")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Role of glycogen synthase kinase-3β inhibitor AZD1080 in ovarian cancer
Authors Chen S, Sun K, Feng M, Sang X, Liu B, Zhao Y
Received 15 December 2015
Accepted for publication 2 February 2016
Published 18 March 2016 Volume 2016:10 Pages 1225—1232
DOI https://doi.org/10.2147/DDDT.S102506
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Shuo Chen, Kai-Xuan Sun, Miao-Xiao Feng, Xiu-Bo Sang, Bo-Liang Liu, Yang Zhao
Department of Gynecology, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China
Background: Glycogen synthase kinase-3β (GSK-3β) is a multifunctional serine/threonine kinase that plays an important role in cancer tumorigenesis and progression. We investigated the role of the GSK-3β inhibitor AZD1080 in ovarian cancer cell lines.
Methods: A2780 and OVCAR3 ovarian cancer cell lines were exposed to AZD1080, after which cell proliferation, cell cycle, invasion, and migration assays were performed. Phalloidin staining was used to observe lamellipodia formation. Reverse transcription polymerase chain reaction and Western blot were used to assess the respective mRNA and protein expression levels of GSK-3β, CDK2, CDK1, cyclin D1, matrix metalloproteinase-9 (MMP9), and Bcl-xL.
Results: AZD1080 exposure suppressed ovarian cancer cell proliferation, invasion, migration, and lamellipodia formation, and induced G1 arrest, which was concentration dependent. AZD1080 also significantly downregulated GSK-3β, CDK2, CDK1, cyclin D1, MMP9, and Bcl-xL expression at both mRNA and protein levels.
Conclusion: Taken together, our results demonstrate that the GSK-3β inhibitor AZD1080 suppresses ovarian cancer development and therefore may indicate a new direction for ovarian cancer treatment.
Keywords: ovarian cancer, GSK-3β inhibitor, AZD1080, tumorigenesis, progression
Introduction
Ovarian cancer is the third most common female reproductive system cancer. The ovarian cancer mortality rate is the highest of the gynecological malignancies due to the lack of effective early diagnostic methods, its chemotherapy resistance, and its ability to metastasize and recurrence. The 5-year survival rate is <30%, making it as a serious threat to the health and lives of women.1,2 Ovarian cancer has numerous malignant transformations and molecular signaling pathways, and consequently, the search for new drugs to treat epithelial ovarian cancer remains a major challenge.
Identification of key kinase isoforms regulating ovarian tumor development, chemoresistance, and metastasis is an important component of ovarian research. Glycogen synthase kinase-3β (GSK-3β) is a highly conserved serine/threonine kinase that may have different functions in different types of cancers.3–5 Recent studies have suggested that the “hyper-activation” of GSK-3β may function as an oncogene in several types of human cancer, including colon cancer,6 oral cancer,7 osteosarcoma,8 and malignant melanoma.9 Of particular interest, it has been reported that the expression of GSK-3β is significantly higher in ovarian carcinoma tissues.10 Overall, it is clear that GSK-3β plays an important role in tumorigenesis and in tumor promotion and progression. Recently, GSK-3β knockdown and GSK-3β inhibitors have been shown to inhibit the proliferation of malignant cells in pancreatic,11 prostatic,12 and colonic13 cancers and in leukemia.14 However, less is known about GSK-3β inhibitors in ovarian cancer. Consequently, we studied the role of the new GSK-3β inhibitor, AZD1080,15 in ovarian cancer.
Materials and methods
Cell culture
The ovarian carcinoma cell lines A2780 and OVCAR3 were obtained from American Type Culture Collection (Manassas, VA, USA). The cells were maintained in Roswell Park Memorial Institute (RPMI) 1640 (OVCAR3) or Dulbecco’s Modified Eagle’s Medium (A2780) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified atmosphere of 5% CO2 at 37°C with or without AZD1080 (Chemicals, Shanghai, People’s Republic of China) treatment at doses of 0.5 μM, 1.0 μM, 2.0 μM, and 4.0 μM. The study was approved by China Medical University Ethics Committee.
Cell viability assay
Cell viability was determined using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells (3×103/well) were seeded directly into 96-well plates and allowed to adhere. At different time points, 20 μL of MTT (5 mg/mL; Sigma-Aldrich Co., St Louis, MO, USA) was added, the plates were incubated at 37°C for 4 hours, the supernatants were removed, and 150 μL of dimethyl sulfoxide (Sigma-Aldrich Co.) was added to each well. The absorbance value (optical density) of each well was measured at 490 nm.
Cell cycle analysis
After incubation at 37°C in 5% CO2, cells were detached by trypsinization, collected, washed twice with phosphate-buffered saline (PBS), and fixed in 500 μL ice-cold ethanol (70%) for at least 2 hours. The cells were again washed twice with PBS, and propidium iodide containing RNase A (BD Biosciences, San Jose, CA, USA) was added and cultivated at 4°C in the dark for 30 minutes. Cell cycle analysis was performed by flow cytometry analysis of propidium iodide staining.
Wound healing assay
Cells (1×106/well) were seeded in six-well culture plates. After they had grown to confluence, the cell monolayer was scratched with a 200-μL pipette tip to create a wound. The cells were then washed with PBS three times and cultured in FBS-free medium with or without AZD1080 treatment. Cells were photographed at 0 hour and 24 hours, and the scratched areas were measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA). The wound healing rate was calculated as follows: wound healing rate = (original wound area - actual wound area at different times)/original wound area ×100%.
Cell invasion assays
Cells (5×104) were resuspended in FBS-free Dulbecco’s Modified Eagle’s Medium and seeded into the top chambers of Matrigel-coated Transwell inserts (BD Biosciences). The bottom compartments of the chambers contained 10% FBS as a chemoattractant. After 48-hour incubation at 37°C in 5% CO2, the cells on the upper surface of the membranes were wiped away, and cells on the lower surface of the membranes were washed with PBS, fixed in 4% paraformaldehyde, and stained with crystal violet to quantify the extent of invasion.
Real-time reverse transcription polymerase chain reaction
Total RNA was extracted from the ovarian carcinoma cell lines using TRIzol (Takara, Kyoto, Japan). Real-time reverse transcription polymerase chain reaction (RT-PCR) was performed using 2 μg total RNA using avian myeloblastosis virus (AMV) reverse transcriptase and random primers (Takara). 18S rRNA was used as the internal control. Complementary DNA (cDNA) amplification was performed using the SYBR Premix Ex Taq II kit (Takara) according the supplier’s protocol. Briefly, RT-PCR amplification was carried out in a final volume of 20 μL containing 10 μL 2× SYBR Premix Ex Taq, 0.08 μL primer, 0.4 μL ROX reference dye, and 1 μL template cDNA (50 μg/μL). The relative gene expression level (the amount of target normalized to endogenous control gene) was calculated using the comparative threshold cycle (Ct) method (2−ΔΔCt).16
Western blot analysis
Protein assays were performed according to the Bradford method using a Bio-Rad protein assay kit (Bio-Rad Laboratories Inc., Hercules, CA, USA). Denatured proteins were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to Hybond membranes (Amersham, Germany). The membranes were blocked for 1 hour in 5% skimmed milk in Tris-buffered saline with Tween 20 (TBST; 10 mM Tris–HCl, 150 mM NaCl, 0.1% Tween 20). Immunoblotting was performed with the following primary antibodies against GSK-3β, CDK1, CDK2, matrix metalloproteinase-9 (MMP9), cyclin D1, and Bcl-xL (1:500, Proteintech; Proteintech Group, Chicago, IL, USA). The membranes were incubated overnight at 4°C, rinsed with TBST, and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin G antibodies (1:5,000; Dako Denmark A/S, Glostrup, Denmark). After applying ECL Plus detection reagents (Santa Cruz Biotechnology Inc., Dallas, TX, USA), protein bands were visualized using X-ray film (Fujifilm, Tokyo, Japan). The immunoblots were washed with Western blot stripping buffer (pH 2–3; Nacalai, Tokyo, Japan) and were probed using a GAPDH-specific monoclonal antibody (1:2,000; Santa Cruz Biotechnology Inc.).
Immunofluorescence
Cells were grown on glass coverslips, fixed with PBS containing 4% formaldehyde for 15 minutes, and permeabilized with 0.2% Triton X-100 in PBS for 15 minutes at room temperature. After washing with PBS, the cells were incubated overnight at 4°C with Alexa Fluor 594 Phalloidin (Thermo Fisher Scientific, Waltham, MA, USA) to visualize the lamellipodia. Nuclei were stained with 1 mg/mL diaminophenylindole (Sigma-Aldrich Co.) for 5 minutes at room temperature. The coverslips were then mounted with SlowFade Gold antifade reagent (Thermo Fisher Scientific) and observed under a confocal laser microscope (Olympus Corporation, Tokyo, Japan).
Statistical analysis
All experiments were repeated three or more times. The t-test and the Mann–Whitney U-test were used for statistical analysis of the data. A P-value of <0.05 was considered statistically significant. SPSS Version 10 (SPSS Inc., Chicago, IL, USA) was used to analyze all data.
Results
Effects of AZD1080 in ovarian carcinoma cells
The A2780 and OVCAR3 carcinoma cell lines were exposed to 0.5 μM, 1.0 μM, 2.0 μM, and 4.0 μM AZD1080 or dimethyl sulfoxide, and subjected to MTT proliferation assay. A2780 and OVCAR3 cell proliferation was lower following AZD1080 treatment compared with the control cell lines (P<0.05; Figure 1A); AZD1080 suppressed ovarian carcinoma cell proliferation. We performed flow cytometric cell cycle analysis to further investigate the mechanism by which AZD1080 suppresses ovarian carcinoma cell proliferation, and we found that AZD1080 induced G1 arrest in the A2780 and OVCAR3 cells, this being dose dependent (P<0.05; Figure 1B). The wound healing and invasion assays also showed that AZD1080 decreased cell migration and invasion in a concentration-dependent manner (Figures 2 and 3). In addition, AZD1080 exposure suppressed lamellipodia formation in the two cell lines, as indicated by the loss of F-actin structure (Figure 4).
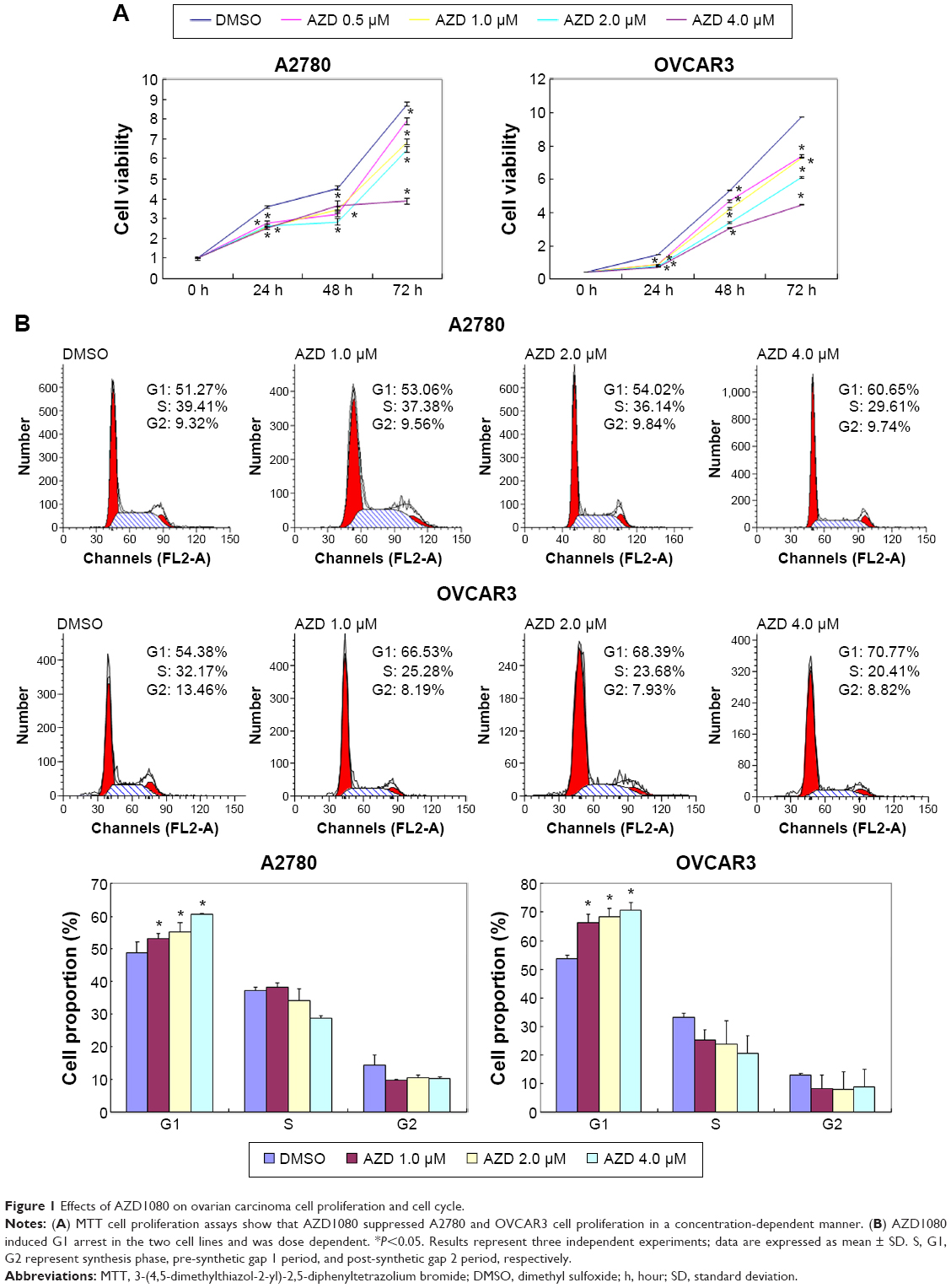 | Figure 1 Effects of AZD1080 on ovarian carcinoma cell proliferation and cell cycle. |
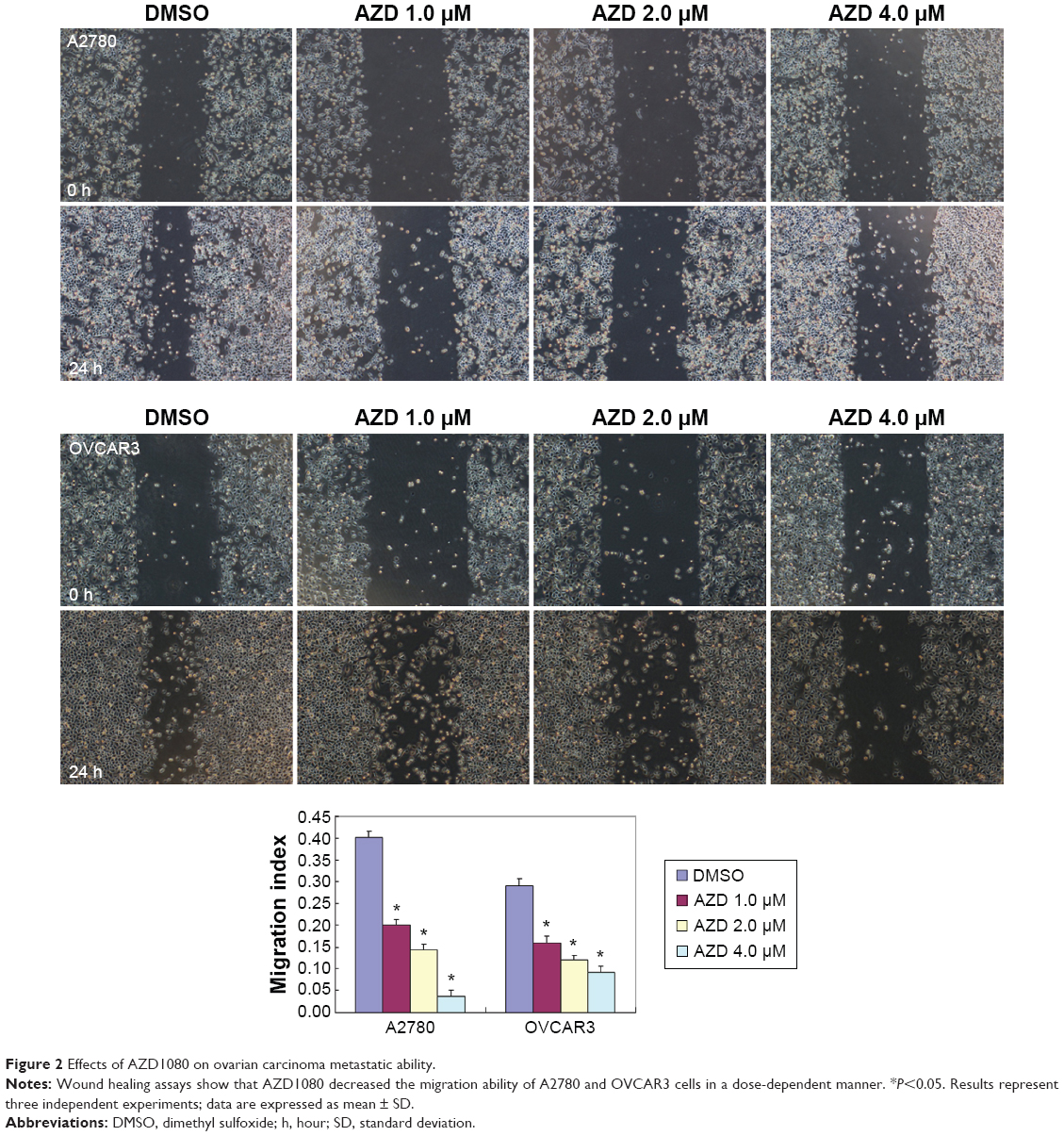 | Figure 2 Effects of AZD1080 on ovarian carcinoma metastatic ability. |
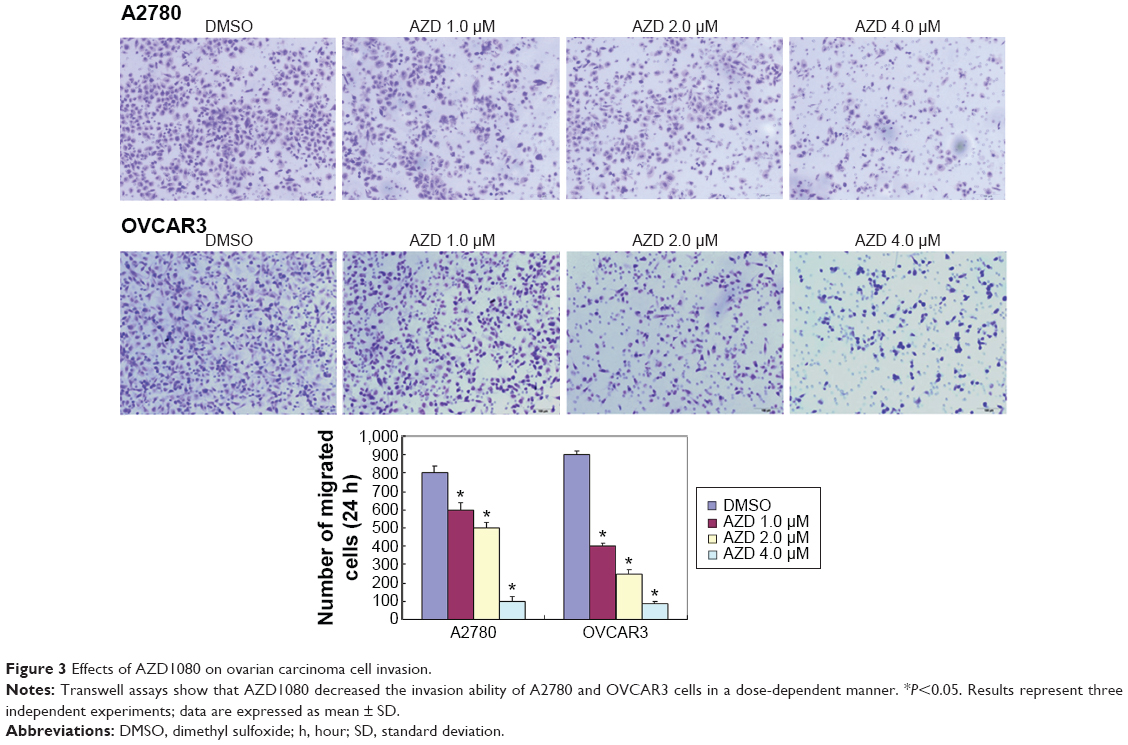 | Figure 3 Effects of AZD1080 on ovarian carcinoma cell invasion. |
 | Figure 4 Effects of AZD1080 on ovarian carcinoma cell lamellipodia formation. |
mRNA and protein expression of phenotype-related molecules in ovarian carcinoma cells after exposure to AZD1080
After AZD1080 treatment, the mRNA expression of GSK-3β, CDK2, CDK1, cyclin D1, MMP9, and Bcl-xL in the two ovarian carcinoma cell lines was lower than that in the control cells (Figure 5A). Western blot analysis (Figure 5B) demonstrated that AZD1080 downregulated GSK-3β, CDK2, CDK1, cyclin D1, MMP9, and Bcl-xL protein expression in the two cell lines, this being dose dependent.
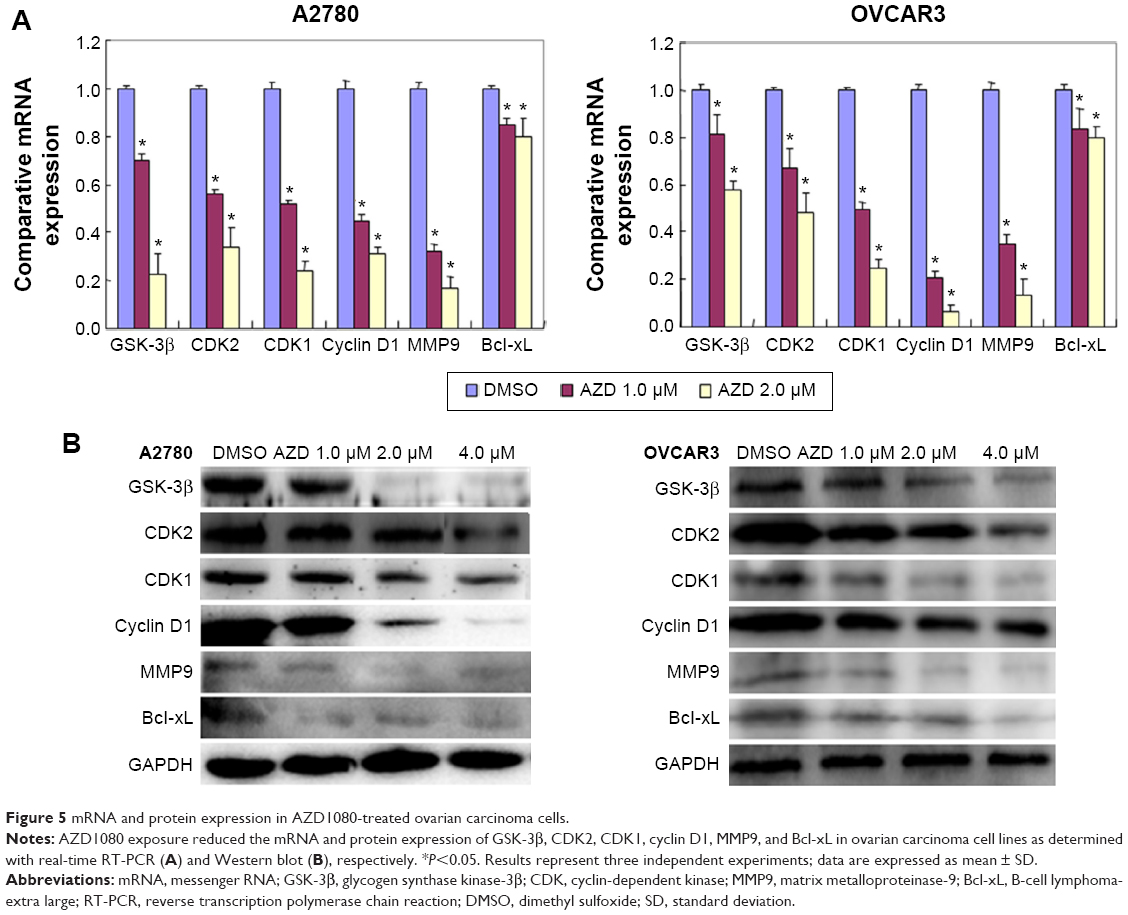 | Figure 5 mRNA and protein expression in AZD1080-treated ovarian carcinoma cells. |
Discussion
An increasing number of studies show that GSK-3β overexpression or aberrant kinase activity can increase cell proliferation and viability and promote cell malignant transformation, leading to tumorigenesis.17–21 Georgievska et al reported that GSK-3β inhibitors may inhibit cell proliferation through modulating cyclin-dependent kinases (CDKs). CDKs bind to cyclins, forming complexes that have protein kinase activity, promoting cell cycle phase transition, initiating DNA synthesis, and regulating cellular transcription and other functions; for example, CDK1 and cyclin B1 accelerate cell division and cell cycle progression via forward regulation.22,23 In prostate cancer, CDK1 activation induces cells to enter the mitosis stage while promoting MMP2 and MMP9 expression in tumor invasion and increasing metastasis.24 In hepatocellular carcinoma, downregulated CDK2 expression increased the proportion of cells in G1 and reduced the expression of cyclin D1.25 Studies have shown that GSK-3β inhibition via the β-catenin signaling pathway leads to depletion of cyclin D1, Bcl-xL, and MMP9.26–30 In short, GSK-3β plays a role in tumorigenesis and in tumor promotion and progression through regulating relevant genes. Therefore, inhibitors aimed at downregulating GSK-3β expression may have a role in treating tumors. Consequently, in this study, we investigated the role of the GSK-3β inhibitor AZD1080 in two ovarian cancer cell lines.
AZD1080, a novel GSK-3β inhibitor, has been reported to play a pivotal role in attenuating the downstream detrimental effects of signaling pathways activated by multiple stimuli relevant to Alzheimer’s disease.15 The selectivity of AZD1080 (at a concentration of 10 μM) was tested against different protein kinases, including GSK-3β, CDK2, and CDK1, and the results indicate, for the first time, that AZD1080 has the ability in humans to inhibit the GSK-3β enzyme at concentrations of 1–10 μmol/kg, offering the possibility of a dose-dependent acute oral treatment. We hypothesized that AZD1080 may inhibit ovarian cancer progression at doses of 0.125–16.0 μM. Our results showed significant reductions in the viability of cancer cells in both ovarian cancer cell lines at a dose of 1.0 μM and above. Therefore, we used doses of 1.0 μM, 2.0 μM, and 4.0 μM in further studies. AZD1080 also significantly decreased the expression of GSK-3β, CDK2 and CDK1, cyclin D1, MMP9, and Bcl-xL mRNA and protein. Following AZD1080 treatment, the cell proliferation of A2780 and OVCAR3 was decreased in a dose-dependent manner. AZD1080 also inhibits filopodia formation and cell invasion and metastasis, which may be subject to CDK regulation, while decreasing MMP9 protein expression.
AZD1080 has considerable potential for treating human cancers. As far as we know, we are the first to report the inhibitory effect of the GSK-3β inhibitor AZD1080 on ovarian cancer cell lines and its role in ovarian tumorigenesis and development. As it is a new potential anticancer drug, further in vitro tests and clinical studies are needed before it can be used to treat ovarian cancer clinically. We are confident that studying GSK-3β inhibitors will provide new ideas for ovarian cancer treatment.
Acknowledgment
This study was supported by grants (81202049, 81472440) from the National Natural Science Foundation of China.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Piek JM, van Diest PJ, Verheijen RH. Ovarian carcinogenesis: an alternative hypothesis. Adv Exp Med Biol. 2008;622:79–87. | ||
Bandera CA. Advances in the understanding of risk factors for ovarian cancer. J Reprod Med. 2005;50(6):399–406. | ||
Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2(10):769–776. | ||
Doble BW, Woodgett JR. GSK-3: tricks of the trade for a multi-tasking kinase. J Cell Sci. 2003;116(Pt 7):1175–1186. | ||
Kotliarova S, Pastorino S, Kovell LC, et al. Glycogen synthase kinase-3 inhibition induces glioma cell death through c-MYC, nuclear factor-kappaB, and glucose regulation. Cancer Res. 2008;68(16):6643–6651. | ||
Salim T, Sjölander A, Sand-Dejmek J. Nuclear expression of glycogen synthase kinase-3beta and lack of membranous beta-catenin is correlated with poor survival in colon cancer. Int J Cancer. 2013;133(4):807–815. | ||
Mishra R. Glycogen synthase kinase 3 beta: can it be a target for oral cancer. Mol Cancer. 2010;9(1):144. | ||
Tang QL, Xie XB, Wang J, et al. Glycogen synthase kinase-3β, NF-κB signaling, and tumorigenesis of human osteosarcoma. J Natl Cancer Inst. 2012;104(10):749–763. | ||
Madhunapantula SV, Sharma A, Gowda R, Robertson GP. Identification of glycogen synthase kinase 3alpha as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 2013;26(6):886–899. | ||
Rask K, Nilsson A, Brännström M, et al. Wnt-signalling pathway in ovarian epithelial tumours: increased expression of beta-catenin and GSK3beta. Br J Cancer. 2003;89(7):1298–1304. | ||
Marchand B, Tremblay I, Cagnol S, Boucher MJ. Inhibition of glycogen synthase kinase-3 activity triggers an apoptotic response in pancreatic cancer cells through JNK-dependent mechanisms. Carcinogenesis. 2012;33(3):529–537. | ||
Zhu Q, Yang J, Han S, et al. Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo. Prostate. 2011;71(8):835–845. | ||
Shakoori A, Mai W, Miyashita K, et al. Inhibition of GSK-3 beta activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci. 2007;98(9):1388–1393. | ||
Holmes T, O’Brien TA, Knight R, et al. Glycogen synthase kinase-3beta inhibition preserves hematopoietic stem cell activity and inhibits leukemic cell growth. Stem Cells. 2008;26(5):1288–1297. | ||
Georgievska B, Sandin J, Doherty J, et al. AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J Neurochem. 2013;125(3):446–456. | ||
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. | ||
Chou CH, Chou AK, Lin CC, et al. GSK3β regulates Bcl2L12 and Bcl2L12A anti-apoptosis signaling in glioblastoma and is inhibited by LiCl. Cell Cycle. 2012;11(3):532–542. | ||
Li T, Lai Q, Wang S, et al. MicroRNA-224 sustains Wnt/β-catenin signaling and promotes aggressive phenotype of colorectal cancer. J Exp Clin Cancer Res. 2016;35(1):21. | ||
Marchand B, Tremblay I, Cagnol S, Boucher MJ. Inhibition of glycogen synthase kinase-3 activity triggers an apoptotic response in pancreatic cancer cells through JNK-dependent mechanisms. Carcinogenesis. 2012;33(3):529–537. | ||
Shakoori A, Ougolkov A, Yu ZW, et al. Deregulated GSK3beta activity in colorectal cancer: its association with tumor cell survival and proliferation. Biochem Biophys Res Commun. 2005;334(4):1365–1373. | ||
Mazor M, Kawano Y, Zhu H, Waxman J, Kypta RM. Inhibition of glycogen synthase kinase-3 represses androgen receptor activity and prostate cancer cell growth. Oncogene. 2004;23(47):7882–7892. | ||
Cazales M, Schmitt E, Montembault E, Dozier C, Prigent C, Ducommun B. CDC25B phosphorylation by Aurora-A occurs at the G2/M transition and is inhibited by DNA damage. Cell Cycle. 2005;4(9):1233–1238. | ||
Lindqvist A, Kallstrom H, Lundgren A, Barsoum E, Rosenthal CK. Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J Cell Biol. 2005;171(1):35–45. | ||
Chang WL, Yu CC, Chen CS, Guh JH. Tubulin-binding agents down-regulate matrix metalloproteinase-2 and -9 in human hormone-refractory prostate cancer cells – a critical role of Cdk1 in mitotic entry. Biochem Pharmacol. 2015;94(1):12–21. | ||
Shi XN, Li H, Yao H, et al. In silico identification and in vitro and in vivo validation of anti-psychotic drug fluspirilene as a potential CDK2 inhibitor and a candidate anti-cancer drug. PLoS One. 2015;10(7):e0132072. | ||
Chien AJ, Moore EC, Lonsdorf AS, et al. Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci U S A. 2009;106(4):1193–1198. | ||
Lin P, Liu J, Ren M, et al. Idebenone protects against oxidized low density lipoprotein induced mitochondrial dysfunction in vascular endothelial cells via GSK3β/β-catenin signalling pathways. Biochem Biophys Res Commun. 2015;465(3):548–555. | ||
Campa VM, Baltziskueta E, Bengoa-Vergniory N, et al. A screen for transcription factor targets of glycogen synthase kinase-3 highlights an inverse correlation of NFκB and androgen receptor signaling in prostate cancer. Oncotarget. 2014;5(18):8173–8187. | ||
Gao X, He Y, Gao LM, et al. Ser9-phosphorylated GSK3β induced by 14-3-3ζ actively antagonizes cell apoptosis in a NF-κB dependent manner. Biochem Cell Biol. 2014;92(5):349–356. | ||
Bao H, Ge Y, Peng A, Gong R. Fine-tuning of NFκB by glycogen synthase kinase 3β directs the fate of glomerular podocytes upon injury. Kidney Int. 2015;87(6):1176–1190. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.