothelial cells damage.
Furthermore, GGA-induced HSP22 expression and inhibited NF-kB and ICAM-1 expression in mice arteries, and by which mechanism GGA inhibited formation of atherosclerosis. However, this protective effect of GGA on the development of atherosclerosis was not significantly affected in the HSP22-knockout mice, suggesting HSP22 may not be involved in mediating the protective of GGA in the formation of atherosclerosis in the mice. While the mechanisms remain to be further investigated, discrepancy between the results of in vitro (HSP22 mediating GGA effect on endothelial cells) and in vivo (HSP22-KO could not block GGA effect) studies on the role of HSP22 in mediating GGA effect suggested that molecules other than HSP22 might also be involved in mediating GGA effect in the development of atherosclerosis.
Studies have indicated that chronic inflammation plays an important role in the development of atherosclerosis.27,30 In the current study, therefore, we explored NF-kB and ICAM-1 expression in response to GGA. We found that GGA significantly inhibited expression of NF-kB and ICAM-1 expression in the WT mice, but not in the HSP22-knockout mice, suggesting GGA may inhibit inflammatory reaction through HSP22.
ROS is believed not only induce endothelial cell injury, but also initiate inflammatory reaction and by which mechanism it is involved in the development of atherosclerosis.31 Here, we found that GGA significantly inhibited ROS in the endothelial cells, which was attenuated in the cells lacking HSP22, suggesting HSP22 mediates the inhibitory effect of GGA on ROS formation.
NO plays a role in regulating vascular function, smooth muscle cell contractility, and inflammatory cells infiltration into the vascular wall.32 eNOS is one of the three enzymes that synthesize NO. Studies demonstrated that HSP90 is necessary for eNOS activation.33,34 In this regard, vascular endothelial growth factor stimulated the combination of HSP90, protein kinase B (Akt), and eNOS to form HSP90-eNOS-Akt complex, which leads to NO synthesis.27 Here, we reported that GGA did not significantly affect eNOS level although eNOS expression was reduced in the HSP22-knockout mice, suggesting eNOS may not be involved in mediating the GGA effect in the development of atherosclerosis.
In the animal models of the current study, high-lipid diet resulted in a significant increase of plasma lipoproteins. Interestingly, GGA could not block the increase of lipoproteins in the animals regardless of WT or HSP22-KO mice. These findings suggested that GGA could not directly reduce lipoproteins. However, GGA could significantly reduce area of atherosclerotic plaque in WT of the animal models of atherosclerosis as evidenced by Oil Red O staining and H&E histological staining, however, the inhibitory effect of GGA on atherosclerotic plaque size was not significantly attenuated in the HSP22-knockout mice, suggesting HSP22 may not be associated with the atherosclerotic plaque formation.
Taken together, the current study demonstrated that GGA-induced HSP22 in HCAECs in vitro cell culture of HCAECs and in vivo animal artery tissues. HSP22 mediated the inhibitory effect of GGA on ox-LDL-induced apoptosis of HCAECs as well as expression of NF-kB and ICAM-1 expression in HCAECs. Consistent with the in vitro cell culture results, studies with animal models of atherosclerosis also revealed that GGA significantly inhibited NF-kB and ICAMP-1 expression in WT mice, but not in the HSP22-knockout mice. Furthermore, GGA significantly reduced the size of atherosclerotic plaque in the WT animals, but not in the HSP22-knockout mice. These findings suggested that HSP22 mediate the effect GGA on ox-LDL-induced apoptosis and inflammation of the endothelial cells, and by which mechanism, GGA may modulate the development and stability of atherosclerosis.
Availability of data and material
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This research was approved by the Ethics Committee of The Second Affiliated Hospital of Nanchang University. All methods were carried out in accordance with relevant guidelines and regulations. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Acknowledgment
This work was supported by research grant from the National Natural Science Foundation of China (No. 8166020210).
Author contributions
Ren Gong and Yan-Qing Wu contributed to the conception and design of the study; Xi-Yong Li contributed to the acquisition of data; Huai-Jing Chen and Cong-Cong Xu performed the experiments; Hai-Yang Fang and Jian Xiang contributed to the analysis of data; Ren Gong wrote the manuscript. All authors reviewed and approved the final version of the manuscript. All authors contributed to the data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspect of the work.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Han NK, Jeong YJ, Pyun BJ, Lee YJ, Kim SH, Lee HJ. Geranylgeranylacetone ameliorates intestinal radiation toxicity by preventing endothelial cell dysfunction. Int J Mol Sci. 2017;18(10). doi:10.3390/ijms18102103
2. Hua C, Ju WN, Jin H, Sun X, Zhao G. Molecular chaperones and hypoxic-ischemic encephalopathy. Neural Regen Res. 2017;12(1):153–160. doi:10.4103/1673-5374.199008
3. Bakthisaran R, Tangirala R, Rao CM. Small heat shock proteins: role in cellular functions and pathology. Biochim Biophys Acta. 2015;1854(4):291–319. doi:10.1016/j.bbapap.2014.12.019
4. He D, Song X, Li L. Geranylgeranylacetone protects against cerebral ischemia and reperfusion injury: HSP90 and eNOS phosphorylation involved. Brain Res. 2015;1599:150–157. doi:10.1016/j.brainres.2014.12.019
5. Katsuno M, Sang C, Adachi H, et al. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102(46):16801–16806. doi:10.1073/pnas.0506249102
6. Suzuki S, Maruyama S, Sato W, et al. Geranylgeranylacetone ameliorates ischemic acute renal failure via induction of Hsp70. Kidney Int. 2005;67(6):2210–2220. doi:10.1111/j.1523-1755.2005.00326.x
7. Cao W, Li M, Li J, Li C, Xu X, Gu W. Geranylgeranylacetone ameliorates lung ischemia/reperfusion injury by HSP70 and thioredoxin redox system: NF-kB pathway involved. Pulm Pharmacol Ther. 2015;32:109–115. doi:10.1016/j.pupt.2015.02.009
8. Ooie T, Takahashi N, Saikawa T, et al. Single oral dose of geranylgeranylacetone induces heat-shock protein 72 and renders protection against ischemia/reperfusion injury in rat heart. Circulation. 2001;104(15):1837–1843.
9. Umegaki E, Kuramoto T, Kojima Y, et al. Geranylgeranylacetone, a gastromucoprotective drug, protects against NSAID-induced esophageal, gastroduodenal and small intestinal mucosal injury in healthy subjects: a prospective randomized study involving a comparison with famotidine. Intern Med. 2014;53(4):283–290. doi:10.2169/internalmedicine.53.1572
10. Zhong JM, Wu SY, Bai J, et al. Antidepressant effect of geranylgeranylacetone in a chronic mild stress model of depression and its possible mechanism. Exp Ther Med. 2012;4(4):627–632. doi:10.3892/etm.2012.669
11. Shinohara T, Takahashi N, Kohno H, et al. Mitochondria are targets for geranylgeranylacetone-induced cardioprotection against ischemia-reperfusion in the rat heart. Am J Physiol Heart Circ Physiol. 2007;293(3):H1892–H1899. doi:10.1152/ajpheart.00493.2007
12. Takahashi N, Wakisaka O, Yoshimatsu H, Saikawa T. Induction of heat shock proteins prevents the arrhythmogenic substrate for atrial fibrillation. Int J Hyperthermia. 2009;25(8):641–646. doi:10.3109/02656730903070949
13. Sakabe M, Shiroshita-Takeshita A, Maguy A, et al. Effects of a heat shock protein inducer on the atrial fibrillation substrate caused by acute atrial ischaemia. Cardiovasc Res. 2008;78(1):63–70. doi:10.1093/cvr/cvn019
14. Brundel BJ, Shiroshita-Takeshita A, Qi X, et al. Induction of heat shock response protects the heart against atrial fibrillation. Circ Res. 2006;99(12):1394–1402. doi:10.1161/01.RES.0000252323.83137.fe
15. Yamamoto K, Sarukawa M, Ito T, Aoki H, Ichida M, Shimada K. An anti-ulcer drug, geranylgeranylacetone, suppresses inducible nitric oxide synthase in cultured vascular smooth muscle cells. J Hypertens. 2005;23(10):1847–1853.
16. Chen Q, Xiang J, Gong R, et al. Atorvastatin downregulates HSP22 expression in an atherosclerotic model in vitro and in vivo. Int J Mol Med. 2019;43(2):821–829. doi:10.3892/ijmm.2018.4015
17. Nefedova VV, Sudnitsyna MV, Gusev NB. Interaction of small heat shock proteins with light component of neurofilaments (NFL). Cell Stress Chaperones. 2017;22(4):467–479. doi:10.1007/s12192-016-0757-6
18. Bouhy D, Juneja M, Katona I, et al. A knock-in/knock-out mouse model of HSPB8-associated distal hereditary motor neuropathy and myopathy reveals toxic gain-of-function of mutant Hspb8. Acta Neuropathol. 2018;135(1):131–148. doi:10.1007/s00401-017-1756-0
19. Mambula SS, Calderwood SK. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J Immunol. 2006;177(11):7849–7857. doi:10.4049/jimmunol.177.11.7849
20. Lee YJ, Lee HJ, Choi SH, et al. Soluble HSPB1 regulates VEGF-mediated angiogenesis through their direct interaction. Angiogenesis. 2012;15(2):229–242. doi:10.1007/s10456-012-9255-3
21. Rashed E, Lizano P, Dai H, et al. Heat shock protein 22 (Hsp22) regulates oxidative phosphorylation upon its mitochondrial translocation with the inducible nitric oxide synthase in mammalian heart. PLoS One. 2015;10(3):e0119537. doi:10.1371/journal.pone.0119537
22. Mymrikov EV, Seit-Nebi AS, Gusev NB. Large potentials of small heat shock proteins. Physiol Rev. 2011;91(4):1123–1159. doi:10.1152/physrev.00023.2010
23. Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40(2):253–266. doi:10.1016/j.molcel.2010.10.006
24. Kitahata H, Nozaki J, Kawahito S, Tomino T, Oshita S. Low-dose sevoflurane inhalation enhances late cardioprotection from the anti-ulcer drug geranylgeranylacetone. Anesth Analg. 2008;107(3):755–761. doi:10.1213/ane.0b013e31817f0e61
25. Hirakawa T, Rokutan K, Nikawa T, Kishi K. Geranylgeranylacetone induces heat shock proteins in cultured guinea pig gastric mucosal cells and rat gastric mucosa. Gastroenterology. 1996;111(2):345–357.
26. Xu Q, Metzler B, Jahangiri M, Mandal K. Molecular chaperones and heat shock proteins in atherosclerosis. Am J Physiol Heart Circ Physiol. 2012;302(3):H506–H514. doi:10.1152/ajpheart.00646.2011
27. Jiang Y, Jiang LL, Maimaitirexiati XM, Zhang Y, Wu L. Irbesartan attenuates TNF-alpha-induced ICAM-1, VCAM-1, and E-selectin expression through suppression of NF-kappaB pathway in HUVECs. Eur Rev Med Pharmacol Sci. 2015;19(17):3295–3302.
28. Kampinga HH, Hageman J, Vos MJ, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14(1):105–111. doi:10.1007/s12192-008-0068-7
29. De Maio A. Heat shock proteins: facts, thoughts, and dreams. Shock. 1999;11(1):1–12.
30. Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49–62. doi:10.1038/nrm2083
31. Papatheodorou L, Weiss N. Vascular oxidant stress and inflammation in hyperhomocysteinemia. Antioxid Redox Signal. 2007;9(11):1941–1958. doi:10.1089/ars.2007.1750
32. Siragusa M, Frohlich F, Park EJ, Schleicher M, Walther TC, Sessa WC. Stromal cell-derived factor 2 is critical for Hsp90-dependent eNOS activation. Sci Signal. 2015;8(390):ra81. doi:10.1126/scisignal.aaa2819
33. Xi W, Satoh H, Kase H, Suzuki K, Hattori Y. Stimulated HSP90 binding to eNOS and activation of the PI3-Akt pathway contribute to globular adiponectin-induced NO production: vasorelaxation in response to globular adiponectin. Biochem Biophys Res Commun. 2005;332(1):200–205. doi:10.1016/j.bbrc.2005.04.111
34. Fujimura N, Jitsuiki D, Maruhashi T, et al. Geranylgeranylacetone, heat shock protein 90/AMP-activated protein kinase/endothelial nitric oxide synthase/nitric oxide pathway, and endothelial function in humans. Arterioscler Thromb Vasc Biol. 2012;32(1):153–160. doi:10.1161/ATVBAHA.111.237263
Supplementary materials
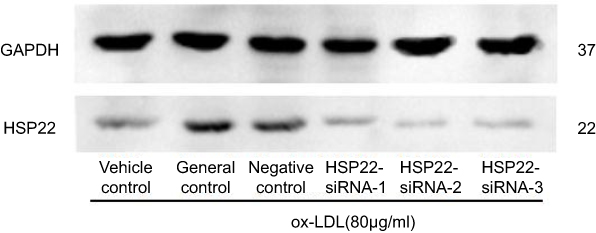 |
Figure S1 Representative image of HSP22 suppression by siRNA. HCAECs were transfected with HSP22-siRNA or non-specific siRNA as described in the Materials and methods section. Level of HSP22 was examined by immunoblotting as described in the Materials and methods section. |
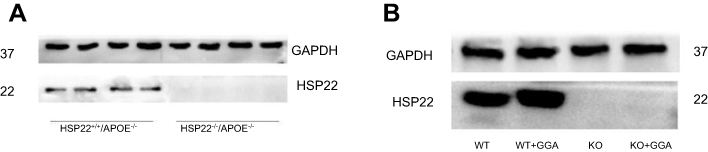 |
Figure S2 HSP22 expression in wild type and HSP22-knockout mice. (A) Representative image of HSP22 expression in wild type and HSP22-knockout mice. (B) Effect of GGA on HSP22 expression in wild type and HSP22-knockout mice. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.