")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Role of GRP78 inhibiting artesunate-induced ferroptosis in KRAS mutant pancreatic cancer cells
Authors Wang K , Zhang Z , Wang M, Cao X, Qi J, Wang D, Gong A, Zhu H
Received 25 December 2018
Accepted for publication 15 May 2019
Published 2 July 2019 Volume 2019:13 Pages 2135—2144
DOI https://doi.org/10.2147/DDDT.S199459
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Kang Wang,1,* Zhengyang Zhang,1,* Ming Wang,1 Xiongfeng Cao,1 Jianchen Qi,1 Dongqing Wang,1,2 Aihua Gong,3 Haitao Zhu1,2
1Central Laboratory of Medical Imaging, Affiliated Hospital of Jiangsu University, Zhenjiang, 212001, People’s Republic of China; 2Department of Radiology, Affiliated Hospital of Jiangsu University, Zhenjiang 212001, People’s Republic of China; 3School of Medicine, Jiangsu University, Zhenjiang 212013, People’s Republic of China
*These authors contributed equally to this work
Objective: To investigate the exact role of GRP78 in artesunate-induced ferroptosis in KRAS mutant pancreatic cancer cells.
Methods: Artesunate-induced KRAS mutant human pancreatic cancer cells (AsPC-1 and PaTU8988) ferroptosis was confirmed by fluorescent staining experiments and CCK8. Western blot and short-hairpin RNA-based knockdown assays were conducted to detect GRP78 activity and its role in artesunate-induced ferroptosis.
Results: Artesunate induced AsPC-1 and PaTU8988 cell death in ferroptosis manner, rather than necrosis or apoptosis. In addition, artesunate increased the mRNA and protein levels of GRP78 in a concentration-dependent manner in AsPC-1 and PaTU8988 cells. Knockdown GRP78 enhanced artesunate-induced ferroptosis of pancreatic cancer cells in vitro and in vivo.
Conclusion: Combining artesunate with GRP78 inhibition may be a novel maneuver for effective killing of KRAS mutant pancreatic ductal adenocarcinoma cells.
Keywords: ferroptosis, GRP78, artesunate, pancreatic cancer
Introduction
Despite technical advances in radiation delivery and the advent of new agents, overall survival remains poor in pancreatic ductal adenocarcinoma (PDAC) patients, especially in tumor cells bearing constitutively active oncogenic KRAS. Many cytokines and chemokines are the downstream targets of mutant KRAS that result in resistance to apoptosis and cell death.1 Thus, the efficient strategies to kill KRAS mutant pancreatic cancer cells remain to be resolved. Several researches revealed that KRAS mutant cancer cells dependent on more glutamine and glucose metabolism to sustain cancer cell survival.2 Based on the metabolism and redox treatment may improve the sensitivity of KRAS mutant pancreatic cancer cells to treatment.
Ferroptosis is a novel form of regulated cell death (RCD), which is caused by the iron-dependent accumulation of lipid hydroperoxides. Different from the other types of RCD, ferroptosis is driven by loss of activity of the lipid repair enzyme glutathione peroxidase 4 (GPX4) and subsequent accumulation of lipid-based reactive oxygen species (ROS) at the biochemical level. Moreover, ferroptosis is characterized by the presence of smaller than normal mitochondria with condensed mitochondrial membrane densities, reduction or vanishing of mitochondria crista, and outer mitochondrial membrane rupture at the morphology level.1 It was reported that ferroptosis inducers can specifically kill KRAS mutant tumor cells.3–5 Several small molecules, such as erastin, sulfasalazine, sorafenib, and artesunate, have been identified that they can specially induce cancer cells ferroptosis.6–8
Among these inducers, artesunate is an Artemisinin derivative and often used as a first-line drug for the treatment of malaria.9 Previous studies confirmed that artesunate activated apoptosis or necroptosis in a cancer type-dependent manner. It was recently shown that artesunate can also trigger cancer cells ferroptosis by inducing excessive accumulation of ROS in the cells. Eling et al found artesunate could specially activate the ferroptosis in KRAS mutant pancreatic cancer cells.2 However, the precise mechanisms for artesunate inducing KRAS mutant pancreatic cancer cells ferroptosis are still to be elucidated.
78-kDa Glucose-regulated protein 78 (GRP78) is one of the most active molecular chaperone components in the endoplasmic reticulum of cancer cells and is overexpressed in different kinds of cancers.10,11 Moreover, GRP78 is correlated with tumor progression. The expression of GRP78 may be related to resistance against anticancer therapy in which apoptosis signaling is involved. Studies also have shown that the level of GRP78 is highly associated with the resistance of pancreatic cancer to the ferroptosis inducers, erastin, and sulfasalazine, indicating that GRP78 plays an important role in anti-ferroptosis.12 Therefore, we are interested in if GRP78 is also involved in the artesunate-induced KRAS mutant pancreatic cancer cells ferroptosis.
To test this hypothesis, we used the Lipid Peroxidation Assay and confirmed that artesunate triggered KRAS mutant pancreatic cancer cells ferroptosis in vitro and in vivo. We found that inhibiting GRP78 by shRNA enhanced the effect of artesunate inducing pancreatic cancer cell ferroptosis.
Materials and methods
Cell culture and agents
Human pancreatic cancer cell lines (PaTU8988 and AsPC-1) were obtained from the Cell Bank of the China Academy of Sciences (Shanghai, China). PaTU8988 cells were cultured in high glucose Dulbecco’s modified Eagle medium (DMEM) containing 10% fetal bovine serum and antibiotics (100 units/mL penicillin, 100 mg/mL streptomycin). AsPC-1 cells were cultured in Roswell Park Memorial Institute 1640 (RPMI-1640) containing 10% fetal bovine serum and antibiotics (100 units/mL penicillin, 100 mg/mL streptomycin). They were maintained at 37°C 5% CO2 and saturated humidity. Artesunate (#HY-N0193), Z-VAD-FMK (#HY-16658), Necrosulfonamide (#HY-100573), Ferrostatin-1 (Fer-1, #HY-100), and Deferoxamine mesylate (DFO, #HY-B0988) were obtained from MedChemExpress (MCE).
Cell counting kit-8 (CCK8) assays
Pancreatic cancer cells were treated with artesunate (20 µM) with or without the following death inhibitor: i) Fer-1 (1 µM); ii)DFO (100 nM); iii) ZVAD-FMK (1 µM); and iv) necrosulfonamide (0.5 µM) for 24 hrs. CCK8 was carried out according to the manufacturer’s instructions. The absorbance (OD value) of each well was measured at 450 nm with a microplate reader.
Lipid peroxidation assay
Pancreatic cancer cells were treated with or without artesunate (20 µM) and Fer-1 (1 µM). The relative malondialdehyde (MDA) concentration was assessed using a Lipid Peroxidation Assay Kit (#ab118970; Abcam), and the experiments were carried out according to the manufacturer’s instructions.
Lipid component fluorescent staining
Wide-field fluorescence microscopy was performed using 60× objective Zeiss inverted fluorescence microscope. Cells were seeded in 8-well microscope slides for live cell imaging or fixation in PBS containing 4% paraformaldehyde. For live cell imaging, cells were stained with tetramethylrhodamine methyl ester (TMRM, 50 nM) for 20 mins at 37°C or stained with BODIPY C11 (#D2861, Invitrogen) for 30 mins at 37°C. Staining with Alexa Fluor 546 human transferrin (HTF546, 5 μg/ml) was used to monitor the uptake of whole transferrin in viable cells over a specified period of time.
For immunofluorescence, fixed cells were permeabilized with 0.3% PBS, blocked with 3% BSA, and incubated with antibodies against cytochrome C or SMAC for 2 hrs at room temperature. Fluorescence staining was performed for 1 hr at room temperature using a cross-adsorbed secondary antibody Alexa Fluor 488 or Alexa Fluor 546. Images of representative cells were captured using the Z-axis scan function. The acquired images were analyzed and prepared by using Image J and Fiji. The degree of lipid peroxidation was determined by dividing the average green intensity [oxidized BODIPY C11] by the average red intensity per cell [reduced BODIPY C11].
Cell transfection and viral infection
AsPC-1 and PaTU8988 cells (5×105 cells per well) were grown in a 6-well plate overnight followed by transfection with pcmv6-GRP78 or pcmv6-vector with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cancer cells RNAi and gene transfection were performed as previously described.12 The following specific shRNA sequences were used:
The human GRP78-shRNA1:
5ʹ-GTACCGGAGATTCAGCAACTGGTTAAAGCTCGAGCTTTAACCAGTTGCTGAATCTTTTTTTG-3ʹ
The human GRP78-shRNA2:
5ʹ-CCGGGAAATCGAAAGGATGGTTAATCTCGAGATTAACCATCCTuTCGATTTCTTTTTG-3ʹ
Quantitative real-time PCR (qRT-PCR)
AsPC-1 and PaTU8988 cells were cultured in 6-well plates in different concentrations of artesunate (0, 10, 20, and 40 µM). Total RNA was extracted from cultured cancer cells using the RNeasy Kit (Qiagen). For mRNA analysis, cDNA was synthesized from 1 μg total RNA using the RevertAid RT Reverse Transcription Kit (Thermo Fisher Scientific). SYBR Green-based real-time PCR was subsequently performed in triplicate using the SYBR Green master mix (Thermo Fisher Scientific) on an Applied Biosystems StepOnePlus real-time PCR machine (Thermo Fisher Scientific). For analysis, the threshold cycle (Ct) values for each gene were normalized to those of GAPDH. The following gene-specific primers were used:
GRP78 (5ʹ-CTGTCCAGGCTGGTGTGCTCT-3ʹ, 5ʹ-CTTGGTAGGCACCACTGTGTTC-3ʹ).
Western blot
Protein concentrations were determined by bicinchoninic acid (BCA) method. Western blot assay was performed as described previously. The antibodies were GRP78 (#ab32618, Abcam), GAPDH (#ab9485, Abcam), and mouse anti-Flag (Sigma). Secondary antibody (either anti-rabbit or anti-mouse) was purchased from Boster Biotechnology Company (China). The ECL luminescent reagent was developed, and the bands were analyzed using BandScan Image J software.
Xenograft tumor models
Animal studies were approved by the Committee on the Use of Live Animals for Teaching and Research of Jiangsu University. Four-week-old female BALB/c nude mice were purchased from the Animal Center of Yangzhou University and maintained in Animal Center of Jiangsu University in compliance with the Guideline for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
AsPC-1 cells (1×106) with a control or GRP78 shRNA transfection were injected into right subcutaneous flank of nude mice (five mice per group). The nude mice were randomized into two groups and treated with DMSO or artesunate (30 mg/kg/i.p.), respectively. Artesunate was administered every two days. The tumor growth speed and volume were monitored every two days until the end point at day 35. All the tumor size and weight in the artesunate-treated groups were measured by using a caliper and an electronic balance. Tumor volume was calculated using the formula length×width2×π/6. Fresh tumor weight were measured following the mice were euthanized.
Statistical analysis
All data are presented as the mean±standard error of the mean (SEM). Two-tailed Mann–Whitney U tests were used to compare the statistical differences between the treatment groups. The significances of differences between groups were analyzed using Student’s t-tests, one-way analysis of variance (ANOVA), or two-way ANOVA. P<0.05 was considered to reflect a statistically significant difference. All the experiments were repeated at least three times.
Results
Artesunate induced ferroptosis in KRAS mutant pancreatic cancer cells
To examine the role of artesunate inducing cell death, KRAS mutated pancreatic cancer cells (AsPC-1 and PaTU8988) were treated with 20 µM artesunate. CCK8 assay showed artesunate dramatically induced cell death (Figure 1A), and the effect can be reversed by Fer-1 (ferroptosis-specific inhibitor), rather than ZVAD-FMK (apoptosis inhibitor) and necrosulfonamide (necrosis inhibitor) (Figure 1A). These results indicate that the anticancer activity of artesunate depends on the induction of ferroptosis but not apoptosis or necrosis.
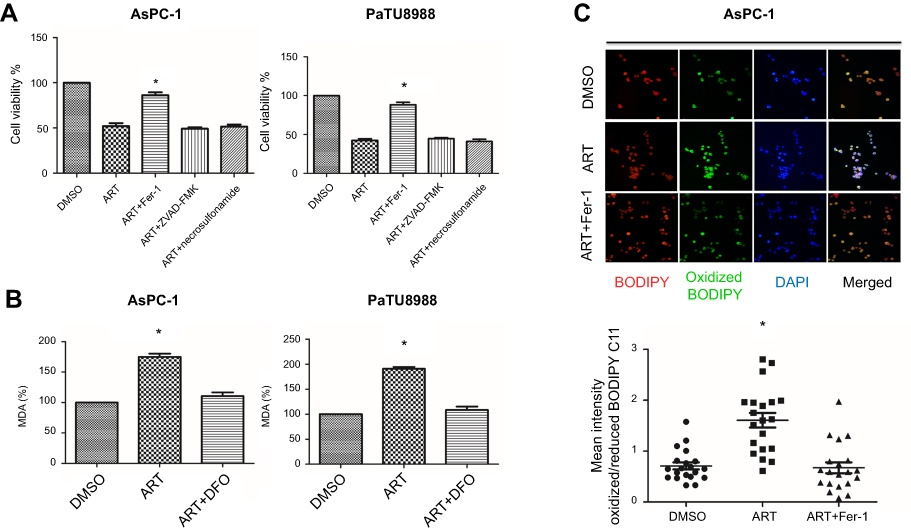 |
Figure 1 Artesunate induced ferroptosis in KRAS mutant pancreatic cancer cells in vitro. (A) AsPC-1 and PaTU8988 were treated with DMSO (control), ART (artesunate, 20 µM), ART (20 µM) + Fer-1 (ferrostatin-1, ferroptosis-specific inhibitor), ART (20 µM) + ZVAD-FMK (apoptosis inhibitor) and ART (20 µM) + necrosulfonamide (necrosis inhibitor) for 24 hrs. Cell viability was accessed by CCK8 assay. (B) AsPC-1 and PaTU8988 were treated with DMSO (control), ART (20 µM), ART (20 µM) + DFO (deferoxamine mesylate, ferroptosis-specific inhibitor) for 24 hrs. The level of MDA was assayed. (C) AsPC-1 and PaTU8988 were treated DMSO (control), ART (20 µM), ART (20 µM) + Fer-1 for 24 hrs. Immunofluorescence assays were performed to access level of oxidized C11-BODIPY. Red: reduced BODIPY C11; Blue: oxidized BODIPY C11. Scale bar, 50 μm. Lipid peroxidation is presented as single cell quantification of oxidized vs reduced BODIPY C11 fluorescence intensity. Twenty cells per condition were analyzed from three independent experiments. ART represents for artesunate. Experiments were repeated three times, and the data are expressed as the mean±SEM. *P<0.05 relative to control, **P<0.01, ***P<0.001. Statistical analysis was performed using Student’s t-test. Abbreviations: DMSO, dimethylsulfoxide; MDA, malondialdehyde; Fer-1, Ferrostatin-1; DFO, Deferoxamine mesylate. |
To further confirm the role of artesunate inducing KRAS mutated pancreatic cancer cells ferroptosis, we accessed MDA concentration and lipid oxidized level in AsPC-1, which are indicator of ferroptosis. Compared to the control group, artesunate induced higher concentration MDA (Figures 1B and S1A) and higher level lipid peroxidation (Figure 1C). Also, these can be reversed by DFO (iron chelator) and Fer-1.
Artesunate increases the expression of GRP78 in KRAS mutant pancreatic cancer cells
GRP78 plays an important role in cell survival. We next access the expression level of GPR78 in AsPC-1 and PaTU8988 following artesunate treatment. qRT-PCR showed that mRNA expression level of GRP78 increased in a concentration-dependent manner following 24-hrsartesunate treatment in AsPC-1 and PaTU8988 (Figure 2A). The similar result can be acquired at the protein level of GRP78 by Western blot in AsPC-1 and PaTU8988 (Figure 2B). These results indicate that artesunate could induce the expression of GRP78 at the mRNA and protein level in KRAS mutant pancreatic cancer cells.
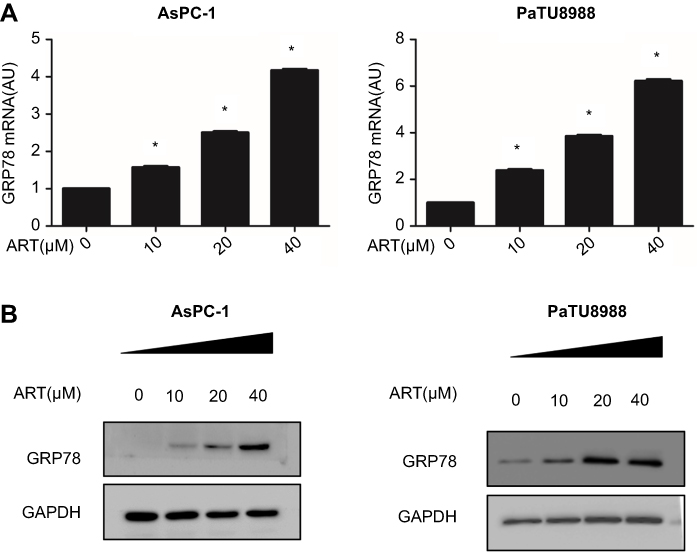 |
Figure 2 Expression level of GRP78 in artesunate treated KRAS mutant pancreatic cancer cells. (A) qRT-PCR analysis the mRNA expression level of GRP78 in AsPC-1 and PaTU8988 cancer cells after 24 hrs treated with various concentrations ART (0, 10, 20, and 40 µM). GAPDH was detected as a loading control. (B) Western blot analysis of protein expression levels of GRP78 in AsPC-1 and PaTU8988 cancer cells after 24 hrs treated with various concentrations ART (0, 10, 20, and 40 µM). GAPDH was detected as a loading control. ART represents for artesunate. Experiments were repeated three times, and the data are expressed as the mean±SEM. *P<0.05 relative to control, **P<0.01. Statistical analysis was performed using Student’s t-test. |
GRP78 mediates artesunate-induced ferroptosis resistance in KRAS mutant pancreatic cancer cells in vitro
To elucidate the functional role of GRP78 in artesunate-induced ferroptosis resistance, overexpression (Flag-GRP78) and two stable GRP78 knockdown cell clones (GRP78 shRNA1 and shRNA2) were established and highly silencing efficiency were confirmed by Western blot (Figure 3A and C) and qRT-PCR (Figure 3B and D). Silencing GRP78 in pancreatic cancer cells substantially inhibited the cell viability and colony forming ability in the presence of artesunate, while overexpression GRP78 rescued this phenomenon (Figure 4A and B). Compared to the control group, the concentrations of MDA level are higher in the shGRP78 group and lower in GRP78 overexpression group in the presence of artesunate (Figure 4C).
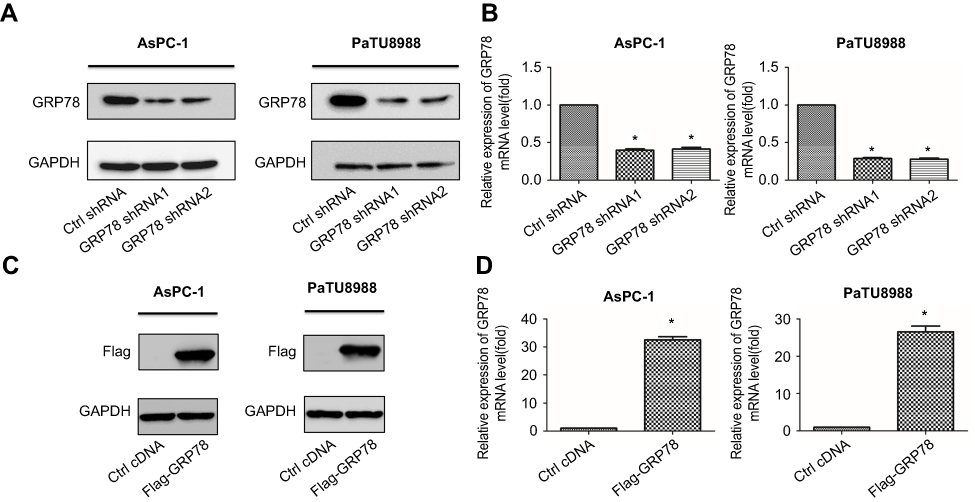 |
Figure 3 Knockdown or overexpression of GRP78 in KRAS mutant pancreatic cancer cells. (A) Western blot analysis of shRNA-mediated knockdown of GRP78 protein expression in AsPC-1 and PaTU8988. GAPDH expression was detected as a loading control. (B) qRT-PCR analysis of shRNA-mediated knockdown of GRP78 mRNA expression in AsPC-1 and PaTU8988. GAPDH expression was detected as a loading control. (C) Western blot analysis of overexpression efficiency of GRP78 protein expression in AsPC-1 and PaTU8988. GAPDH expression was detected as a loading control. (D) qRT-PCR analysis of overexpression efficiency of GRP78 mRNA expression in AsPC-1 and PaTU8988. GAPDH expression was detected as a loading control. Experiments were repeated three times, and the data are expressed as the mean±SEM. *P<0.05 relative to control. Statistical analysis was performed using Student’s t-test. |
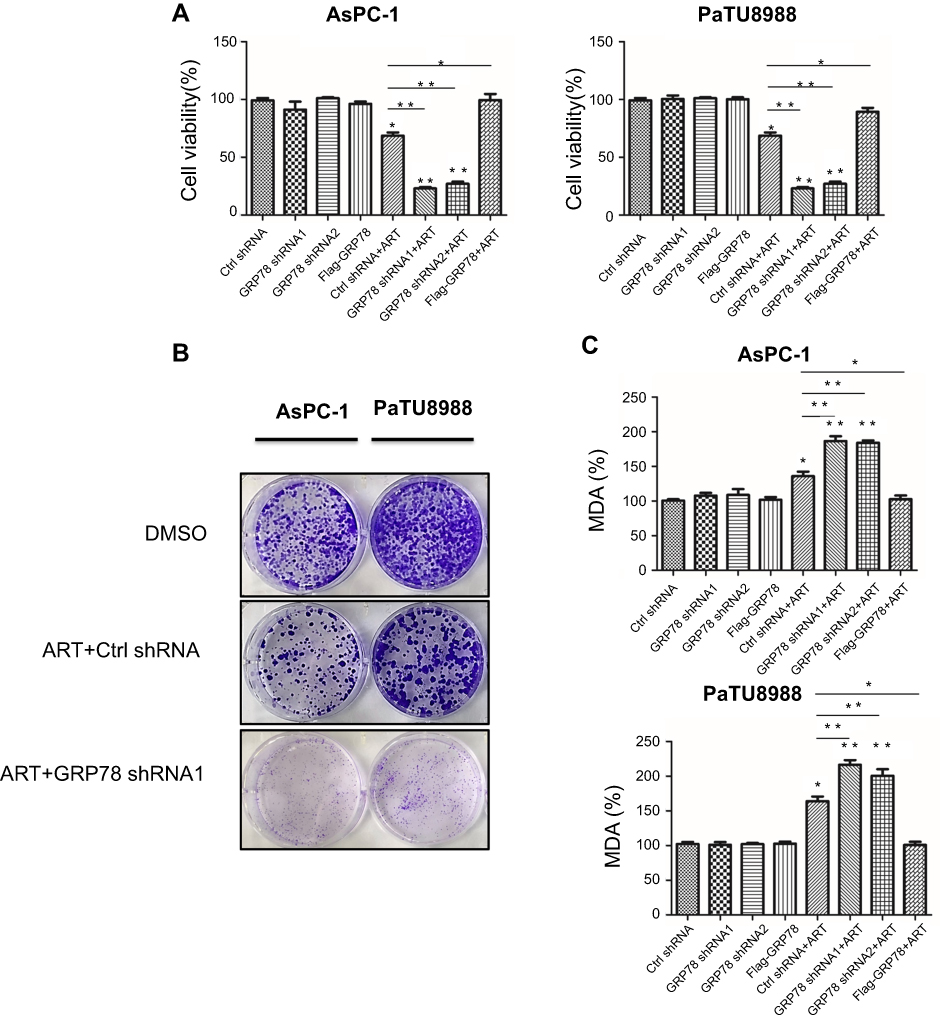 |
Figure 4 GRP78 mediates ferroptosis resistance in KRAS mutant pancreatic cancer cells in vitro. (A) AsPC-1 (Ctrl shRNA), GRP78 knockdown AsPC-1 (GRP78 shRNA1 and GRP78 shRNA2), GRP78 overexpression AsPC-1 (Flag-GRP78), PaTU8988 (Ctrl shRNA), GRP78 knockdown PaTU8988 (GRP78 shRNA1 and GRP78 shRNA2), and GRP78 overexpression PaTU8988 (Flag-GRP78) cancer cells were treated with or without ART (20 µM). Cell viability was accessed by CCK8 assay. (B) Colony-forming assay analysis of the colony forming ability of AsPC-1 (Ctrl shRNA), GRP78 knockdown AsPC-1 (GRP78 shRNA1), PaTU8988 (Ctrl shRNA), and GRP78 knockdown PaTU8988 (GRP78 shRNA1) cancer cells were treated with or without ART (20 µM). (C) AsPC-1 (Ctrl shRNA), GRP78 knockdown AsPC-1 (GRP78 shRNA1 and GRP78 shRNA2), GRP78 overexpression AsPC-1 (Flag-GRP78), PaTU8988 (Ctrl shRNA), GRP78 knockdown PaTU8988 (GRP78 shRNA1 and GRP78 shRNA2), and GRP78 overexpression PaTU8988 (Flag-GRP78) cancer cells were treated with or without ART (20 µM). The level of MDA was assayed. ART represents for artesunate. Experiments were repeated three times, and the data are expressed as the mean±SEM. *P<0.05 relative to control, **P<0.01. Statistical analysis was performed using Student’s t-test. Abbreviations: DMSO, dimethylsulfoxide; MDA, malondialdehyde; Fer-1, Ferrostatin-1; DFO, Deferoxamine mesylate. |
GRP78 mediates artesunate-induced ferroptosis resistance in KRAS mutant pancreatic cancer cells in vivo
To validate our in vitro observation that GRP78 mediates ferroptosis resistance, stably GRP78 knockdown AsPC-1 (GRP78–AsPC-1) cells were used to establish the subcutaneously nude mice. Compared to the control group (Crtrl shRNA + DMSO), artesunate treatment significantly slows down the tumor growth rate (Figure 5A). Compared to the Crtrl shRNA + artesunate group, GRP78 shRNA1+ artesunate group exhibited lower tumor growth rates (Figure 5A). Moreover, the tumor weight was lower (Figure 5B) and the tumor volume was smaller in the GRP78 shRNA1+ artesunate group than the Crtrl shRNA + artesunate group. These results suggested that GRP78 negatively regulated artesunate induced ferroptosis in vivo.
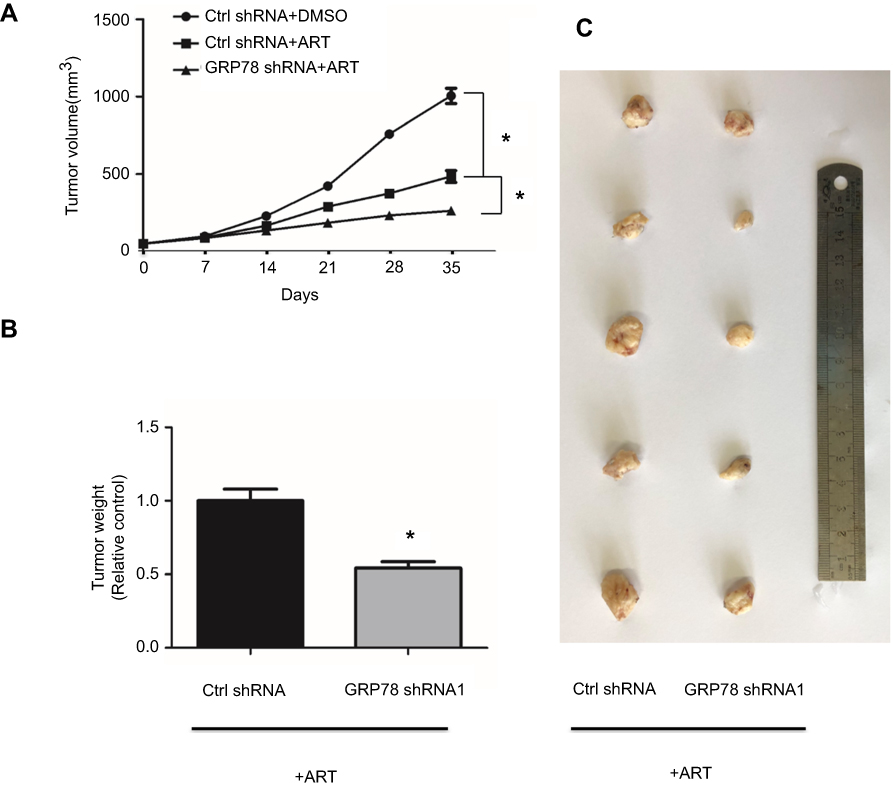 |
Figure 5 Knockdown GRP78 enhanced artesunate-induced ferroptosis in AsPC-1 cells in vivo. Cultured GRP78 knockdown AsPC-1(GRP78 shRNA1) cells and control cancer cells were transplanted subcutaneously with 1×106 cells/mouse into the right subcutaneous flank of nude mice (five mice per group). The nude mice were randomized into two groups and treated with DMSO or artesunate (30 mg/kg/i.p.), respectively. The tumor-growth rate (A), tumor weight (B), and tumor volume (C) were measured at 35 d post-injection. ART represents for artesunate. *P<0.05 relative to control. Statistical analysis was performed using Student’s t-test. Abbreviation: DMSO, dimethylsulfoxide. |
Discussion
In summary, in this study, we found that artesunate induced KRAS mutant pancreatic cancer cells ferroptosis as well as the elevation of GRP78. Furthermore, knockdown GRP78 enhanced the sensitivity of KRAS mutant pancreatic cancer cells to artesunate-induced ferroptosis; therefore, combining ART with GRP78 inhibitor may be a novel maneuver for effective killing KRAS mutant PDAC cells.
PDAC is an extremely heterogeneous and universally fatal disease.13 As the most frequently mutated gene, KRAS mutant occurred in 95% PDAC.14,15 Throughout the progressive accumulation of mutations, KRAS continues to drive PDAC development. Constitutive activation of KRAS results in pancreatic cancer cells' unlimited proliferation, resistance to apoptosis, and metastasis. In addition to the abovementioned biology, KRAS also has driven the alteration of glucose and glutamine utilization that contribute to PDAC progression. It was reported that KRAS mutant PDAC uses a unique form of glutamine metabolism to regulate redox balance.16 Thus, the appreciated connections between KRAS and dysregulated glutamine metabolism signaling in PDAC may provide attractive novel directions for targeted therapy. In this study, we chose AsPC-1 and PaTu8988 as the study model for their similarly in KRAS mutant, and more aggressive and cell-death resistance to the traditional chemotherapy and radiotherapy. AsPC1 cell line was derived from metastatic human pancreatic carcinoma, while PaTu8988 cell line was of pancreatic ductal cell origin. In vitro and in vivo results show that AsPC-1 and/or PaTu8988 cells were sensitive to artesunate-induced ferroptosis.
Ferroptosis is a newly discovered programmed cell death, could be triggered by the accumulation of glutamate, polyunsaturated fatty acid-phospholipids, or by depletion of endogenous inhibitors of ferroptosis, such as GSH, NADPH, GPX4.2,17 It was reported that erastin has the ability to reduce glutathione (GSH) level by directly inhibiting cystine/glutamate antiporter system Xc− with activation of the ER stress response and ferroptosis.15 Also, Buthionine sulfoximine (BSO) inhibits GSH synthesis with decreased GPX activity and increased ROS levels, which results in ferroptosis in KRAS-mutated cells.18 Therefore, inducing ferroptosis represents a potential therapeutic treatment to kill such as KRAS-mutated pancreatic cancer cells. artesunate is a water-soluble derivative of Artemisinin.19 Recent studies have shown that Artemisinin and its derivatives can be used not only to treat malaria but also to kill tumor cells, which can cause excessive accumulation of iron ions and oxygen-free radicals in tumor cells, leading to the accumulation of lipids in tumor cells. The component undergoes a peroxidation reaction, resulting in the death of tumor cells, thereby achieving the effect of inhibiting tumor growth.2 Our study found that artesunate can induced KRAS-mutated pancreatic cancer cells death in a ferroptosis manner depending on accumulation oxidizing lipid components, which is consist to the former research. Moreover, we further confirmed that GRP78 negatively regulated artesunate-induced ferroptosis.
As one of the most active endoplasmic reticulum chaperone protein, GRP78 is overexpressed in different kinds of cancers and involved in tumorigenesis, metastasis, and angiogenesis.10 GRP78 conferred breast cancer cells resistance BIK-mediated apoptosis. Overexpression of GRP78 decreases the sensitivity of glioma cells to cisplatin-induced cell death. Zhu et al reported that GRP78 formed complex with GPX4 in pancreatic cancer cells which further mediating erastin induced ferroptosis resistance. Our study firstly confirmed that GRP78 could also conferred KRAS mutant pancreatic cancer cells resistance to artesunate induced ferroptosis. The exact mechanism needs further investigation.
Conclusion
In this study, we provide the evidence that artesunate induced ferroptosis in human KRAS mutant PDAC cells. Inhibition of the GRP78 increased artesunate sensitivity and reversed the ferroptosis resistance in KRAS mutant PDAC cells (Figure 6). More in vivo and clinical investigations should be conducted to explore this promising artesunate based and GRP78 inhibition combination cancer therapy for patients with oncogenic KRAS mutation.
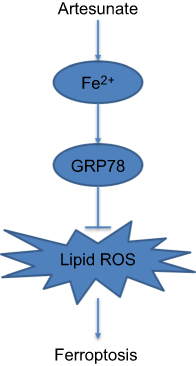 |
Figure 6 Flow diagram representation of GRP78 positively regulates artesunate-induced ferroptosis. |
Abbreviation list
GRP78, 78-kDa glucose-regulated protein; CCK8, Cell Counting Kit-8; RCD, regulated cell death; GPX4, glutathione peroxidase 4; DMEM, dulbecco’s modified eagle medium; MDA, malondialdehyde; DMSO, dimethylsulfoxide; BCA, bicinchoninic acid; PBS, phosphate buffer saline; BSA, bovine serum albumin; PDAC, pancreatic ductal adenocarcinoma.
Ethics approval and consent to participate
All animal studies were approved by the Committee on the Use of Live Animals for Teaching and Research of the Jiangsu University.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (grant number 81502663), the Social Development Foundation of Jiangsu Province (grand number BE2018691), Young Medical Talents of Jiangsu (grand number QNRC20168339), Postgraduate Innovation Project of Jiangsu Province (grand number KYCX17_1802), Six talent peals project of Jiangsu Province (grand number WSW-039), and Six for one project of Jiangsu Province (grand numbers LGY2018093).
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
2. Eling N, Reuter L, Hazin J, Hamacherbrady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517–532. doi:10.18632/oncoscience.160
3. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
4. Yang WS, Stockwell BR. Ferroptosis: death by Lipid Peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi:10.1016/j.tcb.2015.10.014
5. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi:10.1038/nature14344
6. Song X, Zhu S, Chen P, et al. AMPK-Mediated BECN1 Phosphorylation Promotes Ferroptosis by Directly Blocking System Xc(-) Activity. Curr Biol. 2018;28(15):2388–99 e5. doi:10.1016/j.cub.2018.05.094
7. Kim SE, Zhang L, Ma K, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol. 2016;11(11):977–985. doi:10.1038/nnano.2016.164
8. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–262. doi:10.1016/j.redox.2016.12.010
9. Vandewynckel YP, Laukens D, Geerts A, et al. Therapeutic effects of artesunate in hepatocellular carcinoma: repurposing an ancient antimalarial agent. Eur J Gastroenterol Hepatol. 2014;26(8):861–870. doi:10.1097/MEG.0000000000000066
10. Bi X, Zhang G, Wang X, et al. Endoplasmic Reticulum Chaperone GRP78 Protects Heart From Ischemia/Reperfusion Injury Through Akt Activation. Circ Res. 2018;122(11):1545–1554. doi:10.1161/CIRCRESAHA.117.312641
11. Tian N, Gao Y, Wang X, et al. Emodin mitigates podocytes apoptosis induced by endoplasmic reticulum stress through the inhibition of the PERK pathway in diabetic nephropathy. Drug Des Devel Ther. 2018;12:2195–2211. doi:10.2147/DDDT.S167405
12. Zhu S, Zhang Q, Sun X, et al. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017;77(8):2064–2077. doi:10.1158/0008-5472.CAN-16-1979
13. Perusina Lanfranca M, Thompson JK, Bednar F, et al. Metabolism and epigenetics of pancreatic cancer stem cells. Semin Cancer Biol. 2018. doi:10.1016/j.semcancer.2018.09.008
14. Dolma S, Lessnick SL, Hahn WC, Stockwell RB. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–296.
15. Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523. doi:10.7554/eLife.02523
16. Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi:10.1038/nature12040
17. Xie Y, Hou W, Song X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–379. doi:10.1038/cdd.2015.158
18. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
19. Tyagi RK, Gleeson PJ, Arnold L, et al. High-level artemisinin-resistance with quinine co-resistance emerges in P. falciparum malaria under in vivo artesunate pressure. BMC Med. 2018;16(1):181. doi:10.1186/s12916-018-1156-x
Supplementary material
 |
Figure S1 (A) AsPC-1 and PaTU8988 were treated DMSO (control), ART (20 µM), ART (20 µM) + Fer-1 for 24 hrs. The level of MDA was assayed. (B) The protein expression level of GAPDH in AsPC-1 and PaTU8988 treated with or without artesunate. ART represents for artesunate. *P<0.05 relative to control. Statistical analysis was performed using Student’s t-test. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.