")
Back to Journals » Journal of Inflammation Research » Volume 14
Role of Ferroptosis in Lung Diseases
Authors Xu W, Deng H, Hu S , Zhang Y , Zheng L, Liu M, Chen Y, Wei J, Yang H , Lv X
Received 18 February 2021
Accepted for publication 22 April 2021
Published 20 May 2021 Volume 2021:14 Pages 2079—2090
DOI https://doi.org/10.2147/JIR.S307081
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Wenting Xu,1,2,* Huimin Deng,2,* Song Hu,2,3,* Yiguo Zhang,2,3 Li Zheng,1,2 Meiyun Liu,2 Yuanli Chen,2 Juan Wei,2 Hao Yang,2 Xin Lv1– 3
1Department of Anesthesiology, Fuyang Hospital of Anhui Medical University, Fuyang, Anhui, 236000, People’s Republic of China; 2Department of Anesthesiology, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai, 200433, People’s Republic of China; 3Graduate School, Wannan Medical College, Wuhu, AnHui, 241002, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xin Lv; Hao Yang
Department of Anesthesiology, Shanghai Pulmonary Hospital, Tongji University School of Medicine, 507 Zhengmin Road, Shanghai, 200433, People’s Republic of China
Email [email protected]; [email protected]
Abstract: Ferroptosis is a new type of programmed cell death characterized by intracellular iron accumulation and lipid peroxidation that leads to oxidative stress and cell death. The metabolism of iron, lipids, and amino acids and multiple signalling pathways precisely regulate the process of ferroptosis. Emerging evidence has demonstrated that ferroptosis participates in the occurrence and progression of various pathological conditions and diseases, such as infections, neurodegeneration, tissue ischaemia-reperfusion injury and immune diseases. Recent studies have also indicated that ferroptosis plays a critical role in the pathogenesis of acute lung injury, chronic obstructive pulmonary disease, pulmonary fibrosis, pulmonary infection and asthma. Herein, we summarize the latest knowledge on the regulatory mechanism of ferroptosis and its association with iron, lipid and amino acid metabolism as well as several signalling pathways. Furthermore, we review the contribution of ferroptosis to the pathogenesis of lung diseases and discuss ferroptosis as a novel therapeutic target for various lung diseases.
Keywords: ferroptosis, metabolic networks and pathways, signalling pathways, lung diseases
Introduction
Ferroptosis is a newly identified type of iron-dependent programmed cell death that was first reported by Dixon et al in 2012.1 As a new type of programmed cell death, ferroptosis is different from apoptosis, autophagy, necrosis, pyrolysis and other forms of cell death in terms of morphology and biochemistry. The morphological characteristics of ferroptosis mainly manifest as intact cytomembranes, cell volume shrinkage, increased mitochondrial membrane density, reduced or even absent mitochondrial cristae, mitochondrial membrane crumpling and outer membrane rupture, and normal nucleus size without concentrated chromatin.1,6 Biochemically, the mechanisms underlying ferroptosis are believed to be mainly related to intracellular iron accumulation, glutathione depletion, glutathione peroxidase 4 inactivation and lipid peroxidation.2–5 Recently, numerous studies have shown that ferroptosis can be inhibited by iron chelators,6 lipid peroxidation inhibitors,7 and lipophilic antioxidants8 and can be tightly regulated by several signalling pathways, such as the nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1) signalling pathway.9 Therefore, ferroptosis is characterized by the perturbation of intricate metabolic networks and signalling pathways, which we will describe in detail in the following sections.
Mechanisms of Ferroptosis
The regulatory mechanisms of ferroptosis are complicated and involve a variety of metabolic networks and signalling pathways (Figure 1).
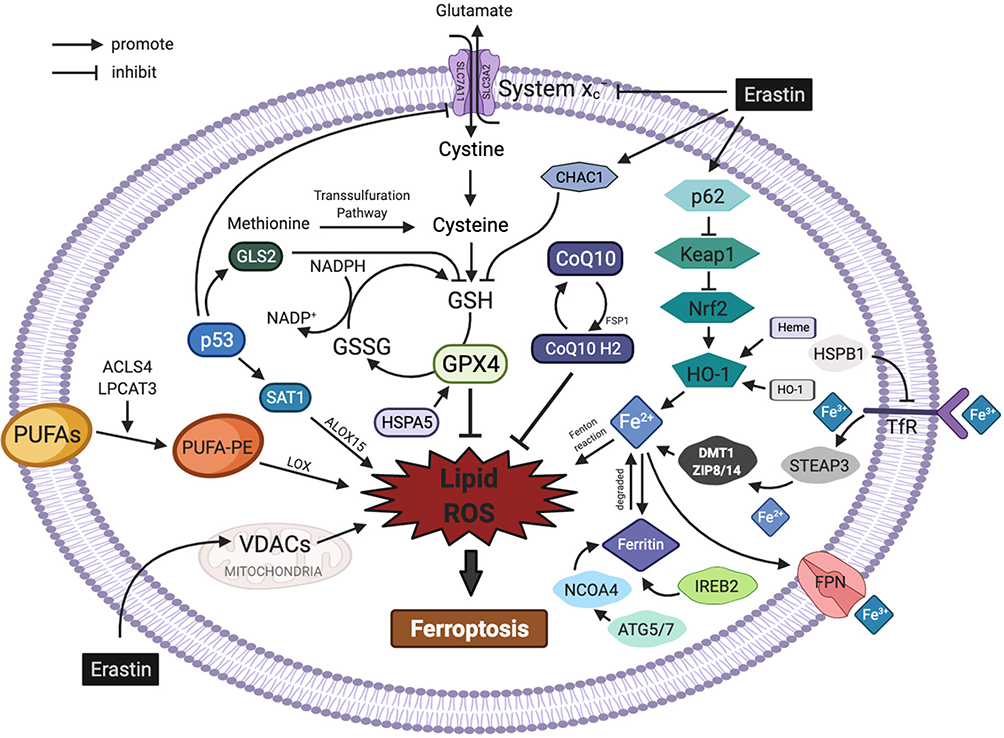 |
Figure 1 Regulatory pathways of ferroptosis. The figure shows the regulatory pathways of ferroptosis, which can be roughly divided into two categories: the first category involves metabolic pathways associated with ferroptosis, such as abnormal iron, lipid and amino acid pathways; the second category involves signalling pathways associated with ferroptosis, such as the p53, nuclear factor erythroid 2-related factor 2, heat shock proteins, haem oxygenase-1, nuclear receptor coactivator 4 and mevalonate pathways. In addition, erastin acts on multiple pathways to induce ferroptosis. |
Metabolic Pathways of Ferroptosis
Abnormal Iron Metabolism
Iron is necessary for the accumulation of lipid peroxides and the process of ferroptosis. Therefore, the proteins involved in the binding, transport, dissociation and storage of iron all play an important role in the regulation of ferroptosis. Under normal physiological conditions, intracellular iron is always in dynamic homeostasis through absorption and metabolism. Dietary iron is mainly absorbed by intestinal epithelial cells in the form of Fe3+ and enters cells through the transferrin receptor (TfR) on the membrane after binding to transferrin. Subsequently, Fe3+ in cells is reduced to Fe2+ by six-transmembrane epithelial antigen of prostate 3 (STEAP3), and then, Fe2+ is released into the cytoplasmic unstable iron pool via divalent metal transporter 1 (DMT1) or zinc-iron regulatory protein family 8/14 (ZIP8/14). Abnormal expression or dysfunction of these related proteins will imbalance the intracellular concentration of iron ions and cause iron overload. Excess Fe2+ produced in the cell can directly catalyse lipid reactive oxygen species (ROS) generation through the Fenton reaction, resulting in the continuous accumulation of intracellular lipid ROS and triggering ferroptosis.3 Cellular iron accumulation can be detected by calcein-AM staining or Perls’ Prussian blue staining.10,11 Recent studies have also shown that in addition to proteins, genes involved in iron metabolism can also lead to ferroptosis. For example, knockout of the ferroportin (FPN) gene in neuroblastoma cells promotes erastin-induced ferroptosis by inhibiting iron metabolism.12 In contrast, suppression of iron-responsive element-binding protein 2, a key regulator of iron metabolism, significantly inhibits erastin-induced ferroptosis.1 Iron regulatory protein 2 (IREB2), HO-1, and recombinant human CDGSH iron sulfur domain 1 (CDSD1) can mediate ferroptosis by modulating the level of intracellular iron.1,13,14
Abnormal Lipid Metabolism
Lipid ROS accumulation is a hallmark feature of ferroptosis. ROS are a class of partially reduced oxygen-containing molecules, including peroxides, superoxide anions and free radicals, that are essential for maintaining the steady state of cells and tissues.15 Most ferroptosis-associated ROS are derived from the Fenton and Haber–Weiss reactions16 and subsequently interact with polyunsaturated fatty acids (PUFAs) on lipid membranes to form lipid ROS and then cause ferroptosis when large amounts of lipid ROS accumulate in cells.17 The levels of lipid ROS can be determined using BODIPY-C11 dye.18,19 Lipoxygenases (LOXs) may be involved in the formation of iron-dependent lipid ROS by catalysing the oxidation of PUFAs into lipid hydroperoxides and forming toxic lipid-free radicals in the presence of a large number of iron ions in the cytoplasm, which may eventually lead to cell damage.20 Meanwhile, protons beside PUFAs can be transferred by these toxic lipid-free radicals and then start a new round of lipid oxidation reactions, which eventually lead to more severe oxidative damage.21 Both cytoplasmic ROS eliminators (N-acetylcysteine) and mitochondria-targeted antioxidants can mitigate erastin- and its analogue-induced ferroptosis by blocking ROS production.22 Nicotinamide adenine dinucleotide phosphate (NADPH), an important intracellular reductant, can also reduce the sensitivity of ferroptosis by eliminating lipid hydroperoxides, especially in the central nervous system (CNS).1,23
Lipids are vital for maintaining the morphology and function of cells, including the biomembrane composition, energy storage and signal transmission. The metabolism of lipids is crucial for the development of ferroptosis. Studies have shown that ferroptosis is a type of cell death caused by the excessive oxidation of phospholipids containing PUFAs.20 PUFAs are important components of the lipid bilayer of cytomembranes and are also the molecular basis for the fluidity and deformability of the cytomembrane.24 However, the carbon–carbon double bonds in PUFAs are unstable and can be the targets of lipid peroxidation reactions, which provide a material basis for the occurrence of ferroptosis.21 Dixon et al identified two enzymes that play key roles in the synthesis of PUFAs in ferroptosis by screening haploid cell lines: acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3).25 When these two genes were knocked out, PUFA synthesis was decreased, leading to the inhibition of ferroptosis.20,26 In contrast, arachidonic acid or other PUFAs enhanced the effects of ferroptosis inducers, such as RAS-selective lethal (RSL3), on lipid peroxides and ferroptosis.21
The core process of ferroptosis is the accumulation of lipid peroxidation products. The intracellular lipid peroxidation process can be divided into two general types: non-enzymatic lipid peroxidation and enzymatic lipid peroxidation. Non-enzymatic lipid peroxidation, also known as lipid autoxidation, is a free radical-mediated chain reaction, and enzymatic lipid peroxidation refers to the generation of various lipid hydroperoxides catalysed by LOXs. The reduction of LOX expression is also effective in ameliorating ferroptosis induced by erastin.20,21 Continued oxidative reactions and depletion of PUFAs may alter the fluid structure of the cytomembrane, thus increasing membrane permeability and eventually leading to cell death.27
Abnormal Amino Acid Metabolism
Ferroptosis caused by abnormal amino acid metabolism is primarily associated with unusual metabolism of glutathione (GSH). Glutathione peroxidase 4 (GPX4), cystine-glutamate antiporter (system xc-), the sulfur-transfer pathway and a series of genes and regulatory factors related to GSH biosynthesis and degradation can all participate in the regulation of ferroptosis. Reverse transcription-polymerase chain reaction (RT-PCR), immunoblotting and flow cytometry can be used to analyse relevant genes, and protein expression and GSH levels can be determined using an intracellular glutathione (GSH) detection assay kit.27
GSH is the core material for amino acid metabolism during ferroptosis and is synthesized from cysteine, glutamate and glycine.5 Cystine is reduced to cysteine after entering cells to participate in the synthesis of GSH. GPX4 is an important enzyme for scavenging lipid oxygen-free radicals. Once GPX4 is activated, GSH can reduce toxic lipid hydroperoxides (L-OOH) to non-toxic lipid alcohols (L-OH),28 indicating that GSH is a significant protective metabolite that prevents ferroptosis. In this process, GPX4 works as a key enzyme and GSH works as a reducing agent to mediate the reduction reaction of lipid peroxide and negatively regulate ferroptosis. Cysteine, one of the major components of the synthesis of GSH, can enter cells in two main ways: one way depends on system xc- and the other way depends on the sulfur-transfer pathway. Damage to the above-mentioned two ways has a certain promoting effect on the development and progression of ferroptosis.
Cysteine is one of the raw materials for the synthesis of GSH. One of the main ways cysteine enters cells is through system xc-, which is a heteromeric amino acid transporter consisting of two subunits: solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2). The membrane transporter x catalyses the exchange of cysteine (which is imported into the intracellular space) and glutamate (which is exported into the extracellular space) across the membrane in a 1:1 ratio.29 Intracellular cystine can be converted into cysteine, which is crucial for GSH synthesis. When the function of system xc- is impaired or there is insufficient intracellular cysteine, the synthesis of GSH is reduced, causing ferroptosis. A previous study suggested that the expression of system xc- could be suppressed by interferon-γ (IFNγ),30 and IFNγ can be produced by a variety of immune cells, such as CD8+ T cells. Therefore, ferroptosis can be regulated by immune cells, but the specific mechanism is still unclear.31
GPX4, a phospholipid-hydroperoxide glutathione peroxidase, contains an efficient selenocysteine unit that can increase its peroxidase activity,32,33 which can attenuate the toxicity of lipid peroxides and maintain the stability of the lipid bilayer of the cytomembrane, indicating that it is a central regulator of ferroptosis. Two molecules of GSH can be oxidized into glutathione (GSSG) by the selenoenzyme GPX4, and donating electrons reduces toxic L-OOH to non-toxic L-OH.28 GPX4 overexpression inhibits ROS production and lipid peroxidation,29 while a decrease in GPX4 activity or expression results in the accumulation of intracellular lipid peroxide, which leads to ferroptosis.34 Recent studies have shown that knockout of GPX4 can activate macrophages and microglia in the kidney and hippocampus, trigger an inflammatory response, and ultimately lead to organ damage.35 Interleukin-4 (IL)-4 and IL-13 can inhibit GPX4 expression, which coincides with increased expression of arachidonic acid lipoxygenases 15 (ALOX15) in a variety of tissues, such as lung, thus permitting robust production of key inflammatory intermediates.36 Therefore, the impaired activity of GPX4 may contribute to immune surveillance in ferroptosis. However, GPX4 knockout can also damage tissues in a necrotic, apoptotic, or pyroptotic manner.37–39 Therefore, further study of GPX4 will be of great practical significance.
Cysteine is a critical non-essential amino acid in the process of cell proliferation. Previous studies have demonstrated that the sulfur-transfer pathway is important for the synthesis of cysteine through transferring a sulfur atom from methionine to serine.40 Cysteine is not only a raw material for protein synthesis but also a key substrate for GSH antioxidation to protect cells against oxidative damage. When cysteine is deficient, cells obtain cysteine through both system xc- and the sulfur-transfer pathway to restore the intracellular GSH level and protect cells against oxidative damage.40 Therefore, upregulating genes related to the sulfur-transfer pathway can inhibit cellular ferroptosis.41
Signalling Pathways Associated with Ferroptosis
p53
p53 is one of the two major ferroptosis signalling molecules in the nucleus and a cancer suppressor gene that can be activated by oxidative stress. Recent studies have shown that p53 has a complex dual role in the regulation of ferroptosis through either transcriptional or post-translational mechanisms.42,43 On the one hand, p53 is involved in ferroptosis as a transcriptional repressor of SLC7A11. SLC7A11 is an essential component of system xc-, and p53 can inhibit cysteine uptake by downregulating SLC7A11 to reduce GPX activity and GSH synthesis, subsequently causing intracellular ROS accumulation and cellular ferroptosis.44 On the other hand, p53 can trigger ferroptosis by enhancing the expression of glutaminase 2 (GLS2) and spermidine/spermine N1-acetyltransferase 1 (SAT1). The former participates in ferroptosis by decreasing the level of GSH and increasing the levels of cellular ROS,45 and the latter promotes the expression and activation of 15-LOX (an iron-binding enzyme) to promote PUFA oxidation and lipid peroxidation.46 In addition, p53 has been reported to inhibit ferroptosis by directly inhibiting the activity of dipeptidyl peptidase-4 or promoting the expression of cyclin-dependent kinase inhibitor 1A.47
Nuclear Factor Erythroid 2-Related Factor 2, Nrf2
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor in the cellular antioxidant response and a major ferroptosis signalling molecule in the nucleus. Under normal physiological conditions, Nrf2 binds to Kelch-like-ECH-associated protein 1 (Keap1) in the cytoplasm and is degraded through the ubiquitin-proteasome pathway. Under oxidative stress, Nrf2 mediates detoxification by dissociating from the cytoplasmic inhibitor Keap1 and then translocates into the nucleus, activating Nrf2 transcriptional genes and protecting cells against oxidative stress.48 The activation of Nrf2 reduces iron absorption, limits ROS production and enhances the cellular antioxidant capacity.48–51 Therefore, Nrf2 can inhibit ferroptosis. In addition, GSH catalytic and regulatory-dependent subunits—glutamate cysteine ligase catalytic/glutamate cysteine ligase modifier subunit, glutathione synthetase and SLC7A11—are also transcriptional targets of Nrf2. Activation of the Keap1-Nrf2 signalling pathway promotes system xc- activation and GPX4 expression and accelerates cystine-glutamate transport, thereby scavenging accumulated lipid peroxides and inhibiting ferroptosis.44,52
Heat Shock Proteins, HSPs
Heat shock proteins are a class of highly conserved stress proteins that widely exist in organisms. Heat shock protein-β1 (HSPB1) is a negative regulator of ferroptosis in cancer cells.53 Erastin can stimulate the expression of heat shock factor 1 (HSF1)-dependent HSPB1 in cancer cells.53 Downregulation of HSF1 and HSPB1 enhances erastin-induced ferroptosis. In contrast, heat shock pretreatment and HSPB1 overexpression inhibit erastin-induced ferroptosis.53 Protein kinase c-mediated phosphorylation of HSPB1 protects organisms against ferroptosis damage by reducing production of iron-mediated ROS.53 Inhibition of the HSF1-HSPB1 pathway and HSPB1 phosphorylation can increase the anticancer activity of erastin in a human xenograft mouse tumour model.53 The above-mentioned results reveal that HSPB1 exerts an inhibitory effect on ferroptosis.53 In addition to HSPB1, heat shock protein A5 (HSPA5), located in the endoplasmic reticulum, has also been shown to bind and stabilize GPX4, which greatly increases the cellular antioxidant capacity and may be associated with the negative regulation of ferroptosis.54
Haem Oxygenase-1, HO-1
HO-1 is an intracellular enzyme that catalyses the oxidation of haem to generate ferrous iron, carbon monoxide (CO) and biliverdin. Over-activation of HO-1 also leads to the accumulation of large amounts of Fe2+ in the cytoplasm and promotes ROS production, which ultimately causes ferroptosis.13 The HO-1 inhibitor zinc protoporphyrin IX prevents erastin-induced ferroptosis in cancer cells. Haem- and CO-releasing molecules (CORM) promote ferroptosis by increasing HO-1 expression and membrane lipid peroxidation,55 suggesting that HO-1 can mediate ferroptosis.
Nuclear Receptor Coactivator 4, NCOA4
Recent studies have reported that autophagy can regulate the sensitivity of cells to ferroptosis inducers by affecting iron metabolism. Mancias et al found that nuclear receptor coactivator 4 (NCOA4), a selective cargo receptor, could specifically recognize ferritin and transport it to lysosomes, and then, lysosomes released iron ions from ferritin into the cytoplasm and generated oxygen-free radicals, which caused oxidative damage through the Fenton reaction.56–58 Downregulating NCOA4 expression or silencing autophagy-related genes (ATGs) (eg, ATG3, ATG5, ATG7, and ATG13) can inhibit ferroptosis by reducing ferritin degradation, iron accumulation, and lipid peroxidation.56 Further investigations have shown that the activation of ATG5-ATG7-NCOA4 autophagy pathway promotes ferroptosis, which may be a potential therapeutic target for ferroptosis-related diseases.57
Mevalonate Pathway
Shimada et al found that isopentenyl pyrophosphate (IPP) and antioxidant coenzyme Q10 (CoQ10), two essential products of the mevalonate pathway, were both involved in the regulation of ferroptosis.59 IPP is a chemical substance produced by decarboxylation after the activation of mevalonate that biosynthesizes cholesterol, isopentenylates selenocysteine transferase, and produces the antioxidant CoQ10.60 In addition, IPP can regulate GPX4 synthesis by regulating the maturation of selenocysteine tRNA.61 CoQ10, a lipophilic radical-trapping antioxidant, can be inhibited by myristoylated ferroptosis suppressor protein 1 (FSP1). The inhibition of CoQ10 induces ferroptosis by decreasing the cellular antioxidant capacity and increasing lipid peroxides.62 Cells with FSP1 knockout are more susceptible to ferroptosis inducers, and this process can be rescued by overexpression of FSP1.63
Prostaglandin-Endoperoxide Synthase 2, PTGS2
Prostaglandin-endoperoxide synthase 2 (PTGS2) is a well-known gene that encodes cyclooxygenase-2 (COX-2), and its function is to metabolize arachidonic acid (AA) into prostaglandins. As the most upregulated gene in BJeLR cells following treatment with ferroptosis inducers, such as erastin or RSL3,64 PTGS2 is considered a biomarker of ferroptosis. However, recent studies have reported that downregulation of PTGS2 expression prevents ferroptotic neuronal death in a traumatic brain injury mouse model,65 and PTGS2 inhibitors (eg, indomethacin) fail to affect ferroptotic cell death.64 Therefore, the exact function of PTGS2 in ferroptosis needs to be further studied.
ChaC Glutathione-Specific Gamma-Glutamylcyclotransferase 1, CHAC1
ChaC glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1) has γ-glutamylcyclotransferase activity and reduces intracellular GSH levels by digesting glutathione into 5-oxoproline and cysteinylglycine dipeptide.66 RNA sequencing has shown that CHAC1 is the most upregulated gene after treatment with erastin and sorafenib in vitro.67 Knockdown of CHAC1 rescues the cystine starvation-induced reduction in glutathione (GSH) levels and cell death.68 Moreover, CHAC1 degradation of GSH enhances cystine starvation-induced necroptosis and ferroptosis through the activated GCN2-eIF2α-ATF4 pathway in TNBC cells.68 Several studies have also reported the upregulation of CHAC1 expression in tumour cells treated with the ferroptosis inducers erastin or artesunate.47,69 These results suggest that increased expression of CHAC1 may be an effective biomarker of ferroptosis.
Ferroptosis and Lung Diseases
Previous studies have proven that ferroptosis plays an essential role in the occurrence and progression of various pathological processes and diseases, including intracerebral haemorrhage,9 ischaemic stroke,70 sepsis,71 cancer,72 and myocardial infarction.73 More recently, several studies have indicated that ferroptosis is a potential therapeutic target for various lung diseases, such as acute lung injury, chronic obstructive pulmonary disease, pulmonary fibrosis, pulmonary infection and asthma (Table 1). In this review, we summarize the association between ferroptosis and diverse lung diseases.
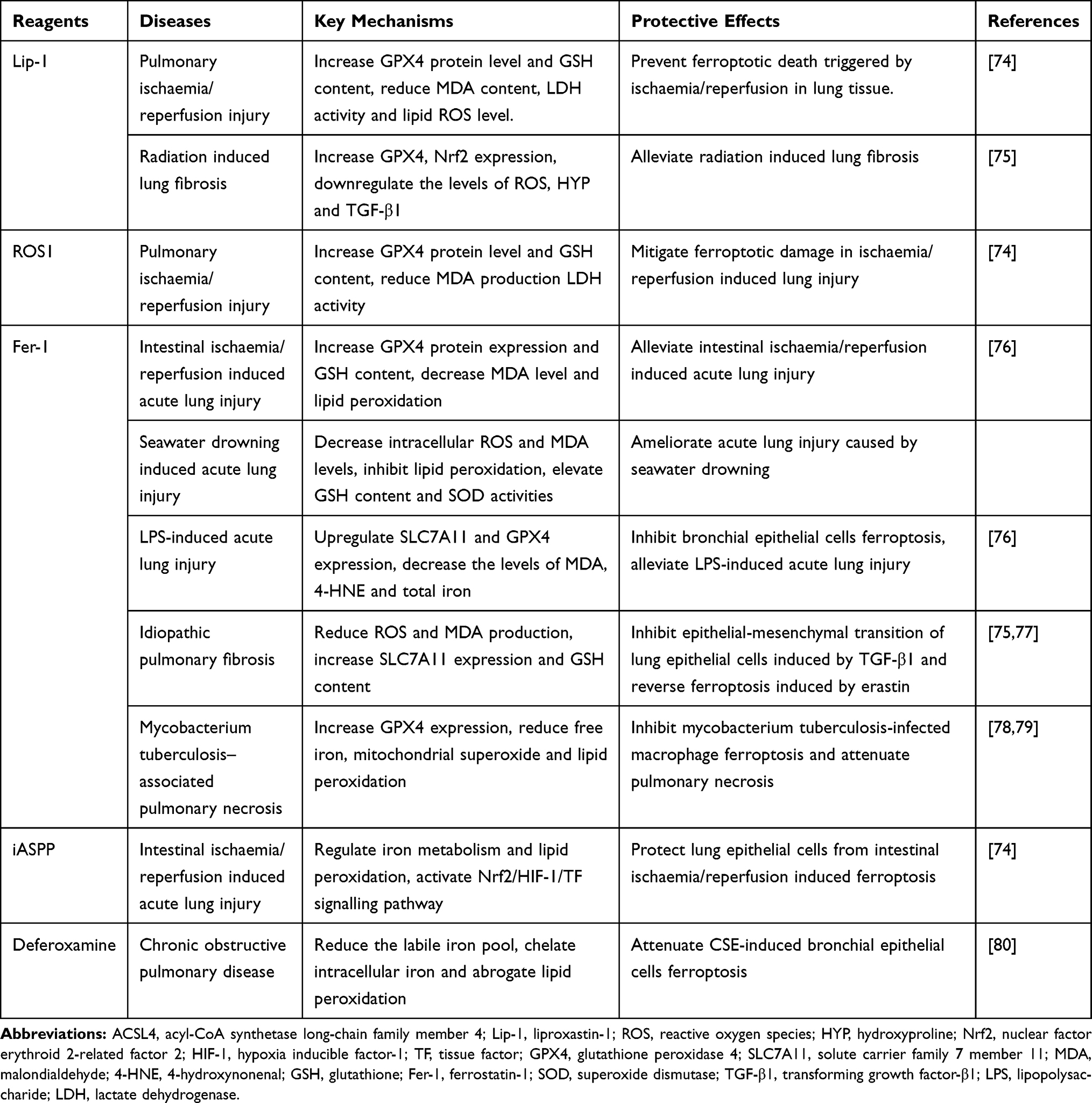 |
Table 1 Treatment Strategy for Ferroptosis in Lung Diseases |
Acute Lung Injury, ALI
ALI is an acute hypoxic respiratory insufficiency or respiratory failure caused by diffuse pulmonary interstitial and alveolar oedema. Recently, numerous studies have shown that ferroptosis is involved in the initiation and progression of ALI.81 It has been shown that the pulmonary tissue of oleic acid-induced ALI mice presents ferroptosis-related features, such as iron overload, decreased GSH and downregulation of GPX4 expression, suggesting that ferroptosis may occur in the lung tissue of ALI mice.82 Inhibitor of apoptosis stimulating protein of P53 (iASPP), the only inhibitor of the apoptosis stimulating protein of p53 (ASPP) family, can effectively inhibit ferroptosis and attenuate ALI induced by intestinal ischaemia/reperfusion, and this protective effect is mediated by the activation of the Nrf2/HIF-1/TF signalling pathway.74 Lipopolysaccharide, a component of the outer wall of gram-negative bacteria, is composed of lipids and polysaccharides and can reduce the activation of the human bronchial epithelial cell line BEAS-2B, and this effect can be rescued by the ferroptosis inhibitor ferrostatin-1 (fer-1). In vivo experiments have further proven that fer-1 has a therapeutic effect on lipopolysaccharide-induced ALI and can downregulate the levels of the proinflammatory cytokines IL-6 and tumour necrosis factor-α (TNF-α) in bronchoalveolar lavage fluid and then alleviate ferroptosis.76 These ferroptosis-related characteristics occur in lung tissue, suggesting that ferroptosis is involved in the development and progression of ALI and that inhibiting ferroptosis can effectively attenuate the pathological level of ALI.
Chronic Obstructive Pulmonary Disease, COPD
COPD is a chronic respiratory disease that leads to the dilation of distal bronchioles accompanied by destruction of the tracheal wall. The morbidity and mortality rates of COPD are among the highest of all diseases worldwide.83 Smoking is an independent risk factor of COPD. Previous studies have demonstrated that unstable iron accumulation and increased lipid peroxidation accompanied by non-apoptotic cell death occur in lung epithelial cells during exposure to cigarette smoke (CS). This process is negatively regulated by GPX4, suggesting that ferroptosis may be involved in the process of COPD.80 Treatment of lung epithelial cells with deferoxamine and fer-1 can reduce the lipid peroxidation induced by CS extract, and inhibiting GPX4 has the same effect.80 Therefore, the inhibition of lipid peroxidation by GPX4 and antioxidants (such as fer-1) and the chelation of unstable intracellular iron by deferoxamine may be effective means to prevent ferroptosis and the progression of COPD.
Another study showed that CS exposure could increase the lavage concentration of iron and ferritin, serum ferritin level and non-heme iron concentration in lung tissue. Exposure of cultured respiratory epithelial cells to cigarette smoke condensates in vitro could cause similar iron accumulation and oxidative stress responses and increase IL-8 release. The authors also found that compared with those in healthy non-smokers, serum iron and ferritin concentrations were increased in smokers. Deposition of iron and other particulate matter in the lungs of smokers could interfere with oxidative stress by regulating iron homeostasis.84
Radiation-Induced Lung Injury, RILI
Radiation-induced lung injury (RILI) includes acute radiation pneumonitis and radiation-induced lung fibrosis (RILF), which most commonly occur after radiotherapy of chest tumours. It is traditionally believed that cell death in RILF is caused by direct radiation damage to DNA.85 However, ionizing radiation not only damages DNA but also induces the production of ROS, causing inflammation and fibrosis.86,87 Therefore, other types of cell death in addition to apoptosis may occur in RILF.
ROS accumulation is the main cause of ferroptosis and the major trigger of the inflammatory response in RILF.88 Studies have shown that the expression of GPX4 in the lung tissue of RILF mice is lower than that of normal mice and that the level of ROS is increased. Ferroptosis inhibitors can reduce collagen deposition in the lungs of RILF mice after radiation, thereby alleviating RILF.89 The ferroptosis inhibitor liproxstatin-1, which downregulates TNF-α, IL-6, IL-10 and transforming growth factor-β1 (TGF-β1) by activating the Nrf2 pathway, may be an effective treatment for RILF.89
In the lung tissue of acute RILI mice, the expression of GPX4 is downregulated. Ferroptosis inhibitors can significantly alleviate radiation-induced histopathological changes in mouse lung tissues,90 suggesting that ferroptosis is involved in acute RILI and that ionizing radiation-induced ROS may be the original trigger factor of ferroptosis in acute RILI. At the same time, ferroptosis can also affect the level of inflammatory cytokines during acute RILI.90
Pulmonary Fibrosis, PF
Pulmonary fibrosis (PF) is a fatal fibrous lung disease. It has been shown that the lncRNA zinc finger antisense 1 (ZFAS1) is upregulated in bleomycin-induced PF rat lung tissue and in TGF-β1-induced human foetal lung fibroblast cells and is positively correlated with the expression of solute carrier family 38 member 1 (SLC38A1), suggesting that the lncRNA ZFAS1 is an important regulator of lipid peroxidation. Knockdown of lncRNA ZFAS1 can significantly alleviate the fibroblast activation, inflammation and lipid peroxidation induced by TGF-β1. Experiments have shown that inhibition of the lncRNA ZFAS1 can significantly abolish the lipid peroxidation induced by bleomycin, thus inhibiting ferroptosis and alleviating PF.91
Idiopathic pulmonary fibrosis (IPF) is a refractory and irreversible progressive pulmonary fibrosis disease. The pathological features of IPF include lung epithelial injury, fibroblast proliferation and extracellular matrix deposition in the lung parenchyma.92,93 It is known that the differentiation of fibroblasts into myofibroblasts is involved in the pathogenesis of IPF.94,95 Previous experiments have used TGF-β1-induced human foetal lung fibroblast cells to study the relationship between ferroptosis and PF and the role of GPX4 in this process. The results of those studies indicate that the ferroptosis inducer erastin accelerates the differentiation of fibroblasts into myofibroblasts by promoting lipid peroxidation and inhibiting the expression of GPX4, while fer-1 can inhibit PF and ferroptosis by reducing lipid peroxidation and enhancing the expression of GPX4.75
Paraquat is an inexpensive and highly effective herbicide that can cause acute lung failure, pulmonary fibrosis and a series of other important organ diseases. The toxicity of paraquat is caused by the formation of intracellular high-energy oxygen-free radicals and lipid peroxidation. Studies suggest that ferroptosis is related to PF caused by paraquat, and applying ferroptosis inhibitors may be an effective therapy to reduce paraquat poisoning.77,96
In addition, ferroptosis is also related to the fibrosis of other organs. Inhibition of ferroptosis by fer-1 can improve myocardial fibrosis in mice, and application of fer-1 or knockout of p53 to inhibit ferroptosis of hepatic stellate cells can aggravate liver fibrosis in mice.97,98
Infectious Lung Diseases
Necroptosis can control infection by inhibiting the bacterial load and inflammation. However, ferroptosis plays an adverse role in bacterial infection-related lung injury and bacterial septicemia.99 Aeruginosa oxidizes arachidonic acid-phosphatidylethanolamine in the host cytomembrane by secreting lipoxygenase and triggers ferroptosis in host bronchial epithelial cells.79 Mycobacterium tuberculosis (Mtb) can cause the death of macrophages, as represented by the decreased contents of GSH and GPX4 in dead cells and the increased contents of free iron, mitochondrial superoxide and lipid peroxidation, suggesting that Mtb can cause ferroptosis. In addition, fer-1 can significantly reduce Mtb-induced macrophage injury, lung tissue necrosis and the bacterial burden of Mtb in mice, which are believed to be related to reduced GPX4 expression and enhanced lipid peroxidation.78 Fast accumulation of lipid peroxides in GPX4-deficient T cells can cause ferroptosis.100 T cells lacking GPX4 cannot effectively prevent viral and parasitic infections. These studies indicate that GPX4 and ferroptosis are crucial to the response of T cells to antiviral and antiparasitic infections.100
Asthma
The role of ferroptosis in asthma remains poorly understood. The phosphatidylethanolamine-binding protein 1/15 lipoxygenase (PEBP1/15-LO) complex is one of the main regulators of ferroptosis and can stimulate IL-13/IL-4 to induce helper T cell inflammation. The co-localization level of PEBP1 and 15-LO in human airway epithelial cells (HAECs) of asthmatic patients has been shown to be higher than that in normal patients, suggesting that ferroptosis may occur in HAECs of asthmatic patients. RSL3 can significantly promote the accumulation of oxidized phosphatidyl ethanolamine and ferroptosis in IL-13-treated HAECs; otherwise, blocking PEBP1 in HAECs can reduce sensitivity to ferroptosis. Therefore, inhibition of ferroptosis in HAEC is an effective treatment for asthma.101 In addition, a recent study proved that acupuncture was effective in treating asthma by markedly reducing the IgE and IL-17A levels in BALF and increasing the IL-4, IL-5 and IL-13 levels in serum, and the mechanism could be associated with the regulation of ferroptosis.102
Allergic Airway Inflammation
Eosinophils are critical for allergic disorders, and promoting eosinophil death effectively attenuates allergic airway inflammation. A previous study showed that treatment with ferroptosis-inducing agents (FINs) triggered eosinophil death in vivo and eventually relieved eosinophilic airway inflammation in mice.103 Furthermore, FINs exerted a synergistic effect with dexamethasone to induce eosinophil death in vitro and to alleviate allergic airway inflammation in vivo, suggesting that FINs are promising therapeutic drugs for allergic airway inflammation.103
Conclusion and Outlook
Ferroptosis is a new type of regulated cell death that has received increasing attention and interest since it was first reported in 2012.1 An increasing number of studies have shown that ferroptosis plays a vital role in a variety of pathophysiological processes, but its complex regulatory mechanism remains elusive. This review summarizes the latest research progress in ferroptosis, showing that in addition to the metabolic pathways of iron ions, amino acids and lipids, many signal regulators, such as Nrf2 and p53, are also involved in the regulation of ferroptosis. Current studies have shown that ferroptosis is involved in many pathological changes in various tissues and organs. Among these changes, the negative regulation of ferroptosis in tumours has been widely studied. In addition, ferroptosis plays a role in neurological diseases, infectious diseases and ischaemia-reperfusion injury. Due to the special state of physiological hyperoxia of the lung tissue, it may be of great value to study the role of ferroptosis in lung injury. Current studies have shown that ferroptosis is crucial to lung diseases, such as ALI and COPD, and that the application of ferroptosis inhibitors can alleviate these lung injuries, as mentioned above. However, the mechanisms of these inhibitors are not yet clear. In-depth study of the pathophysiological mechanism of ferroptosis and lung injury will provide new ideas for the treatment and prevention of related diseases. Despite the remarkable achievements in animal studies on the association between ferroptosis and lung injury, many issues regarding the biological behaviour of ferroptosis need to be clarified before it can be applied to the clinical management of lung injury.
Funding
This work was supported by the National Natural Science Foundation of China [grant numbers: 81871601, 82000085], the Development Fund for Anesthesiology by Shanghai Pulmonary Hospital (2021), the Young Elite Scientist Sponsorship Program by CAST (2018QNRC001), the Basic Research Program for Young Elite Scientist by Shanghai Association for the Study of Pain (2018SASP01), the Research Program for Young Scientist by Shanghai Society of Anesthesiology (2019SSA).
Disclosure
The authors have declared that no competing interests exist.
References
1. Dixon S, Lemberg K, Lamprecht M, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi:10.1016/j.cell.2012.03.042
2. Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014:360438. doi:10.1155/2014/360438
3. Dixon S, Stockwell B. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi:10.1038/nchembio.1416
4. Shah R, Margison K, Pratt D. The potency of diarylamine radical-trapping antioxidants as inhibitors of ferroptosis underscores the role of autoxidation in the mechanism of cell death. ACS Chem Biol. 2017;12:2538–2545. doi:10.1021/acschembio.7b00730
5. Stockwell B, Friedmann Angeli J, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–285. doi:10.1016/j.cell.2017.09.021
6. Sampilvanjil A, Karasawa T, Yamada N, et al. Cigarette smoke extract induces ferroptosis in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2020;318:H508–H518. doi:10.1152/ajpheart.00559.2019
7. Birsen R, Larrue C, Decroocq J, et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica. 2021. doi:10.3324/haematol.2020.259531
8. Yan H, Zou T, Tuo Q, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49.
9. Ma H, Wang X, Zhang W, et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the Nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxid Med Cell Longev. 2020;2020:9067610. doi:10.1155/2020/9067610
10. Bebber C, Thomas E, Stroh J, et al. Ferroptosis response segregates small cell lung cancer (SCLC) neuroendocrine subtypes. Nat Commun. 2021;12:2048. doi:10.1038/s41467-021-22336-4
11. Rui T, Wang H, Li Q, et al. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J Pineal Res. 2021;70:e12704. doi:10.1111/jpi.12704
12. Geng N, Shi B, Li S, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. 2018;22:3826–3836. doi:10.26355/eurrev_201806_15267
13. Chang L, Chiang S, Chen S, Yu Y, Chou R, Chang W. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124–137. doi:10.1016/j.canlet.2017.12.025
14. Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun. 2016;478:838–844. doi:10.1016/j.bbrc.2016.08.034
15. Brandes R, Weissmann N, Schröder K. Nox family NADPH oxidases: molecular mechanisms of activation. Free Radic Biol Med. 2014;76:208–226.
16. Shen Z, Liu T, Li Y, et al. Fenton-reaction-acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano. 2018;12:11355–11365. doi:10.1021/acsnano.8b06201
17. Florean C, Song S, Dicato M, Diederich M. Redox biology of regulated cell death in cancer: a focus on necroptosis and ferroptosis. Free Radic Biol Med. 2019;134:177–189. doi:10.1016/j.freeradbiomed.2019.01.008
18. Wang Y, Quan F, Cao Q, et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J Adv Res. 2021;28:231–243. doi:10.1016/j.jare.2020.07.007
19. Wiernicki B, Dubois H, Tyurina YY, et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020;11:1–11. doi:10.1038/s41419-020-03118-0
20. Kagan V, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi:10.1038/nchembio.2238
21. Yang W, Kim K, Gaschler M, Patel M, Shchepinov M, Stockwell B. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–75. doi:10.1073/pnas.1603244113
22. DeHart D, Fang D, Heslop K, Li L, Lemasters J, Maldonado E. Opening of voltage dependent anion channels promotes reactive oxygen species generation, mitochondrial dysfunction and cell death in cancer cells. Biochem Pharmacol. 2018;148:155–162. doi:10.1016/j.bcp.2017.12.022
23. Wang Y, Chang S, Wu Q, et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci. 2016;8:308. doi:10.3389/fnagi.2016.00308
24. Richard D, Kefi K, Barbe U, Bausero P, Visioli F. Polyunsaturated fatty acids as antioxidants. Pharmacol Res. 2008;57:451–455. doi:10.1016/j.phrs.2008.05.002
25. Dixon S, Winter G, Musavi L, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–1609. doi:10.1021/acschembio.5b00245
26. Doll S, Proneth B, Tyurina Y, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98. doi:10.1038/nchembio.2239
27. Yan B, Ai Y, Sun Q, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2021;81:355–369.e10. doi:10.1016/j.molcel.2020.11.024
28. Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197–211. doi:10.1016/0005-2760(82)90150-3
29. Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018;38:12. doi:10.1186/s40880-018-0288-x
30. Sato H, Fujiwara K, Sagara J, Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages by bacterial lipopolysaccharide. Biochem J. 1995;310:547–551. doi:10.1042/bj3100547
31. Wang W, Green M, Choi J, et al. CD8 T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–274. doi:10.1038/s41586-019-1170-y
32. Brütsch SH, Wang C, Li L, et al. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid Redox Signal. 2015;22:281–293. doi:10.1089/ars.2014.5967
33. Melchers J, Diechtierow M, Fehér K, et al. Structural basis for a distinct catalytic mechanism in Trypanosoma brucei tryparedoxin peroxidase. J Biol Chem. 2008;283:30401–30411. doi:10.1074/jbc.M803563200
34. Kim S, Kang S, Joo J, et al. Characterization of ferroptosis in kidney tubular cell death under diabetic conditions. Cell Death Dis. 2021;12:160. doi:10.1038/s41419-021-03452-x
35. Proneth B, Conrad M. Ferroptosis and necroinflammation, a yet poorly explored link. Cell Death Differ. 2019;26:14–24. doi:10.1038/s41418-018-0173-9
36. Schnurr K, Borchert A, Kuhn H. Inverse regulation of lipid-peroxidizing and hydroperoxyl lipid-reducing enzymes by interleukins 4 and 13. FASEB J. 1999;13:143–154. doi:10.1096/fasebj.13.1.143
37. Canli Ö, Alankuş Y, Grootjans S, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–148. doi:10.1182/blood-2015-06-654194
38. Kang R, Zeng L, Zhu S, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24:97–108.e4. doi:10.1016/j.chom.2018.05.009
39. Ran Q, Liang H, Ikeno Y, et al. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J Gerontol a Biol Sci Med Sci. 2007;62:932–942. doi:10.1093/gerona/62.9.932
40. McBean G. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids. 2012;42:199–205. doi:10.1007/s00726-011-0864-8
41. Hayano M, Yang W, Corn C, Pagano N, Stockwell B. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270–278. doi:10.1038/cdd.2015.93
42. Green D, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458:1127–1130. doi:10.1038/nature07986
43. Khoo K, Hoe K, Verma C, Lane D. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217–236.
44. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi:10.1038/nature14344
45. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107:7455–7460. doi:10.1073/pnas.1001006107
46. Ou Y, Wang S, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113:E6806–E6812. doi:10.1073/pnas.1607152113
47. Xie Y, Zhu S, Song X, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–1704. doi:10.1016/j.celrep.2017.07.055
48. Sun X, Niu X, Chen R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64:488–500. doi:10.1002/hep.28574
49. Chen G, Wei J, Lv X. Research progress of nuclear factor-erythroid 2 related factor 2 in acute lung injury. Chin Crit Care Med. 2018;30:270–274. doi:10.3760/cma.j.issn.2095-4352.2018.03.016
50. Dodson M, Castro-Portuguez R, Zhang D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi:10.1016/j.redox.2019.101107
51. Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–184. doi:10.1002/hep.28251
52. Chen D, Tavana O, Chu B, et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell. 2017;68(1):224–232.e4. doi:10.1016/j.molcel.2017.09.009
53. Sun X, Ou Z, Xie M, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34:5617–5625. doi:10.1038/onc.2015.32
54. Zhu S, Zhang Q, Sun X, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77:2064–2077. doi:10.1158/0008-5472.CAN-16-1979
55. Kwon M, Park E, Lee S, Chung S. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. 2015;6:24393–24403. doi:10.18632/oncotarget.5162
56. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–1032. doi:10.1038/cr.2016.95
57. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–1428. doi:10.1080/15548627.2016.1187366
58. Mancias J, Wang X, Gygi S, Harper J, Kimmelman A. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–109. doi:10.1038/nature13148
59. Shimada K, Skouta R, Kaplan A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503. doi:10.1038/nchembio.2079
60. Moosmann B, Behl C. Selenoproteins, cholesterol-lowering drugs, and the consequences: revisiting of the mevalonate pathway. Trends Cardiovasc Med. 2004;14:273–281. doi:10.1016/j.tcm.2004.08.003
61. Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172:409–422.e21. doi:10.1016/j.cell.2017.11.048
62. Bersuker K, Hendricks J, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692. doi:10.1038/s41586-019-1705-2
63. Doll S, Freitas F, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. doi:10.1038/s41586-019-1707-0
64. Yang W, SriRamaratnam R, Welsch M, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi:10.1016/j.cell.2013.12.010
65. Xiao X, Jiang Y, Liang W, et al. miR-212-5p attenuates ferroptotic neuronal death after traumatic brain injury by targeting Ptgs2. Mol Brain. 2019;12:1–12. doi:10.1186/s13041-019-0501-0
66. Kumar A, Tikoo S, Maity S, et al. Mammalian proapoptotic factor ChaC1 and its homologues function as γ-glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep. 2012;13:1095–1101. doi:10.1038/embor.2012.156
67. Dixon SJ, Patel DN, Welsch M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi:10.7554/eLife.02523
68. Chen MS, Wang SF, Hsu CY, et al. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2α-ATF4 pathway. Oncotarget. 2017;8:114588–114602. doi:10.18632/oncotarget.23055
69. Wang N, Zeng GZ, Yin JL, et al. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s lymphoma. Biochem Biophys Res Commun. 2019;519:533–539. doi:10.1016/j.bbrc.2019.09.023
70. Cui Y, Zhang Y, Zhao X, et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav Immun. 2021;93:312–321. doi:10.1016/j.bbi.2021.01.003
71. Li N, Wang W, Zhou H, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–318. doi:10.1016/j.freeradbiomed.2020.08.009
72. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021.
73. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. 2021;11:3052–3059. doi:10.7150/thno.54113
74. Li Y, Cao Y, Xiao J, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020;27:2635–2650. doi:10.1038/s41418-020-0528-x
75. Gong Y, Wang N, Liu N, Dong H. Lipid peroxidation and GPX4 inhibition are common causes for myofibroblast differentiation and ferroptosis. DNA Cell Biol. 2019;38:725–733. doi:10.1089/dna.2018.4541
76. Liu P, Feng Y, Li H, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Biol Lett. 2020;25:10. doi:10.1186/s11658-020-00205-0
77. Sun L, Dong H, Zhang W, et al. SLC7A11Lipid peroxidation, GSH depletion, and inhibition are common causes of EMT and ferroptosis in A549 cells, but different in specific mechanisms. DNA Cell Biol. 2021;2:172–183. doi:10.1089/dna.2020.5730
78. Amaral E, Costa D, Namasivayam S, et al. Mycobacterium tuberculosisA major role for ferroptosis in -induced cell death and tissue necrosis. J Exp Med. 2019;216:556–570. doi:10.1084/jem.20181776
79. Dar H, Tyurina Y, Mikulska-Ruminska K, et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest. 2018;128:4639–4653. doi:10.1172/JCI99490
80. Yoshida M, Minagawa S, Araya J, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10:3145. doi:10.1038/s41467-019-10991-7
81. Li J, Lu K, Sun F, et al. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 2019;19:96. doi:10.1186/s12967-021-02745-1
82. Zhou H, Li F, Niu J, et al. Ferroptosis was involved in the oleic acid-induced acute lung injury in mice. Sheng li Xue Bao. 2019;71:689–697.
83. Boucherat O, Morissette M, Provencher S, Bonnet S, Maltais F. Bridging lung development with chronic obstructive pulmonary disease. Relevance of developmental pathways in chronic obstructive pulmonary disease pathogenesis. Am J Respir Crit Care Med. 2016;193:362–375. doi:10.1164/rccm.201508-1518PP
84. Ghio A, Hilborn E, Stonehuerner J, et al. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am J Respir Crit Care Med. 2008;178:1130–1138. doi:10.1164/rccm.200802-334OC
85. Ogura A, Oowada S, Kon Y, et al. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009;277:64–71. doi:10.1016/j.canlet.2008.11.021
86. Lee J, Krochak R, Blouin A, et al. Dietary flaxseed prevents radiation-induced oxidative lung damage, inflammation and fibrosis in a mouse model of thoracic radiation injury. Cancer Biol Ther. 2009;8:47–53. doi:10.4161/cbt.8.1.7092
87. Terasaki Y, Ohsawa I, Terasaki M, et al. Hydrogen therapy attenuates irradiation-induced lung damage by reducing oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2011;301:L415–26. doi:10.1152/ajplung.00008.2011
88. Basit F, van Oppen L, Schöckel L, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716. doi:10.1038/cddis.2017.133
89. Li X, Duan L, Yuan S, Zhuang X, Qiao T, He J. Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. J Inflamm. 2019;16:11. doi:10.1186/s12950-019-0216-0
90. Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun. 2019;519:240–245. doi:10.1016/j.bbrc.2019.08.165
91. Yang Y, Tai W, Lu N, et al. lncRNA ZFAS1 promotes lung fibroblast-to-myofibroblast transition and ferroptosis via functioning as a ceRNA through miR-150-5p/SLC38A1 axis. Aging. 2020;12:9085–9102. doi:10.18632/aging.103176
92. Kobayashi K, Araya J, Minagawa S, et al. Involvement of PARK2-mediated mitophagy in idiopathic pulmonary fibrosis pathogenesis. J Immunol. 2016;197:504–516. doi:10.4049/jimmunol.1600265
93. Minagawa S, Araya J, Numata T, et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L391–401. doi:10.1152/ajplung.00097.2010
94. King T, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet. 2011;378:1949–1961. doi:10.1016/S0140-6736(11)60052-4
95. Wynn T, Ramalingam T. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi:10.1038/nm.2807
96. Rashidipour N, Karami-Mohajeri S, Mandegary A, et al. Where ferroptosis inhibitors and paraquat detoxification mechanisms intersect, exploring possible treatment strategies. Toxicology. 2020;433-434:152407. doi:10.1016/j.tox.2020.152407
97. Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116:2672–2680. doi:10.1073/pnas.1821022116
98. Wang L, Zhang Z, Li M, et al. P53-dependent induction of ferroptosis is required for artemether to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. IUBMB Life. 2019;71:45–56. doi:10.1002/iub.1895
99. Zhu H, Santo A, Jia Z, Robert LY. GPx4 in bacterial infection and polymicrobial sepsis: involvement of ferroptosis and pyroptosis. Reactive Oxygen Species. 2019;7:154–160. doi:10.20455/ros.2019.835
100. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm G, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–568. doi:10.1084/jem.20140857
101. Wenzel S, Tyurina Y, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628–641.e26. doi:10.1016/j.cell.2017.09.044
102. Tang W, Dong M, Teng F, et al. TMT-based quantitative proteomics reveals suppression of SLC3A2 and ATP1A3 expression contributes to the inhibitory role of acupuncture on airway inflammation in an OVA-induced mouse asthma model. Biomed Pharmacother. 2021;134:111001. doi:10.1016/j.biopha.2020.111001
103. Wu Y, Chen H, Xuan N, et al. Induction of ferroptosis-like cell death of eosinophils exerts synergistic effects with glucocorticoids in allergic airway inflammation. Thorax. 2020;75(11):918–927. doi:10.1136/thoraxjnl-2020-214764
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.