")
Back to Journals » OncoTargets and Therapy » Volume 13
Role of Focal Adhesion Kinase in Head and Neck Squamous Cell Carcinoma and Its Therapeutic Prospect
Received 1 July 2020
Accepted for publication 8 September 2020
Published 9 October 2020 Volume 2020:13 Pages 10207—10220
DOI https://doi.org/10.2147/OTT.S270342
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Yuxi Zhang,1 Xinchen Sun2
1The First School of Clinical Medicine, Nanjing Medical University, Nanjing, Mainland China; 2Department of Radiation Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Mainland China
Correspondence: Xinchen Sun Department of Radiation Oncology
The First Affiliated Hospital of Nanjing Medical University, Nanjing, Mainland China
Tel + 86 178 2606 0286
Email [email protected]
Abstract: Head and neck cancers are one of the most prevalent cancers globally. Among them, head and neck squamous cell carcinoma (HNSCC) accounts for approximately 90% of head and neck cancers, which occurs in the oral cavity, oral pharynx, hypopharynx and larynx. The 5-year survival rate of HNSCC patients is only 63%, mainly because about 80– 90% of patients with advanced HNSCC tend to suffer from local recurrence or even distant metastasis. Despite the more in-depth understanding of the molecular mechanisms underlying the occurrence and progression of HNSCC in recent years, effective targeted therapies are unavailable for HNSCC, which emphasize the urgent demand for studies in this area. Focal adhesion kinase (FAK) is an intracellular non-receptor tyrosine kinase that contributes to oncogenesis and tumor progression by its significant function in cell survival, proliferation, adhesion, invasion and migration. In addition, FAK exerts an effect on the tumor microenvironment, epithelial–mesenchymal transition, radiation (chemotherapy) resistance, tumor stem cells and regulation of inflammatory factors. Moreover, the overexpression and activation of FAK are detected in multiple types of tumors, including HNSCC. FAK inhibition can induce cell cycle arrest and apoptosis, significantly decrease cell growth, invasion and migration in HNSCC cell lines. In this article, we mainly review the research progress of FAK in the occurrence, development and metastasis of HNSCC, and put forward the prospects for the therapeutic targets of HNSCC.
Keywords: head and neck squamous cell carcinoma, focal adhesion kinase, targeted therapy, oncogenesis, cancer metastasis
Introduction
Over 650,000 individuals are diagnosed with head and neck cancer annually worldwide and over 50% of patients die from this disease, making it the sixth leading cause of cancer-related deaths globally as a motley collection of malignancies.1,2 Head and neck squamous cell carcinoma (HNSCC) accounts for approximately 90% of all head and neck cancers that arise in the oral cavity, oral pharynx, hypopharynx and larynx.3 Alcohol and tobacco consumption is associated with the tumorigenesis of over 70% HNSCC,4,5 and the pathogenesis of HNSCC is also related to the infection of human papilloma virus (HPV).6,7
Surgical resection remains the preferential therapy for oral carcinoma8 and advanced throat carcinoma.9 However, due to the complex anatomical structure and various vital organs, the operation on head and neck tissue is generally difficult, usually causing the failure of radical resection. Radiotherapy is one of the main therapeutic modalities for the management of HNSCC. Yet, because of the complexity of radiotherapy target volume of the region and multiple adjacent organs in danger, more-precisely targeted radiotherapy should be explored to mitigate the long-term adverse effects of radiation, such as severe pain and delayed healing and nonunion.10 Cisplatin-based chemotherapy can be simultaneously administered with definitive radiotherapy, accompany by adjuvant radiotherapy, or as induction chemotherapy. However, the toxicity of high-dose cisplatin is enormous, and the significant survival benefits of induced chemotherapy have been rarely revealed by relevant studies.11–13 Anti-epidermal growth factor receptor (EGFR) therapy (cetuximab) can improve the cure rate and simultaneously decrease the recurrence rate and mortality of HNSCC;14 nevertheless, no other molecular targeted therapy has been reported to prolong overall survival of patients. During the past few decades, the most notable progress in HNSCC treatment is the emergence of immunotherapy. To be specific, immunotherapy targeting programmed cell death 1(PD-1) has been recently approved for treating platinum-resistant HNSCC patients with recurrence and metastasis, which might hopefully extend tumor remission in certain patients with less toxicity than conventional chemotherapy.15,16
Therefore, the main challenge in the treatment of tumor in the complex anatomy of the head and neck regions is to achieve high cure rates while simultaneously maintaining essential structures and functions, as vital structures and functions are also affected by tumor itself and the subsequent treatment. Organ preservation should be sufficiently considered, and all therapeutic approaches should be attempted. Meanwhile, based on the latest data, the global survival rate of HNSCC has only slightly increased in recent years. The 5-year survival rate of HNSCC patients is only 63%, mainly because approximately 80–90% of patients with advanced HNSCC tend to have local recurrence or distant metastasis.17 Therefore, it is urgent exploit novel therapeutic approaches for better survival outcomes.
FAK, a non-receptor protein tyrosine kinase with 125KDA in size, was first described in 1992 as a member of the protein tyrosine kinase (PTK) family.18 PTK2, which encodes FAK, is mapped on human chromosome 8.19 FAK can be triggered by extracellular signals such as integrin-extracellular matrix (ECM) binding and some growth factors, including G protein-coupled receptor agonists,20 cytokines,21 epidermal growth factor,22 and hepatocyte growth factor.23 Therefore, FAK is a multi-functional regulator of cell signal between tumor cells and tumor microenvironment.
FAK consists of an N-terminal FERM domain, a central kinase domain, a following proline-rich region and a C-terminal focal adhesion targeting (FAT) domain. The most notable role of FERM domain is the autophosphorylation site of Y397. Moreover, the FERM domain (368–375) includes the binding sites with the Src SH3. Finally, it consists of a highly conserved sequence that binds to the p-Met receptor,24 phosphatidylinositol lipids (PLs),25,26 and the C-terminal FAT domain via intramolecular interactions.27 The FAK kinase domain includes the ATP binding site with the activation loop from residues 564–585. The proline-rich C-terminal region (686–917) contains the sites interacting with p130Cas,28 and activating proteins of small GTPase ASAP and GRAF.29,30 The FAT domain (922–1050), comprising extreme C-terminal, interacts with other adhesive proteins, such as paxillin31 and talin,32,33 and thereby targeting FAK with focal adhesion complexes through its scaffolding protein function. The proline-rich sequences before the FAT domain (910–921) contains the phosphorylation sites of extracellular regulated protein kinase (ERK), S910.34 Further, C-terminal FAK includes two major phosphorylation sites Y925 and Y861, which are both activated by Src kinases (Figure 1). Once activated by integrin-ECM engagement or growth factors, Y397 autophosphorylates the site into a high-affinity binding site to SH2 domain of Src. In FAK, Src interacts with pY397 through its SH2 domain and to the linker between the FERM and kinase domains via the SH3 domain. In turn, Src phosphorylates several tyrosines Y576 and Y577 in FAK activation zone, subsequently inducing the complete catalysis of FAK activity.
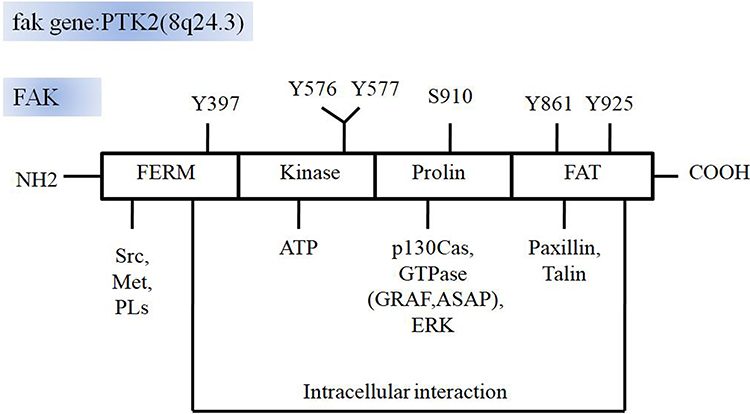 |
Figure 1 The structure of FAK. PTK2 encodes FAK and is mapped on human chromosome 8q24.3. FAK consists of a FERM domain, a kinase domain, a proline-rich region and a FAT domain. FERM domain involves the Y397 autophosphorylation site and binding sites with the Src, Met and PLs, as well as the intramolecular interactions with FAT domain. The FAK kinase domain includes the ATP binding site with the activation loop. The proline-rich region contains the sites interacting with p130Casand activating proteins of small GTPase ASAP and GRAF. The FAT domain can interact with other adhesive proteins, such as paxillin and talin. The proline-rich sequences before the FAT domain contains ERK phosphorylation sites S910 and another two major phosphorylation sites Y925 and Y861. |
Based on the well-known effects of FAK on diverse biological processes of cell survival, proliferation, adhesion, invasion and migration, overexpression and increased activity of FAK have been suggested to play a crucial role in tumor occurrence and development. Hence, FAK is a potential anti-tumor therapy target, particularly in HNSCC, known as an invasive tumor. This article aims to review the research progress of FAK in the tumorigenesis, development and metastasis of HNSCC, and to propose the prospects for the therapeutic targets of HNSCC.
Linkages Between FAK and HNSCC
In 2009, Xia J et al found that the expression of FAK in normal mucosa, dysplasia epithelium of varying degrees and oral squamous cell carcinoma (OSCC) tissues were significantly increased with the progression of multi-stage oncogenesis when inducing rat tongue cancer, which supported the role of FAK in oral carcinogenesis.35 More recently, increased expression of FAK was found in 40 (49%) of 82 laryngeal dysplasia, whose expression was maintained or further enhanced as patient-matched dysplasia subsequently developed into invasive tumors, suggesting the involvement of FAK in malignant transformation of laryngeal cancers and its potential clinical utility as a biomarker for laryngeal cancer.36 Moreover, by analyzing the immunohistochemical staining of nine benign, 19 premalignant, and 19 malignant oral tissues, Chiu YW et al showed that the immunoreactivity of FAK and pY397 was positively correlated with the degree of malignancy. Similarly, the high-invading cell line exhibited higher expression levels of FAK and phosphorylated-Y397 (p-Y397) compared with the low-invading one.37 Besides, the overexpression of p-Y397, FAK Tyr-576 and FAK Tyr-925 has been recently observed in tumor zones and non-neoplastic adjacent epithelial tissue of HNSCC.38 These results indicate that FAK and its phosphorylation form are correlated with tumor invasion and metastasis. Interestingly, there was an exception. In another study including48 mobile tongues squamous cell carcinoma (SCC) tissue samples, high FAK expression was significantly associated with high grade of tumor differentiation and prolonged disease-free survival of patients, which might suggest that FAK overexpression is not limited to aggressive phenotypes but also new events in HNSCC oncogenesis.39
Despite some progress recent decades, the mechanisms underlying the activation and overexpression of FAK in HNSCC remains largely unclarified. Back in 1999, increased expression of FAK gene was detected in many cell lines derived from invasive epithelial tumors, which may contribute to the generally observed elevated protein expression.40 In terms of HNSCC, relevant studies have shown that overexpression of FAK is related to gene amplification both in HPV-negative HNSCC cell lines and clinical tumors.41 Nevertheless, gain of FAK gene copy number are not always associated with increased expression or activity of FAK. In a study of 211 samples of head and neck tissues, FAK protein was overexpressed in compared with corresponding normal mucosa. Additionally, DNA copy number ratios for FAK were higher compared with normal mucosa. However, the high expression of FAK protein was independent of gain at the DNA level, and not all cases displaying FAK gene amplification showed protein overexpression. Similar data were also observed in several HNSCC-derived cell lines, where the mRNA levels of FAK were accurately correlated with its protein expression.42 These results suggest that FAK are involved in the tumorigenesis and development of HNSCC, providing new clues for FAK activation mechanisms instead of amplification genes. Hence, epigenetic mechanisms may also account for the overexpression and increased activity of FAK. Recent studies of human FAK gene promoters suggest that p53 and NF-κB are potential direct inhibitors and activators of promoters, respectively.43,44 However, no correlation was found between p53 positivity and FAK expression in samples from surgically-treated supraglottic larynx SCC (with postoperative radiotherapy in 49 cases).45 On the contrary, a more recent study investigating the expression of FAK and p53 in normal oral mucosa and OSCC revealed that FAK expression was negatively correlated with p53 expression, which supported that FAK activation might participate in p53 down-regulation in OSCC.46 The different results of the above two studies may be due to the differences in tumor location and therapeutic approaches, which require further investigation.
Mechanisms of FAK in Oncogenesis
The overexpression of FAK in precancerous lesions of HNSCC suggests a potential role for FAK in HNSCC oncogenesis. As mentioned before, FAK can be activated by ECM integrins and growth factors in healthy cells, and multiple promotors and suppressor of tumor transformation can regulate the expression and activation of FAK. The overexpression and abnormal activation of FAK can promote the occurrence of HNSCC by regulating cell survival and proliferation (Figure 2).
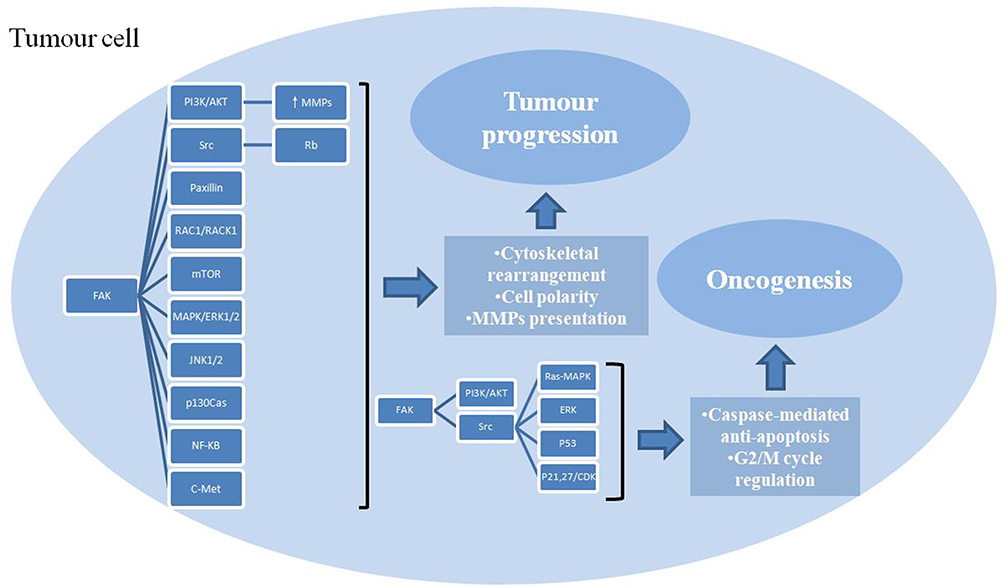 |
Figure 2 Multiple roles of FAK in tumor cells and multiple signaling pathways involved. FAK promotes cell survival and proliferation through caspase-mediated anti-apoptosis and G2/M cycle regulation, enhances invasion and migration via cytoskeletal rearrangement, cell polarity alteration, MMPs presentation, thus leading to oncogenesis and tumor progression. |
The activation of FAK during cell adhesion has been found to protect cells from anoikis, a form of apoptosis induced by cell isolation from ECM, contributing to tumor growth and metastasis.47 In 2012, a study revealed the high expression of p-FAK in patients with tongue OSCC. Importantly, FAK inhibitor TAE226 could significantly suppress the proliferation of human OSCC cells with an apparent structural change of actin fibre and a loss of cell adhesion. Additionally, TAE226 inhibited the expression of p-Y397, causing caspase-mediated apoptosis. Furthermore, TAE226 administration in mice OSCC xenografts suppressed the tumor growth in vivo.48 These results offer strong evidence that FAK is closely related to OSCC and that FAK inhibitors may be potentially be used for effective OSCC treatment.
One of the mechanisms lies in FAK- Src complex formation and its interaction with Ras-GTPase activator protein SH3 domain-binding protein 1(G3BP1), which inhibits apoptosis by regulating Ras/MAPK, TGF-β/Smad, Src/FAK and p53 signaling pathways.49 Another mechanism is that FAK induces the decreased expression of cyclin-dependent kinase (CDK) inhibitors p21 and p27. Simvastatin (a Src inhibitor) was revealed to inhibit cell proliferation, along with increased expression of p21, p27 and activated caspase-3. In contrast, the appearance of p-FAK was decreased, indicating the involvement of FAK in anti-apoptosis by reducing death receptor complex.50
Several studies have shown that suppression of expression and activation of FAK in OSCC cell lines is correlated with caspase-dependent apoptosis and G2/M cell cycle arrest.51,52 The suppression of different domains of FAK could induce apoptosis and cause G2/M cell cycle. Han et al used human oral keratinocytes to assess potential fenretinide-FAK drug-protein interactions and functional consequences on cellular growth regulation. The results showed that the drug-FAK interaction significantly suppressed cell proliferation by inducing apoptosis and G2/M cell cycle blockade.51
Recent researches have also shown that FAK contributes to cell survival by inhibiting the function of p53.46,53–55 Specifically, Zhang Y et al found that apoptosis was increased by blocking FAK with an antisense oligonucleotide and decreased by blocking p53 in SCC cells. Intriguingly, p-FAK was reduced while p53 level was increased in single cells but not aggregates. These findings show that SCC cells may escape suspension-induced anoikis and obtain the metastatic phenotype under the assistance of adjacent cells and the ECM molecule fibronectin.53
Apart from the decisive role in survival, FAK also promotes cell proliferation by stimulating cell cycle progression. Signaling pathways mainly include FAK/PI3K/AKT56–59 and FAK/ERK axis.60,61 A study showed that increased expression of FAK in slow-growing SCC25 cells could increase ERα phosphorylation, transcriptional activity and cell growth rate. Conversely, knockdown of FAK in rapidly growing OECM-1 cells decreased ERα phosphorylation and activity, and delayed cell growth.62 Upregulation of RACK1 protein has been reported to promote the proliferation of nasopharyngeal carcinoma (NPC) via the PI3K/AKT/FAK signaling pathway.56
DecreasedAGO2 protein has been found to retard cell proliferation, arrest cell cycle and induce apoptosis by inhibiting the FAK/PI3K/AKT signaling pathway in FaDu cell line,58 suggesting that AGO2 gene may be involved in malignant phenotype by acting as an oncogene. Antioncogene can also regulate the activation of FAK. CLCA2, a tumor suppressor gene, has been reported to be dysregulated in breast cancer. One recent study has suggested that it also inhibits proliferation in NPC by suppressing the FAK/ERK signaling.61
Mechanisms of FAK in HNSCC Development and Metastasis
Development and metastasis of tumor are a complex process, consisting of detachment, invasion, migration, extravasation, and proliferation of cancer cells at the distal part of the body. FAK can induce morphological changes in cells, which further contributes to cell movement. And the signaling pathway promotes tumor progression and metastasis by regulating cell adhesion, invasion and migration (Figure 2).
One important mechanism lies in the increased expression and activation of metalloproteinases (MMPs). For instance, decreased expression of MMP-2 was involved in the inhibited motility of SCC-9 and SCC-14 cells.63 In another study, knockdown of FAK inhibited the invasion and migration of Tca-8113 by attenuating MMP-2 and MMP-9 activities.64 Similar results are also detected in SCC-4 cells,65,66 SAS cells,67 SCC40 and SCC38 cell lines.68 Patients with OSCC have been reported to elicit a predominance of MMP-9 activity by up-regulating the FAK/PI3K/AKT pathway.69 Inhibition of MMP-2 and MMP-9 signaling may result from the down-regulation of PKC and RhoA by blocking MAPK and FAK/PI3K/AKT signaling pathways and NF-κB, IKK and uPA-mediated pathways.66,67 Besides, overexpression of MMP-11 can also increase OSCC cell migration through the FAK/Src pathway.70 A study concerning FAK-related nonkinase (FRNK) showed that FRNK expression could downregulate the expression of MMP-2 and MMP-9. In contrast, MMP-2 overexpression rescued FRNK-inhibited cell invasion in FRNK-expressing cells.68 Another study found that phosphorylation of FAK and activation of paxillin and SATA1 pathways resulted in overexpression of target genes such as MMP-2 and MMP-26, which was suggestive of cell invasiveness and migration in HNSCC cells.71 Moreover, the expression and activity of MMP14 were found to be attenuated by inhibition of FAK (PF573228) or Src (dasatinib), thus mitigating cell invasion. Further mechanism research showed that Src-FAK/AKT/AP-1 signaling might play a role in MMP14-mediated remodeling of the ECM.72
Metastasis remains a clinically unsolved issue, especially in NPC. A study found that the inhibition of colony formation and motility in NPC cells was correlated with decreased expression of p-FAK and increased expression of E-cadherin.73 In another research, downregulation of E-cadherin along with enhanced cytoskeletal rearrangement and cell polarity was associated with the activation of FAK, thereby promoting cell migration.74 RACK1 protein has been found to play an essential role in increasing the proliferation and invasion of NPC by regulating the PI3K/Akt/FAK signaling pathway.57 Moreover, p-Y397 plays a critical role in FAK-regulated Rac1 activation and invasiveness of OSCC cells.37
Recent research has demonstrated that spreading of endothelial cells in head and neck cancer was altered by inhibiting mTOR, which was associated with augmented p-FAK and enhanced migratory behaviour.75 Abundant researches have revealed that FAK is involved signaling pathways that promote cancer cell adhesion, invasion and migration. EFNB2 specific siRNA transfected into SAS-L1 cells significantly reduced cell adhesion, invasion and migration via the phosphorylation of EGFR, FAK, AKT, ERK1/2, JNK1/2 and p38 pathways.76 A cell cycle control kinase, aurora kinase A, was found to revive dormant tumor cells from G0 phase to active division via activating FAK/PI3K/AKT signaling, thereby promoting invasion and migration in laryngeal cancer.77 Reduction of PTEN activates PI3K-AKT, FAK-p130Cas and Shc-MAPK/ERK1/2 signalings, which drives epithelial to mesenchymal transition (epithelial to mesenchymal transition) and consequently increases the invasion and migration of NPC cells.78 Downregulation of RhoC leads to inactivation of FAK, Src and ERK1/2 signaling pathways in HNSCC, thus reducing cell motility, invasion colonies and stress fibre formation.79 Knockdown of PODXL in the SAS OSCC cell line attenuated tumor invasion and migration by decreasing the phosphorylation of FAK and paxillin.80 Cysteine-rich 61 (Cyr61) is a secretory and matrix-associated protein. Cyr61 has been indicated to enhance the migration of OSCC cells through integrin receptor, FAK, ERK, MEK and NF-κB signaling pathways.81 5ʹ-Nitro-indirubinoxime has been reported to suppress the metastatic ability of human head and neck cancer cells through the inhibition of Integrin β1/FAK/AKT signaling.82 Inhibition of aquaporin 3 caused cell death, which was caused by directly interfering with cell adhesion through FAK-MAPK signaling.83 Suppression of integrin α4 significantly reduced the CS1-regulated cell spreading, which was mediated by FAK.84 A study of diabetic rats indicated that diabetes reduced cell adhesion by altering IRS-1/FAK pathway, thereby increasing the risk of oral cancer.85 The activation of FAK may regulate the hepatocyte growth factor/scatter (HFG/SF)-induced migration of human OSCC cells.86
FAK-Mediated Angiogenesis in Tumor
Anti-angiogenesis therapy is a promising and rational option in cancer treatment. FAK plays a role in promoting cell proliferation, invasion and migration, suggesting that FAK has a potential effect on germination of endothelial cell and angiogenesis in tumor, which composes an indispensable part in tumor metastasis. In the tongue SSC-9 cell line, the phosphorylation of vascular endothelial growth factor receptor 2 (VEGFR2), as well as Src and FAK, downstream of the vascular endothelial growth factor (VEGF)/VEGFR2 pathway was inhibited by Sema3A, which further significantly decreased angiogenesis and drastically suppressed tumor growth.87 FAK inhibitor TAE226 suppressed the growth and angiogenesis of OSCC xenografts in mice.48 FRNK is the primary negative inhibitor of FAK, and its expression decreases FAK phosphorylation and inhibits EGF induced MMP-9 transcription and secretion, thereby decreasing invasion in follicular thyroid carcinoma cells.88 All these studies highlight the role of FAK in promoting cell invasion by increasing angiogenesis.
FAK in Epithelial to Mesenchymal Transition (EMT)
EMT is considered as a prerequisite for tumor cells to acquire an aggressive or migratory phenotype and subsequent metastasis, during which polarized epithelial cells become moving interstitial cells. Therefore, EMT is crucial for the progression and metastasis of cancer.
In Kras/p53 mutant mouse model and human lung cancer cell lines, EMT has been found to activate FAK/Src signaling pathway and improve tumor cell responsiveness to the ECM composition.89 As for HNSCC, CLCA2 has been demonstrated to suppress EMT in NPC through inhibiting FAK/ERK signaling,61 while DLC-1 can induce EMT arrest in NPC by targeting EGFR/AKT/NF-κB pathway.90 Besides, in laryngeal cancer, AURKA induced EMT by activating FAK/PI3K pathway.91 Numb can inhibit EMT in tongue cancer via Notch1/PTEN/FAK signaling pathway.92 Furthermore, Western blot analysis revealed that knockdown of FAK inhibited EMT in tongue cancer cell line.64 The FAK phosphorylation inhibitor PF573228 reversed EMT in V-SAS-LM8 cells.93 Therefore, the activation of FAK is proved to promote EMT in several human HNSCC cells and play a role on EMT-induced tumor progression.
FAK in Tumor Microenvironment
ECM regulates cancer characteristics. Exploration of tumor microenvironment may shed novel light on the understanding of tumor development and metastasis.
In 2017, a study found that morphological changes affecting cell-matrix interaction, and actin cytoskeleton recombination were mainly caused by inhibiting FAK activation and increasing RhoA activity and MLC-2 phosphorylation, thus preventing cell migration.94 In OSCC cells, silencing of AGRIN interferes with tumor cell proliferation, invasion, motility, and tumor formation, and reduces the phosphorylation of FAK, ERK and cyclin D1 proteins.95 Mesenchymal stem cells (MSCs) within the tumor microenvironment play a critical role in tumorigenesis, progression and therapeutic response. IL1-β and CDH1 has been reported to dictate FAK and MAPKK-dependent crosstalk between cancer cells and MSCs.96 In interstitial fibroblast, cancer vascular dissemination and local invasion were enhanced through YAP1-FAK/PXN signaling.97 Carcinoma-associated fibroblasts (CAFs) have also been uncovered to exert an essential impact on the tumorigenesis and progression of OSCC. Knockdown of FAK in CAFs can inhibit OSCC metastasis.98 Connective tissue growth factor (CTGF), a secretory protein binding to integrins, can reduce COX-2 expression through signal transduction pathways such as integrin receptors, FAK, PI3K, AKT, c-Jun and AP-1, thereby inhibiting migration of oral cancer cells.99
Similarly, decreased expression of CTGF could promote cell proliferation, invasion, migration and cell cycle process through FAK/PI3K/AKT, EMT and MMP pathways in NPC.100 HGF/SF factor stimulates mitogenesis and movement of an HNSCC cell line by rapidly increasing the phosphorylation of both FAK and ERK.101 Circulating endothelial cells are released from primary tumors and migrate to the distal sites together with tumor cells. Intriguingly, a study suggested that Bcl-2 overexpression in endothelial cells (EC-Bcl-2) significantly enhanced the expression of adhesion molecules and the binding of tumor cells, which was mainly mediated by E-selectin. Moreover, tumor cells bound to EC-Bcl-2 displayed a tendency of anti-apoptosis by activating the Src/FAK pathways.102
FAK and Inflammatory Factors
Tumor-derived IL-1β, one mediator of the pro-inflammatory cytokine released to tumor culture medium, has been found to be positively regulated by FAK and MAPK signaling and negatively regulated by TGF-β signaling in several human cancer cell lines, including head and neck FaDu cell line.103
TRAF2 plays an essential role in tumor necrosis factor (TNF) during inflammatory and immune responses, which physically interacts with the N-terminal portion of FAK and colocalizes to cell membrane protrusions. This cooperative interaction between TRAF2 and FAK has been confirmed to promote resistance to cell anoikis in human breast cancer cell line. Moreover, analysis of The Cancer Genome Atlas (TCGA) database revealed that co-amplification of TRAF2 and FAK in breast cancer tissues has predictive value for short survival, further supporting their potential role in cancer progression.104
IL-6 has also been reported to promote metastasis in head and neck tumor by inducing EMT via the JAK-STAT3-SNAIL signaling pathway, and STAT3 knockdown significantly reversed IL-6-mediated cell motility by inhibiting FAK activation.105 Inflammation is widely known to have a close relationship with tumor formation and development. At present, researches on the correlation between inflammatory factors and FAK are insufficient, thus, further evidence is warranted to profoundly demonstrate FAK role in inflammation.
Regulation of Cancer Stem Cells (CSCs)
The signal between CSCs and its ECM plays a critical role in the development and maintenance of CSC. Treatment with FAK inhibitor has revealed to decrease cell self-renewal capacities and expression of various putative stem cell markers such as Oct4, Sox2, and Nanog in a dose-dependent manner, indicating a significant role of FAK in the regulation of stemness in HNSCC.106 Another study showed that CSC properties could also be regulated by the activation of the FAK-mediated signaling pathways in oesophageal squamous.107 While mutant EGFRvIII-mediated cross-activation of tyrosine kinase receptor and c-Met could regulate the CSC maintenance and promote tumor recurrence in glioblastoma (GBM).108
Combination with Other Treatments
The Role of FAK and FAK Targeting in Chemotherapy
Chemotherapy can be adopted as concurrent definitive radiochemotherapy, postoperative adjuvant radiochemotherapy or induction chemotherapy. However, due to the emergence of chemoresistance, many patients are not sensitive to chemotherapy and suffer from relapse after these treatments. Therefore, it is necessary to investigate the mechanisms of chemoresistance.
A study has shown that knockdown of KRT16 enhanced the chemosensitivity of OSCC towards 5-fluorouracil. Mechanistically, depletion of KRT16 inhibited the Src/STAT3/FAK/ERK signaling, thereby causing degradation of integrin β5 and c-Met.109 LncRNA KCNQ1OT1 has been reported to regulate cisplatin resistance in tongue cancer via Ezrin/Fak/Src signaling.110 A small-molecule inhibitor targeting miR-21 has recently been found to inhibit CDK5 activity by targeting CDK5RAP1, CDK5 activators p39 and p-FAK, which is likely to be a promising strategy against taxol-induced tumor metastasis.111 Another research has shown that up-regulated expression level of Bmi1 through a FAK-dependent mechanism led to enhanced chemoresistance against cisplatin in OTSCC cells, which adds new insights into the molecular mechanisms underlying OTSCC chemoresistance.112 ECM factors in the tumor microenvironment can also control drug resistance. For instance, a study found that adhesion in the tumor matrix can create an environment in which cisplatin-induced proliferation through integrin β, talin and FAK pathways that regulate NF-kB nuclear activity.113
The Role of FAK and FAK Targeting in Radiation Therapy
Radiotherapy is a commonly used therapeutic strategy for HNSCC, but local failure following radiotherapy remains the leading cause of cancer-related death. So far, cetuximab, an inhibitor of EGFR is the only one targeted, radiosensitizing agent that has been approved for the treatment of HNSCC. However, less than 20% of all eligible patients could gain benefits from the addition of cetuximab to radiotherapy.114 Despite extensive studies, there are still a lack of reliable predictive biomarkers for cetuximab response. Therefore, it is definitively needed to identify new biological pathways related to radioresistance to provide other therapeutic targets.
A recent proteomic analysis showed that the overexpression of FAK was a biomarker for radioresistance in locally advanced HNSCC, and inhibition of FAK radiosensitized HNSCC cells with enhanced G2-M arrest and DNA damage.41 These results suggest that FAK inhibition combined with radiotherapy is worthy of further evaluation for elevating local control rates in HPV-negative HNSCC. Signal of integrin and receptor tyrosine kinases also play an essential role in the therapeutic resistance of malignant tumors. One study has demonstrated that FAK/ERK1-mediated antiradiation precursor signaling is effectively disrupted by integrin β1 and EGFR blockade, suggesting that integrin β1 and EGFR combined targeted therapy is an effective and promising way to overcome radiation resistance.115 Further researches have revealed targeting EGFR/integrin β1 radiosensitized three-dimensional SCC cell cultures, which may be due to attenuated repair of DNA double-strand breaks.116 Combined EGFR/FAK targeting shows more powerful radiation sensitization than either method alone, which may be based on non-overlapping signals downstream.117 Above all, further in-depth clarification is needed on whether EGFR and FAK dual targeting can be reasonably combined with chemoradiotherapy.
Integrin α3 has been reported to be involved in radioresistance in HNSCC cells along with integrin β1, determined by overexpression of the integrin signaling mediator FAK.118 Earlier, Eke I et al found that Integrin β/FAK/cortactin signaling induced cell rounding, leading to radiosensitization in human head and neck cancer,119 implicating that pharmacological targeting integrin β may provide therapeutic benefit to overcome tumor cell resistance to radiotherapy.
Besides, silencing fibronectin extra-domain A (EDA) has also been found to enhance the radiosensitivity of FAK/AKT/JNK pathways in NPC.120 Another study has shown that targeting FAK radiosensitized human HNSCC cells by attenuating AKT1 and MEK1/2 signaling, actively supporting FAK as a relevant molecular target for radiotherapy in HNSCC.121 In 2009, Hehlgans S et al investigated several kinds of cancer cell lines, which revealed a dose-dependent reduction in clonogenic survival. Interestingly, TAE226 only greatly radiosensitized HNSCC cell cultures accompanied by dephosphorylation of FAK, AKT and ERK1/2, which demonstrated FAK inhibitor TAE226 enhanced the radiosensitivity particularly in HNSCC cells.122 However, future studies both in vitro and in vivo should be performed to clarify the specific extent of this approach may be clinically relevant to HNSCC radiotherapy.
Prognostic and Predictive Value of FAK Alterations
A retrospective study of 87 surgically treated patients with hypopharyngeal carcinoma from 2004 to 2012 in Japan found that positive staining of FAK was significantly associated with the number of metastatic lymph nodes. The disease-specific survival rate and the incidence of distant metastasis in FAK positive patients were markedly lower than those in the FAK negative patients, indicating that up-regulation of FAK expression was associated with tumor spreading and poor prognosis in patients with hypopharyngeal carcinoma.123 Another research on laryngeal cancer has revealed that FAK expression was an independent predictor of tumor development, and combined use of FAK and CTTN exhibited superior predictive value, providing the first evidence for their potential clinical application as biomarkers for laryngeal cancer.36 Similar results can be observed in OSCC.124 As one of the most important prognostic factors of cancer, lymph node metastasis is widely studied in HNSCC. Jiang H et al found that the cervical lymph node metastasis of tongue cancer was significantly correlated to FAK expression.125 In terms of patients with clinically negative lymph nodes, Rodrigo JP et al discovered that the combination of E-cadherin and FAK expression could improve the differential diagnosis of patients who were at considerable risk for occult metastases.126 Further study has shown that increased expression of FAK Tyr-576 could identify tumors with more aggressive behavior and epithelial alterations before clinical or histological evaluations.38
Moreover, studies show the potential application of FAK protein expression as a risk marker in tumor. For instance, an immunohistochemical study of 109 patients with precancerous laryngeal lesions supports the applicability of CTTN and FAK as auxiliary indicators of risk stratification.127 In another research enrolling patients with HPV-negative locally advanced HNSCC, gene amplification and mRNA expression of FAK were both greatly associated with worse disease-free survival.44 However, there are some exceptions. In a retrospective study of 224 HNSCC patients treated with primary radiation (chemotherapy) from 1996 to 2005, the expression of FAK and p-FAK was not associated with local control and distant metastasis.128
Similarly, weak FAK expression is also related to lymph node metastasis, recurrence and wretched prognosis in cervical cancer.129 These results suggest that FAK might promote the malignancy of primary neoplasia, yet not definitely leading to distant metastasis. The prognosis potential of FAK may also be related to its therapeutic effects.
Potential Clinical Use of FAK
There are a number of candidates under different stages of pre-clinical and clinical trials and potential efficacy responses in tumor regression and disease stabilization have been observed.
PF-562,271 (also termed VS-6062) is a bisamino pyrimidine derivative as a FAK activity inhibitor. With VS-6062, 31 patients (34%) experienced stable disease (SD) at the first restaging (end of cycle 2). Fifteen of these patients had SD for six or more cycles including two cases of HNSCC.129 Results from the Phase II clinical trials (NCT01951690) for Defactinib (VS‐6063) in patients with KRAS mutant non-small cell lung cancer showed that the drug was generally well tolerated and suitable for long‐term dosing. It is also found that FAK inhibitors need to be combined with other agents as they have not yet shown evidence of single agent activity. Pre-clinical trials for selective FAK inhibitor VS-4718 showed that FAK inhibition enhances immune surveillance by overcoming fibrosis and the immunosuppressive tumor microenvironment in pancreatic ductal adenocarcinoma, thus enabling tumors to respond to immunotherapy.130 Recently, the combination of the FAK inhibitor GSK2256098 and the MEK inhibitor trametinib in recurrent advanced pancreatic ductal adenocarcinoma did not provide significant clinical activity in a phase II trial with the PFS of 1.6 month and OS of 3.6 months.131 In malignant pleural mesothelioma, defactinib in maintenance after first-line chemotherapy in a phase II trial did not provide any benefit either.132 Several clinical trials with defactinib associated with immunotherapy (NCT02758587, NCT03727880, NCT02943317), RAK/MEK inhibitor (NCT03875820), or chemotherapy (NCT02546531) are ongoing.
Conclusions and Future Directions
This review mainly summarizes the role of FAK in cell survival, growth, adhesion, invasion, migration, EMT, and regulation of tumor microenvironment, CSCs, inflammatory factors and radiation (chemotherapy) resistance. These constitute one of the strongest rationales for FAK inhibition as a potential target in HNSCC treatment, whose effects are mediated by changing both tumor and stromal cell biology. Future studies are necessary to confirm whether FAK signaling plays a decisive role in the development and progression of HNSCC or just performs as a combination with other signaling pathways. At present, there are several Phase I studies evaluating the safety and activity of FAK inhibitors, including PF-562,271, Defactinib, VS-4718 and GSK2256098, among solid tumors. However, there is no information about FAK inhibitor in phase II studies in HNSCC patients at present. Future directions include possible combination studies with existing standard and emerging therapies (PD-1/PD-L1, MEK inhibitors and chemotherapy). Therefore, we hope to develop specific FAK inhibitors with clinical significance in HNSCC biology through the regulation of radiation (chemotherapy) resistance, anti-tumor immunity and angiogenesis. Moreover, the role of FAK alteration in predicting HNSCC prognosis is still unclear and more well-designed clinical trials and high-quality Mata analysis are needed. Further research is needed to find predictive biomarkers of response to FAK TKI alone or, probably more promising, in association with another drug. Future research into the FAK-associated pathways will elucidate new therapeutic combinations and biomarkers for patient stratification.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386.
2. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–1953. doi:10.1002/ijc.31937
3. Wyss A, Hashibe M, Chuang SC, et al. Cigarette, cigar, and pipe smoking and the risk of head and neck cancers: pooled analysis in the international head and neck cancer epidemiology consortium. Am J Epidemiol. 2013;178(5):679–690. doi:10.1093/aje/kwt029
4. Maier H, Dietz A, Gewelke U, Heller WD, Weidauer H. Tobacco and alcohol and the risk of head and neck cancer. Clin Investig. 1992;70(3–4):320–327.
5. Blot WJ, McLaughlin JK, Winn DM, et al. Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res. 1988;48(11):3282–3287.
6. Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol. 2011;29(32):4294–4301. doi:10.1200/JCO.2011.36.4596
7. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Human papillomaviruses. IARC Monogr Eval Carcinog Risks Hum. 2007;90:1–636.
8. Chinn SB, Myers JN. Oral cavity carcinoma: current management, controversies, and future directions. J Clin Oncol. 2015;33(29):3269–3276. doi:10.1200/JCO.2015.61.2929
9. Adelstein D, Gillison ML, Pfister DG, et al. NCCN guidelines insights: head and neck cancers, version 2.2017. J Natl Compr Canc Netw. 2017;15(6):761–770. doi:10.6004/jnccn.2017.0101
10. Alterio D, Marvaso G, Ferrari A, Volpe S, Orecchia R, Jereczek-Fossa BA. Modern radiotherapy for head and neck cancer. Semin Oncol. 2019;46(3):233–245. doi:10.1053/j.seminoncol.2019.07.002
11. Haddad R, O’Neill A, Rabinowits G, et al. Induction chemotherapy followed by concurrent chemoradiotherapy (sequential chemoradiotherapy) versus concurrent chemoradiotherapy alone in locally advanced head and neck cancer (PARADIGM): a randomised Phase 3 trial. Lancet Oncol. 2013;14(3):257–264. doi:10.1016/S1470-2045(13)70011-1
12. Cohen EE, Karrison TG, Kocherginsky M, et al. Phase III randomized trial of induction chemotherapy in patients with N2 or N3 locally advanced head and neck cancer. J Clin Oncol. 2014;32(25):2735–2743.
13. Geoffrois L, Martin L, De Raucourt D, et al. Induction chemotherapy followed by cetuximab radiotherapy is not superior to concurrent chemoradiotherapy for head and neck carcinomas: results of the GORTEC 2007-02 phase III randomized trial. J Clin Oncol. 2018;36(31):3077–3083. doi:10.1200/JCO.2017.76.2591
14. Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–1127. doi:10.1056/NEJMoa0802656
15. Cohen EEW, Soulières D, Le Tourneau C, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet. 2019;393(10167):156–167. doi:10.1016/S0140-6736(18)31999-8
16. Levy A, Massard C, Soria JC, Deutsch E. Concurrent irradiation with the anti-programmed cell death ligand-1 immune checkpoint blocker durvalumab: single centre subset analysis from a Phase 1/2 trial. Eur J Cancer. 2016;68:156–162. doi:10.1016/j.ejca.2016.09.013
17. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
18. Guan JL, Shalloway D. Regulation of focal adhesion-associated protein tyrosine kinase by both cellular adhesion and oncogenic transformation. Nature. 1992;358(6388):690–692. doi:10.1038/358690a0
19. Zachary I. Focal adhesion kinase. Int J Biochem Cell Biol. 1997;29(7):929–934.
20. Fan RS, Jácamo RO, Jiang X, Sinnett-Smith J, Rozengurt E. G protein-coupled receptor activation rapidly stimulates focal adhesion kinase phosphorylation at Ser-843. Mediation by Ca2+, calmodulin, and Ca2+/calmodulin-dependent kinase II. J Biol Chem. 2005;280(25):24212–24220. doi:10.1074/jbc.M500716200
21. Schlaepfer DD, Hou S, Lim ST, et al. Tumor necrosis factor-alpha stimulates focal adhesion kinase activity required for mitogen-activated kinase-associated interleukin 6 expression. J Biol Chem. 2007;282(24):17450–17459. doi:10.1074/jbc.M610672200
22. Sieg DJ, Hauck CR, Ilic D, et al. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2(5):249–256. doi:10.1038/35010517
23. Matsumoto K, Matsumoto K, Nakamura T, Kramer RH. Hepatocyte growth factor/scatter factor induces tyrosine phosphorylation of focal adhesion kinase (p125FAK) and promotes migration and invasion by oral squamous cell carcinoma cells. J Biol Chem. 1994;269(50):31807–31813.
24. Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol Cell Biol. 2006;26(13):5155–5167. doi:10.1128/MCB.02186-05
25. Cai X, Lietha D, Ceccarelli DF, et al. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28(1):201–214. doi:10.1128/MCB.01324-07
26. Goñi GM, Epifano C, Boskovic J, et al. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc Natl Acad Sci U S A. 2014;111(31):E3177–E3186. doi:10.1073/pnas.1317022111
27. Brami-Cherrier K, Gervasi N, Arsenieva D, et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014;33(4):356–370. doi:10.1002/embj.201386399
28. Polte TR, Hanks SK. Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc Natl Acad Sci U S A. 1995;92(23):10678–10682. doi:10.1073/pnas.92.23.10678
29. Liu Y, Loijens JC, Martin KH, Karginov AV, Parsons JT. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol Biol Cell. 2002;13(6):2147–2156. doi:10.1091/mbc.e02-01-0018
30. Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol. 1996;16(6):3169–3178. doi:10.1128/MCB.16.6.3169
31. Tachibana K, Sato T, D’Avirro N, Morimoto C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J Exp Med. 1995;182(4):1089–1099. doi:10.1084/jem.182.4.1089
32. Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem. 1995;270(28):16995–16999. doi:10.1074/jbc.270.28.16995
33. Lawson C, Lim ST, Uryu S, Chen XL, Calderwood DA, Schlaepfer DD. FAK promotes recruitment of talin to nascent adhesions to control cell motility. J Cell Biol. 2012;196(2):223–232. doi:10.1083/jcb.2011080781963c
34. Hunger-Glaser I, Salazar EP, Sinnett-Smith J, Rozengurt E. Bombesin, lysophosphatidic acid, and epidermal growth factor rapidly stimulate focal adhesion kinase phosphorylation at Ser-910: requirement for ERK activation. J Biol Chem. 2003;278(25):22631–22643. doi:10.1074/jbc.M210876200
35. Xia J, Lv N, Hong Y, et al. Increased expression of focal adhesion kinase correlates with cellular proliferation and apoptosis during 4-nitroquinoline-1-oxide-induced rat tongue carcinogenesis. J Oral Pathol Med. 2009;38(6):524–529. doi:10.1111/j.1600-0714.2008.00728.x
36. Rodrigo JP, Álvarez-Alija G, Menéndez ST, et al. Cortactin and focal adhesion kinase as predictors of cancer risk in patients with laryngeal premalignancy. Cancer Prev Res (Phila). 2011;4(8):1333–1341. doi:10.1158/1940-6207.CAPR-10-0338
37. Chiu YW, Liou LY, Chen PT, et al. Tyrosine 397 phosphorylation is critical for FAK-promoted Rac1 activation and invasive properties in oral squamous cell carcinoma cells. Lab Invest. 2016;96(3):296–306. doi:10.1038/labinvest.2015.151
38. Flores APC, Dias KB, Hildebrand LC, Oliveira MG, Lamers ML, Sant’Ana Filho M. Focal adhesion kinases in head and neck squamous cell carcinoma. J Oral Pathol Med. 2018;47(3):246–252. doi:10.1111/jop.12674
39. Theocharis S, Kotta-Loizou I, Giaginis C, et al. Expression and clinical significance of concomitant FAK/SRC and p-paxillin in mobile tongue squamous cell carcinoma. Anticancer Res. 2017;37(3):1313–1319.
40. Agochiya M, Brunton VG, Owens DW, et al. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene. 1999;18(41):5646–5653. doi:10.1038/sj.onc.1202957
41. Skinner HD, Giri U, Yang L, et al. Proteomic profiling identifies PTK2/FAK as a driver of radioresistance in HPV-negative head and neck cancer. Clin Cancer Res. 2016;22(18):4643–4650. doi:10.1158/1078-0432.CCR-15-2785
42. Canel M, Secades P, Rodrigo JP, et al. Overexpression of focal adhesion kinase in head and neck squamous cell carcinoma is independent of fak gene copy number. Clin Cancer Res. 2006;12(11):3272–3279. doi:10.1158/1078-0432.CCR-05-1583
43. Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678(2–3):111–125.
44. Golubovskaya VM, Cance WG. Focal adhesion kinase and p53 signaling in cancer cells. Int Rev Cytol. 2007;263:103–153.
45. Rodrigo JP, Cabanillas R, Secades P, Canel M, Chiara MD, Suárez C. Relación entre la expresión de FAK y p53 en los carcinomas epidermoides de laringe [relationship between FAK and P53 expression in squamous cell carcinomas of the larynx]. Acta Otorrinolaringol Esp. 2007;58(8):347–351.
46. Rosado P, Lequerica-Fernández P, Peña I, Alonso-Durán L, de Vicente JC. In oral squamous cell carcinoma, high FAK expression is correlated with low P53 expression. Virchows Arch. 2012;461(2):163–168. doi:10.1007/s00428-012-1283-2
47. George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524(7563):47–53. doi:10.1038/nature14664
48. Kurio N, Shimo T, Fukazawa T, et al. Anti-tumor effect of a novel FAK inhibitor TAE226 against human oral squamous cell carcinoma. Oral Oncol. 2012;48(11):1159–1170. doi:10.1016/j.oraloncology.2012.05.019
49. Zhang CH, Wang JX, Cai ML, Shao R, Liu H, Zhao WL. The roles and mechanisms of G3BP1 in tumour promotion. J Drug Target. 2019;27(3):300–305. doi:10.1080/1061186X.2018.1523415
50. Takeda I, Maruya S, Shirasaki T, et al. Simvastatin inactivates beta1-integrin and extracellular signal-related kinase signaling and inhibits cell proliferation in head and neck squamous cell carcinoma cells. Cancer Sci. 2007;98(6):890–899. doi:10.1111/j.1349-7006.2007.00471.x
51. Han BB, Li S, Tong M, et al. Fenretinide perturbs focal adhesion kinase in premalignant and malignant human oral keratinocytes. Fenretinide’s chemopreventive mechanisms include ECM interactions. Cancer Prev Res (Phila). 2015;8(5):419–430. doi:10.1158/1940-6207.CAPR-14-0418
52. Xi WH, Yang LY, Cao ZY, Qian Y. Tivantinib (ARQ-197) exhibits anti-tumor activity with down-regulation of FAK in oral squamous cell carcinoma. Biochem Biophys Res Commun. 2015;457(4):723–729. doi:10.1016/j.bbrc.2015.01.062
53. Zhang Y, Lu H, Dazin P, Kapila Y. Squamous cell carcinoma cell aggregates escape suspension-induced, p53-mediated anoikis: fibronectin and integrin alphav mediate survival signals through focal adhesion kinase. J Biol Chem. 2004;279(46):48342–48349. doi:10.1074/jbc.M407953200
54. Golubovskaya VM, Finch R, Cance WG. Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280(26):25008–25021. doi:10.1074/jbc.M414172200
55. Lim ST, Chen XL, Lim Y, et al. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29(1):9–22. doi:10.1016/j.molcel.2007.11.031
56. Zhu J, Wu YN, Zhang W, et al. Monocarboxylate transporter 4 facilitates cell proliferation and migration and is associated with poor prognosis in oral squamous cell carcinoma patients. PLoS One. 2014;9(1):e87904. doi:10.1371/journal.pone.0087904
57. Peng H, Gong PG, Li JB, et al. The important role of the receptor for activated C kinase 1 (RACK1) in nasopharyngeal carcinoma progression. J Transl Med. 2016;14(1):131. doi:10.1186/s12967-016-0885-x
58. Zhang Y, Wang B, Chen X, Li W, Dong P. AGO2 involves the malignant phenotypes and FAK/PI3K/AKT signaling pathway in hypopharyngeal-derived FaDu cells. Oncotarget. 2017;8(33):54735–54746.
59. Liu C, Guo T, Sakai A, et al. A novel splice variant of LOXL2 promotes progression of human papillomavirus-negative head and neck squamous cell carcinoma. Cancer. 2020;126(4):737–748. doi:10.1002/cncr.32610
60. Wang GT, Huang S. Baicalein inhibits the proliferation and invasion of oral squamous cell carcinoma and its possible signaling pathway. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2018;34(6):536–540.
61. Qiang YY, Li CZ, Sun R, et al. Along with its favorable prognostic role, CLCA2 inhibits growth and metastasis of nasopharyngeal carcinoma cells via inhibition of FAK/ERK signaling. J Exp Clin Cancer Res. 2018;37(1):34. doi:10.1186/s13046-018-0692-8
62. Chang YL, Hsu YK, Wu TF, et al. Regulation of estrogen receptor α function in oral squamous cell carcinoma cells by FAK signaling. Endocr Relat Cancer. 2014;21(4):555–565. doi:10.1530/ERC-14-0102
63. Yeh CM, Hsieh MJ, Yang JS, et al. Geraniin inhibits oral cancer cell migration by suppressing matrix metalloproteinase-2 activation through the FAK/Src and ERK pathways. Environ Toxicol. 2019;34(10):1085–1093. doi:10.1002/tox.22809
64. Xiao W, Jiang M, Li H, Li C, Su R, Huang K. Knockdown of FAK inhibits the invasion and metastasis of Tca-8113 cells in vitro. Mol Med Rep. 2013;8(2):703–707. doi:10.3892/mmr.2013.1555
65. Kuo CL, Lai KC, Ma YS, Weng SW, Lin JP, Chung JG. Gallic acid inhibits migration and invasion of SCC-4 human oral cancer cells through actions of NF-κB, Ras and matrix metalloproteinase-2 and −9. Oncol Rep. 2014;32(1):355–361. doi:10.3892/or.2014.3209
66. Ho YT, Yang JS, Li TC, et al. Berberine suppresses in vitro migration and invasion of human SCC-4 tongue squamous cancer cells through the inhibitions of FAK, IKK, NF-kappaB, u-PA and MMP-2 and −9. Cancer Lett. 2009;279(2):155–162. doi:10.1016/j.canlet.2009.01.033
67. Lai WW, Hsu SC, Chueh FS, et al. Quercetin inhibits migration and invasion of SAS human oral cancer cells through inhibition of NF-κB and matrix metalloproteinase-2/-9 signaling pathways. Anticancer Res. 2013;33(5):1941–1950.
68. Canel M, Secades P, Garzón-Arango M, et al. Involvement of focal adhesion kinase in cellular invasion of head and neck squamous cell carcinomas via regulation of MMP-2 expression. Br J Cancer. 2008;98(7):1274–1284. doi:10.1038/sj.bjc.6604286
69. Nanda DP, Dutta K, Ganguly KK, et al. MMP-9 as a potential biomarker for carcinoma of oral cavity: a study in eastern India. Neoplasma. 2014;61(6):747–757. doi:10.4149/neo_2014_091
70. Hsin CH, Chou YE, Yang SF, et al. MMP-11 promoted the oral cancer migration and Fak/Src activation. Oncotarget. 2017;8(20):32783–32793. doi:10.18632/oncotarget.15824
71. Huang Y, Sook-Kim M, Ratovitski E. Midkine promotes tetraspanin-integrin interaction and induces FAK-Stat1alpha pathway contributing to migration/invasiveness of human head and neck squamous cell carcinoma cells. Biochem Biophys Res Commun. 2008;377(2):474–478. doi:10.1016/j.bbrc.2008.09.138
72. Srivastava K, Pickard A, McDade S, McCance DJ. p63 drives invasion in keratinocytes expressing HPV16 E6/E7 genes through regulation of Src-FAK signaling. Oncotarget. 2017;8(10):16202–16219. doi:10.18632/oncotarget.3892
73. Liu S-C, Huang C-M, Bamodu OA, et al. Ovatodiolide suppresses nasopharyngeal cancer by targeting stem cell-like population, inducing apoptosis, inhibiting EMT and dysregulating JAK/STAT signaling pathway. Phytomedicine. 2019;56:269–278. doi:10.1016/j.phymed.2018.05.007
74. He JY, Han P, Zhang Y, et al. Overexpression of Nogo receptor 3 (NgR3) correlates with poor prognosis and contributes to the migration of epithelial cells of nasopharyngeal carcinoma patients. J Mol Med (Berl). 2018;96(3–4):265–279. doi:10.1007/s00109-017-1618-1
75. Duarte A, André-Grégoire G, Trillet K, et al. Inhibition of mTOR in head and neck cancer cells alters endothelial cell morphology in a paracrine fashion. Mol Carcinog. 2019;58(1):161–168.
76. Sasabe E, Tomomura A, Tomita R, Sento S, Kitamura N, Yamamoto T. Ephrin-B2 reverse signaling regulates progression and lymph node metastasis of oral squamous cell carcinoma. PLoS One. 2017;12(11):e0188965. doi:10.1371/journal.pone.0188965
77. Yang LY, He CY, Chen XH, Su LP, Liu BY, Zhang H. Aurora kinase A revives dormant laryngeal squamous cell carcinoma cells via FAK/PI3K/Akt pathway activation. Oncotarget. 2016;7(30):48346–48359.
78. Cai L, Ye Y, Jiang Q, et al. Epstein-Barr virus-encoded microRNA BART1 induces tumour metastasis by regulating PTEN-dependent pathways in nasopharyngeal carcinoma. Nat Commun. 2015;6:7353.
79. Islam M, Datta J, Lang JC, Teknos TN. Down regulation of RhoC by microRNA-138 results in de-activation of FAK, Src and Erk1/2 signaling pathway in head and neck squamous cell carcinoma. Oral Oncol. 2014;50(5):448–456. doi:10.1016/j.oraloncology.2014.01.014
80. Lin CW, Sun MS, Wu HC. Podocalyxin-like 1 is associated with tumor aggressiveness and metastatic gene expression in human oral squamous cell carcinoma. Int J Oncol. 2014;45(2):710–718. doi:10.3892/ijo.2014.2427
81. Chuang JY, Yu NY, Chiang IP, Lai CH, Lin CD, Tang CH. Cyr61 increases matrix metalloproteinase-3 expression and cell motility in human oral squamous cell carcinoma cells. J Cell Biochem. 2012;113(6):1977–1986. doi:10.1002/jcb.24066
82. Kim SA, Kwon SM, Kim JA, Kang KW, Yoon JH, Ahn SG. 5ʹ-Nitro-indirubinoxime, an indirubin derivative, suppresses metastatic ability of human head and neck cancer cells through the inhibition of Integrin β1/FAK/Akt signaling. Cancer Lett. 2011;306(2):197–204. doi:10.1016/j.canlet.2011.03.006
83. Kusayama M, Wada K, Nagata M, et al. Critical role of aquaporin 3 on growth of human esophageal and oral squamous cell carcinoma. Cancer Sci. 2011;102(6):1128–1136. doi:10.1111/j.1349-7006.2011.01927.x
84. Kamarajan P, Garcia-Pardo A, D’Silva NJ, Kapila YL. The CS1 segment of fibronectin is involved in human OSCC pathogenesis by mediating OSCC cell spreading, migration, and invasion. BMC Cancer. 2010;10(1):330. doi:10.1186/1471-2407-10-330
85. Goutzanis L, Vairaktaris E, Yapijakis C, et al. Diabetes may increase risk for oral cancer through the insulin receptor substrate-1 and focal adhesion kinase pathway. Oral Oncol. 2007;43(2):165–173. doi:10.1016/j.oraloncology.2006.02.004
86. Kitajo H, Shibata T, Nagayasu H, et al. Rho regulates the hepatocyte growth factor/scatter factor-stimulated cell motility of human oral squamous cell carcinoma cells. Oncol Rep. 2003;10(5):1351–1356.
87. Huang C, Wang Y, Huang JH, Liu W. Sema3A drastically suppresses tumor growth in oral cancer Xenograft model of mice. BMC Pharmacol Toxicol. 2017;18(1):55. doi:10.1186/s40360-017-0163-4
88. Rothhut B, Ghoneim C, Antonicelli F, Soula-Rothhut M. Epidermal growth factor stimulates matrix metalloproteinase-9 expression and invasion in human follicular thyroid carcinoma cells through Focal adhesion kinase. Biochimie. 2007;89(5):613–624. doi:10.1016/j.biochi.2007.01.015
89. Ungewiss C, Rizvi ZH, Roybal JD, et al. The microRNA-200/Zeb1 axis regulates ECM-dependent β1-integrin/FAK signaling, cancer cell invasion and metastasis through CRKL. Sci Rep. 2016;6(1):18652. doi:10.1038/srep18652
90. Huang W, Liu J, Feng X, et al. DLC-1 induces mitochondrial apoptosis and epithelial mesenchymal transition arrest in nasopharyngeal carcinoma by targeting EGFR/Akt/NF-κB pathway. Med Oncol. 2015;32(4):115. doi:10.1007/s12032-015-0564-4
91. Yang L, Zhou Q, Chen X, Su L, Liu B, Zhang H. Activation of the FAK/PI3K pathway is crucial for AURKA-induced epithelial-mesenchymal transition in laryngeal cancer. Oncol Rep. 2016;36(2):819–826. doi:10.3892/or.2016.4872
92. Li JY, Huang WX, Zhou X, Chen J, Li Z. Numb inhibits epithelial-mesenchymal transition via RBP-Jκ-dependent Notch1/PTEN/FAK signaling pathway in tongue cancer. BMC Cancer. 2019;19(1):391. doi:10.1186/s12885-019-5605-5
93. Morita Y, Hata K, Nakanishi M, et al. Cellular fibronectin 1 promotes VEGF-C expression, lymphangiogenesis and lymph node metastasis associated with human oral squamous cell carcinoma. Clin Exp Metastasis. 2015;32(7):739–753. doi:10.1007/s10585-015-9741-2
94. Appert-Collin A, Bennasroune A, Jeannesson P, et al. Role of LRP-1 in cancer cell migration in 3-dimensional collagen matrix. Cell Adh Migr. 2017;11(4):316–326. doi:10.1080/19336918.2016.1215788
95. Rivera C, Zandonadi FS, Sánchez-Romero C, et al. Agrin has a pathological role in the progression of oral cancer. Br J Cancer. 2018;118(12):1628–1638. doi:10.1038/s41416-018-0135-5
96. Al-toub M, Vishnubalaji R, Hamam R, Kassem M, Aldahmash A, Alajez NM. CDH1 and IL1-beta expression dictates FAK and MAPKK-dependent cross-talk between cancer cells and human mesenchymal stem cells. Stem Cell Res Ther. 2015;6(1):135. doi:10.1186/s13287-015-0123-0
97. Liu SC, Hsu T, Chang YS, et al. Cytoplasmic LIF reprograms invasive mode to enhance NPC dissemination through modulating YAP1-FAK/PXN signaling. Nat Commun. 2018;9(1):5105.
98. Min A, Zhu C, Wang J, et al. Focal adhesion kinase knockdown in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis via downregulating MCP-1/CCL2 expression. J Biochem Mol Toxicol. 2015;29(2):70–76. doi:10.1002/jbt.21669
99. Chuang JY, Yang WY, Lai CH, Lin CD, Tsai MH, Tang CH. CTGF inhibits cell motility and COX-2 expression in oral cancer cells. Int Immunopharmacol. 2011;11(8):948–954. doi:10.1016/j.intimp.2011.02.008
100. Zhen Y, Ye Y, Yu X, et al. Reduced CTGF expression promotes cell growth, migration, and invasion in nasopharyngeal carcinoma. PLoS One. 2013;8(6):e64976. doi:10.1371/journal.pone.0064976
101. Fleigel J, Sedwick J, Kornberg LJ. Hepatocyte growth factor/scatter factor stimulates mitogenesis and migration of a head and neck squamous cell carcinoma cell line. Otolaryngol Head Neck Surg. 2002;127(4):271–278. doi:10.1067/mhn.2002.127414
102. Yadav A, Kumar B, Yu JG, Old M, Teknos TN, Kumar P. Tumor-associated endothelial cells promote tumor metastasis by chaperoning circulating tumor cells and protecting them from anoikis. PLoS One. 2015;10(10):e0141602. doi:10.1371/journal.pone.0141602
103. Al-toub M, Almusa A, Almajed M, et al. Pleiotropic effects of cancer cells’ secreted factors on human stromal (mesenchymal) stem cells. Stem Cell Res Ther. 2013;4(5):114. doi:10.1186/scrt325
104. da Silva SD, Xu B, Maschietto M, et al. TRAF2 cooperates with focal adhesion signaling to regulate cancer cell susceptibility to anoikis. Mol Cancer Ther. 2019;18(1):139–146. doi:10.1158/1535-7163.MCT-17-1261
105. Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011;9(12):1658–1667. doi:10.1158/1541-7786.MCR-11-0271
106. Moon JH, Rho YS, Lee SH, et al. Role of integrin β1 as a biomarker of stemness in head and neck squamous cell carcinoma. Oral Oncol. 2019;96:34–41. doi:10.1016/j.oraloncology.2019.07.001
107. Ming XY, Fu L, Zhang LY, et al. Integrin α7 is a functional cancer stem cell surface marker in oesophageal squamous cell carcinoma. Nat Commun. 2016;7(1):13568. doi:10.1038/ncomms13568
108. Vengoji R, Macha MA, Nimmakayala RK, et al. Afatinib and Temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J Exp Clin Cancer Res. 2019;38(1):266. doi:10.1186/s13046-019-1264-2
109. Huang WC, Jang TH, Tung SL, Yen TC, Chan SH, Wang LH. A novel miR-365-3p/EHF/keratin 16 axis promotes oral squamous cell carcinoma metastasis, cancer stemness and drug resistance via enhancing β5-integrin/c-met signaling pathway. J Exp Clin Cancer Res. 2019;38(1):89. doi:10.1186/s13046-019-1091-5
110. Zhang S, Ma H, Zhang D, et al. LncRNA KCNQ1OT1 regulates proliferation and cisplatin resistance in tongue cancer via miR-211-5p mediated Ezrin/Fak/Src signaling. Cell Death Dis. 2018;9(7):742.
111. Ren Y, Zhou X, Yang JJ, et al. AC1MMYR2 impairs high dose paclitaxel-induced tumor metastasis by targeting miR-21/CDK5 axis. Cancer Lett. 2015;362(2):174–182. doi:10.1016/j.canlet.2015.03.038
112. Zhou Y, Zhang L, Pan H, et al. Bmi1 essentially mediates podocalyxin-enhanced cisplatin chemoresistance in oral tongue squamous cell carcinoma. PLoS One. 2015;10(4):e0123208. doi:10.1371/journal.pone.0123208
113. Eberle KE, Sansing HA, Szaniszlo P, Resto VA, Berrier AL. Carcinoma matrix controls resistance to cisplatin through talin regulation of NF-kB. PLoS One. 2011;6(6):e21496. doi:10.1371/journal.pone.0021496
114. Jie HB, Schuler PJ, Lee SC, et al. CTLA-4+ regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75(11):2200–2210. doi:10.1158/0008-5472.CAN-14-2788
115. Eke I, Zscheppang K, Dickreuter E, et al. Simultaneous β1 integrin-EGFR targeting and radiosensitization of human head and neck cancer. J Natl Cancer Inst. 2015;107(2):dju419. doi:10.1093/jnci/dju419
116. Rossow L, Eke I, Dickreuter E, Cordes N. Targeting of the EGFR/β1 integrin connecting proteins PINCH1 and Nck2 radiosensitizes three-dimensional SCC cell cultures. Oncol Rep. 2015;34(1):469–476. doi:10.3892/or.2015.4006
117. Eke I, Cordes N. Dual targeting of EGFR and focal adhesion kinase in 3D grown HNSCC cell cultures. Radiother Oncol. 2011;99(3):279–286. doi:10.1016/j.radonc.2011.06.006
118. Steglich A, Vehlow A, Eke I, Cordes N. α integrin targeting for radiosensitization of three-dimensionally grown human head and neck squamous cell carcinoma cells. Cancer Lett. 2015;357(2):542–548. doi:10.1016/j.canlet.2014.12.009
119. Eke I, Deuse Y, Hehlgans S, et al. β1Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J Clin Invest. 2012;122(4):1529–1540. doi:10.1172/JCI61350
120. Ou J, Pan F, Geng P, et al. Silencing fibronectin extra domain A enhances radiosensitivity in nasopharyngeal carcinomas involving an FAK/Akt/JNK pathway. Int J Radiat Oncol Biol Phys. 2012;82(4):e685–e691. doi:10.1016/j.ijrobp.2011.09.040
121. Hehlgans S, Eke I, Targeting CN. FAK radiosensitizes 3-dimensional grown human HNSCC cells through reduced Akt1 and MEK1/2 signaling. Int J Radiat Oncol Biol Phys. 2012;83(5):e669–e676. doi:10.1016/j.ijrobp.2012.01.065
122. Hehlgans S, Lange I, Eke I, Cordes N. 3D cell cultures of human head and neck squamous cell carcinoma cells are radiosensitized by the focal adhesion kinase inhibitor TAE226. Radiother Oncol. 2009;92(3):371–378. doi:10.1016/j.radonc.2009.08.001
123. Omura G, Ando M, Saito Y, et al. Association of the upregulated expression of focal adhesion kinase with poor prognosis and tumor dissemination in hypopharyngeal cancer. Head Neck. 2016;38(8):1164–1169. doi:10.1002/hed.24176
124. de Vicente JC, Rodrigo JP, Rodriguez-Santamarta T, Lequerica-Fernández P, Allonca E, García-Pedrero JM. Cortactin and focal adhesion kinase as predictors of cancer risk in patients with premalignant oral epithelial lesions. Oral Oncol. 2012;48(7):641–646. doi:10.1016/j.oraloncology.2012.02.004
125. Jiang H, Liu L, Ye J, Liu H, Xing S, Wu Y. Focal adhesion kinase serves as a marker of cervical lymph node metastasis and is a potential therapeutic target in tongue cancer. J Cancer Res Clin Oncol. 2010;136(9):1295–1302. doi:10.1007/s00432-010-0780-4
126. Rodrigo JP, Dominguez F, Suárez V, Canel M, Secades P, Chiara MD. Focal adhesion kinase and E-cadherin as markers for nodal metastasis in laryngeal cancer. Arch Otolaryngol Head Neck Surg. 2007;133(2):145–150. doi:10.1001/archotol.133.2.145
127. MÁ V, Hermida-Prado F, Granda-Díaz R, et al. Immunohistochemical expression of cortactin and focal adhesion kinase predicts recurrence risk and laryngeal cancer risk beyond histologic grading. Cancer Epidemiol Biomarkers Prev. 2018;27(7):805–813.
128. Cordes N, Ney M, Beleites T, et al. Retrospective investigation of the prognostic value of the β1 integrin expression in patients with head and neck squamous cell carcinoma receiving primary radio(chemo)therapy. PLoS One. 2018;13(12):e0209479. doi:10.1371/journal.pone.0209479
129. Infante JR, Camidge DR, Mileshkin LR, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol. 2012;30(13):1527–1533. doi:10.1200/JCO.2011.38.9346
130. Jiang H, Hegde S, Knolhoff BL, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–860. doi:10.1038/nm.4123
131. Aung KL, McWhirter E, Welch S, et al. A phase II trial of GSK2256098 and trametinib in patients with advanced pancreatic ductal adenocarcinoma (PDAC) (MOBILITY-002 trial, NCT02428270). J Clin Oncol. 2018;36(4_suppl):409. doi:10.1200/JCO.2018.36.4_suppl.409
132. Fennell DA, Baas P, Taylor P, et al. Maintenance defactinib versus placebo after first-line chemotherapy in patients with merlin-stratified pleural mesothelioma: COMMAND—a double-blind, randomized, phase II study. J Clin Oncol. 2019;37(10):790–798. doi:10.1200/JCO.2018.79.0543
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.