")
Back to Journals » Journal of Inflammation Research » Volume 15
Role of Ferroptosis in Fibrotic Diseases
Authors Zhou J, Tan Y, Wang R, Li X
Received 14 January 2022
Accepted for publication 2 June 2022
Published 27 June 2022 Volume 2022:15 Pages 3689—3708
DOI https://doi.org/10.2147/JIR.S358470
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Jian Zhou, Yuan Tan, Rurong Wang, Xuehan Li
Department of Anesthesiology, Laboratory of Anesthesia and Critical Care Medicine, West China Hospital, Sichuan University, Chengdu, Sichuan Province, People’s Republic of China
Correspondence: Xuehan Li, Department of Anesthesiology, Laboratory of Anesthesia and Critical Care Medicine, West China Hospital, Sichuan University, No. 37 Guoxue Xiang, Chengdu, Sichuan Province, 610041, People’s Republic of China, Tel +86 18980099133, Email [email protected]
Abstract: Ferroptosis is a unique and pervasive form of regulated cell death driven by iron-dependent phospholipid peroxidation. It results from disturbed cellular metabolism and imbalanced redox homeostasis and is regulated by various cellular metabolic pathways. Recent preclinical studies have revealed that ferroptosis may be an attractive therapeutic target in fibrotic diseases, such as liver fibrosis, pulmonary fibrosis, kidney fibrosis, and myocardial fibrosis. This review summarizes the latest knowledge on the regulatory mechanism of ferroptosis and its roles in fibrotic diseases. These updates may provide a novel perspective for the treatment of fibrotic diseases as well as future research.
Keywords: ferroptosis, liver fibrosis, kidney fibrosis, myocardial fibrosis, pulmonary fibrosis
Introduction
Fibrosis is defined by excessive accumulation of an extracellular matrix (ECM) such as collagen and fibronectin.1 Pathophysiologically, fibrosis is a dysregulated pathologic extension of the normal wound healing response to repetitive or chronic tissue damage, which is characterized by injury, inflammation, myofibroblast activation and migration, and matrix deposition and remodeling.2,3 Common organs involved in fibrosis include the liver (cirrhosis, non-alcoholic steatohepatitis), kidney (chronic kidney disease, renal interstitial fibrosis), heart (heart failure, cardiomyopathy), lung (idiopathic pulmonary fibrosis, cystic fibrosis), and skin (scleroderma),4,5 representing a high incidence rate and significant disease burden. Progressive fibrosis leads to disruption of functional tissue architecture and ultimately organ failure.5,6 Currently available antifibrotic treatments have limited clinical benefits for prevention or regression of disease progression;4 therefore, it is of great significance to explore the effective strategies for fibrosis based on well-established underlying mechanisms. In the past decade, there has been a growing appreciation for the importance of ferroptosis in explaining the pathophysiology of fibrosis and its regression. However, ferroptosis in different cell types may exert distinct influence on fibrosis. The extent to which ferroptosis affects fibrotic diseases is unclear, although several studies have found significant correlations between ferroptosis-associated genes and pathways and fibrotic diseases. Herein, we present the key molecular mechanisms of ferroptosis, describe the crosstalk between ferroptosis and fibrosis-associated signaling pathways, and provide insights into the potential novel therapeutic target of ferroptosis in the context of fibrotic disorders.
Mechanisms of Ferroptosis
Ferroptosis has been a rapidly evolving research area since it was coined in 20127 (see Figure 1). The Nomenclature Committee on Cell Death (NCCD) defines ferroptosis as “a form of regulated cell death (RCD) initiated by oxidative perturbations of the intracellular microenvironment that is under constitutive control by glutathione peroxidase 4 (GPX4) and can be inhibited by iron chelators and lipophilic antioxidants”.8 Subsequent investigations identified a wide range of inducers and inhibitors of ferroptosis (see Table 1). This unique RCD, driven by iron-dependent lipid peroxidation (LPO), is caused by redox imbalance between oxidative damage and antioxidant defense.9 As discussed below, various cellular metabolic pathways have been reported continually in the regulation of ferroptosis at multiple levels, including iron handling, lipid metabolism, mitochondrial activity, redox homeostasis, membrane repair, and intracellular degradation.
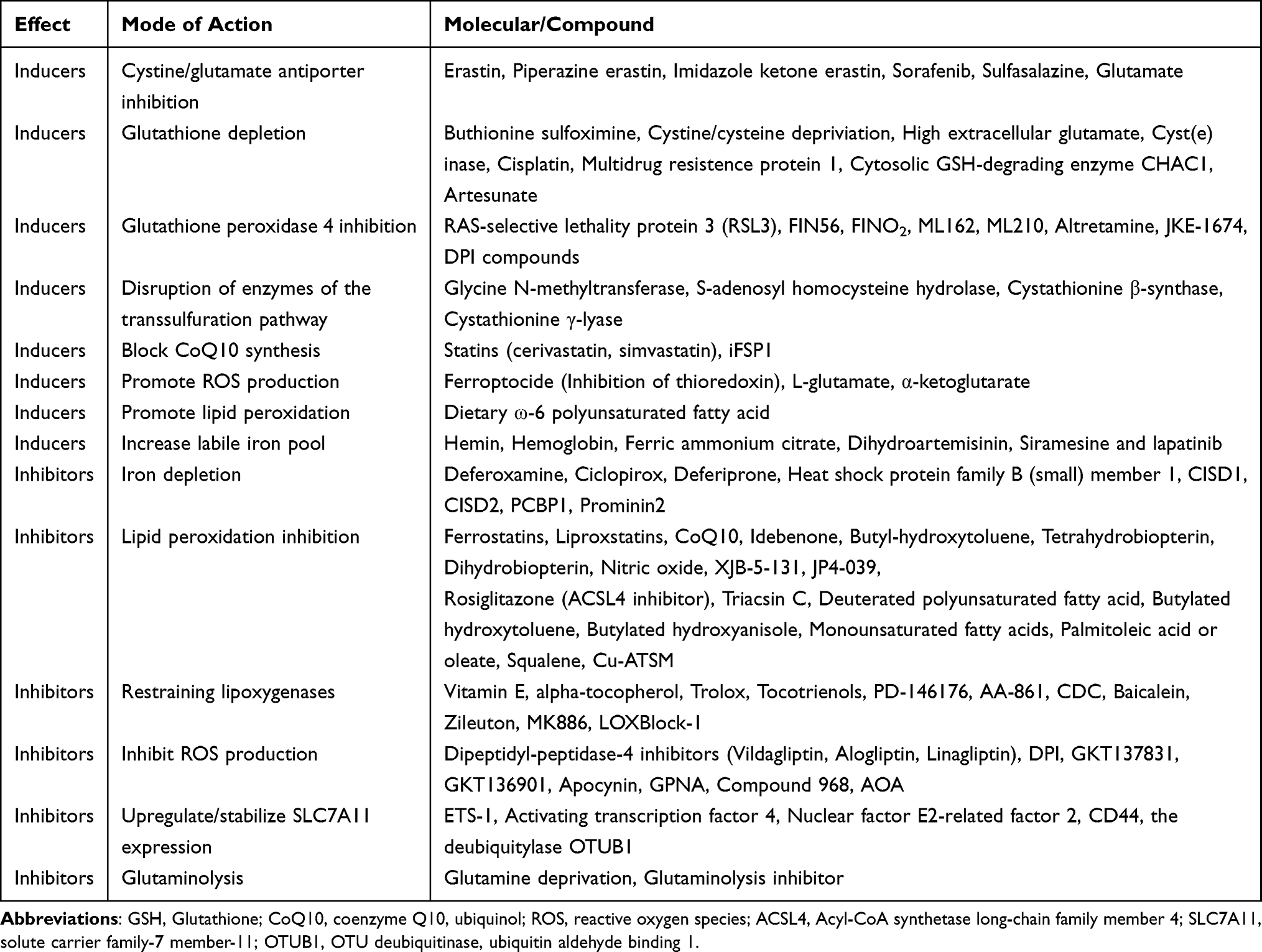 |
Table 1 Inducers and Inhibitors of Ferroptosis |
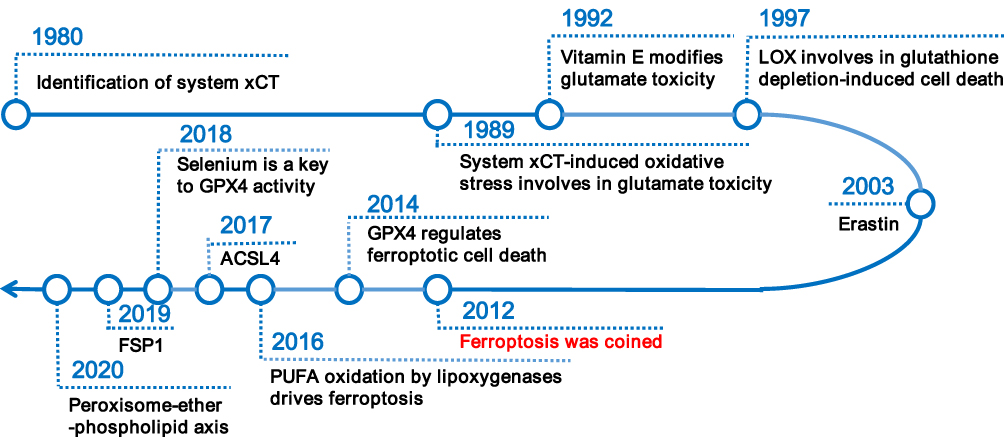 |
Figure 1 Overview of research history of ferroptosis. Abbreviations: xCT, cystine/glutamate antiporter; LOX, lipoxygenase; GPX4, glutathione peroxidase 4; ACSL4, Acyl-CoA synthetase long-chain family member 4; PUFA, polyunsaturated fatty acid; FSP1, ferroptosis suppressor protein 1. |
Iron-Mediated Oxidative Stress
Excessive iron deposition has been reported in fibrotic diseases both in preclinical experimental studies and in clinical samples.10–13 The trace element iron plays central roles in many biological processes of ferroptosis. On the one hand, Fenton reaction relies on iron to amplify phospholipid hydroperoxides (PLOOHs), and PLOOHs further react with iron to yield free radicals and propagate a peroxidation chain reaction;7 on the other hand, iron-dependent enzymes such as lipoxygenases (LOXs), cytochrome P450 oxidoreductase (POR), and NADPH oxidase and many other reactive oxygen species (ROS)-generating processes require iron for catalysis.14 The intracellular labile iron pool (LIP) is carefully orchestrated primarily by iron-regulatory proteins in import, storage, export, and utilization.15,16 Insoluble ferric (Fe3+) from dietary intakes binds to transferrin (Trf) and lactotransferrin, two iron-binding transport proteins that positively regulate iron uptake.17,18 Cellular iron is mainly imported by the plasma membrane protein transferrin receptor (TFRC)-mediated endocytosis and stored within ferritin.19 Specifically, in the acidic environment of endosomes, ferrous reductase STEAP3 metalloreductase reduces Fe3+ to ferrous (Fe2+), the free form of which constitutes LIP.16 Then solute carrier family 11 member 2 (SLC11A2/DMT1) mediates the release of Fe2+ from endosome to cytoplasm. Ferritin heavy chain (FHC) and ferritin light chain constitute the iron-storage protein ferritin, the degradation of which by lysosomes increases LIP. The export of cellular iron is mediated by ferroportin 1 coupled with ceruloplasmin or by ferritin-containing multivesicular bodies and exosomes.19 In extracellular space, Fe2+ is deoxidized Fe3+ to by ferroxidases.16 Conceivably, ferroptosis can be promoted by increasing cellular iron import or releasing ferritin-stored iron; on the contrary, processes that enhance cellular iron export have been shown to suppress ferroptosis (see Figure 2).
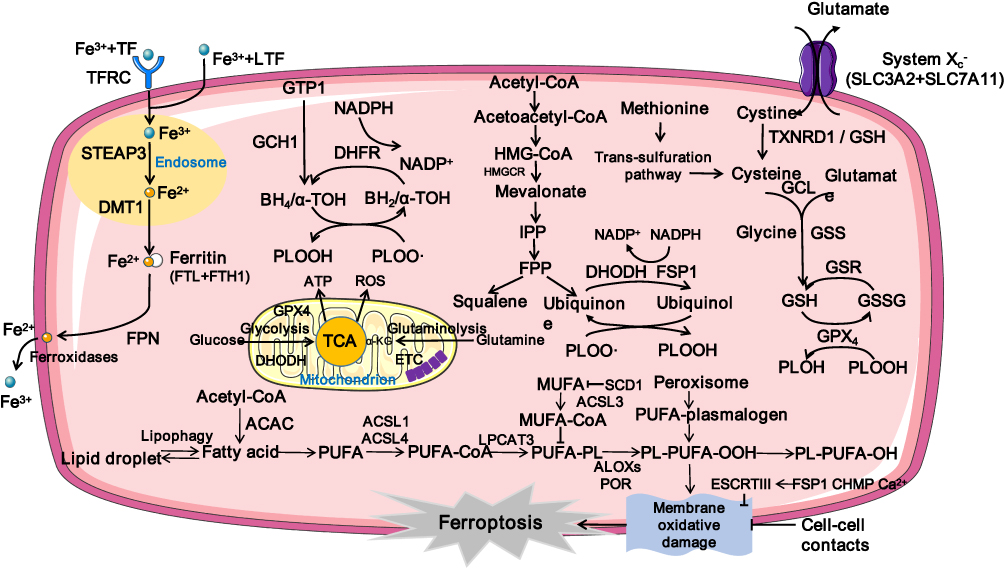 |
Figure 2 Mechanism underlying ferroptosis. Abbreviations: Fe3+, ferric; TF, transferrin; TFRC, transferrin receptor; LTF, lactotransferrin; STEAP3, six-transmembrane epithelial antigen of the prostate 3; Fe2+, ferrous; DMT1, divalent metal transporter 1; FTL, ferritin light chain; FTH1, ferritin heavy chain 1; FPN, Ferroportin; GCH1, GTP cyclohydrolase 1; DHFR, dihydrofolate reductase; PLOOH, dihydroorotate dehydrogenase; PLOO, peroxyl radical; PLOH, phospholipid alcohol; HMG-CoA, hydroxy methylglutaryl coenzyme A reductase inhibitor; HMGCR, HMG-CoA Reductase; IPP, isopentenyl pyrophosphate; FPP, farnesyl pyrophosphate; FSP1, ferroptosis suppressor protein 1; DHODH, dihydroorotate dehydrogenase; SLC7A11, solute carrier family-7 member-11; GCL, glutamate-cysteine ligase; GSS, Glutathione Synthetase; GSR, glutathione-disulfide reductase; GSH, glutathione; GSSG, oxidized glutathione; GPX4, glutathione peroxidase 4; ATP, Adenosine triphosphate; ROS, reactive oxygen species; ETC, electron transport chain; α-KG, α-ketoglutarate; ACAC, Acetyl CoA carboxylase; PUFA, polyunsaturated fatty acid; PUFA-PLs, phospholipids containing polyunsaturated fatty acid chains; MUFA, monounsaturated fatty acid; ACSL1, Acyl-CoA synthetase long-chain family member 1; ACSL3, Acyl-CoA synthetase long-chain family member 3; ACSL4, Acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; POR, cytochrome P450 oxidoreductase; ALOXs, lipoxygenases. |
LPO-Mediated Cytotoxicity
It is now established that uncontrolled LPO is the hallmark of ferroptosis.20,21 The oxidative destruction of lipids requires transition metal iron, ROS and phospholipids containing polyunsaturated fatty acid chains (PUFA-PLs).20 Briefly, initiation of LPO can occur by both non-enzymatic and enzymatic way in PUFA-PLs. Enzymatic LPO is catalyzed by LOXs and POR;22,23 Non-enzymatic LPO is driven by Fenton reaction, a chemical reaction between iron and hydrogen peroxide generating PLOOHs.24 Polyunsaturated fatty acids (PUFAs) such as arachidonic acid and adrenic acid are the main precursors of PLOOHs.25 The adjacent double bond of the bisallylic methylene group in PUFAs makes it more susceptible to be oxidized than other fatty acids (saturated fatty acids [no double bond], monounsaturated fatty acids [1 double bond]) by abating the hydrogen bonding energy.20 Acyl-CoA synthetase long-chain family member 4 (ACSL4) ligates long-chain PUFAs with coenzyme A.26,27 Then lysophosphatidylcholine acyltransferase 3 (LPCAT3) re-esterified them into phospholipids and membranes.26 Therefore, ACSL4 and LPCAT3 are two critical membrane-remodeling enzymes involved in the LPO of ferroptosis (see Figure 2). In addition, a recent study of lipidomic profiling reported that polyunsaturated ether phospholipids synthesized by peroxisomes also acts as substrates for LPO.28 Apart from lipid synthesis, lipid storage and degradation also affect ferroptosis sensitivity. Formation of endoplasmic reticulum-derived lipid droplets isolates PUFA from membrane phospholipids and limits ferroptosis, while lipophagy increases LPO and subsequent ferroptosis.29–31
Antioxidant System-Mediated Detoxification Defenses
Ferroptosis occurs when the prerequisites for ferroptosis exceed the buffering capability of ferroptosis defense systems.9 As discussed below, the cyst(e)ine/glutathione (GSH)/GPX4 axis, the ferroptosis suppressor protein 1 (FSP1)/coenzyme Q10 (CoQ10, coenzyme Q10, ubiquinol) axis, the dihydroorotate dehydrogenase (DHODH)/CoQ10 axis, and the GTP cyclohydrolase 1 (GCH1)/tetrahydrobiopterin (BH4)/dihydrofolate reductase (DHFR) axis are four known antioxidant systems that mediate detoxification of lipid peroxides and protect against ferroptosis (see Figure 2). While the cyst(e)ine/GSH/GPX4 axis locates both in cytoplasm and in mitochondria, the FSP1/CoQ10 axis mainly cooperates with cytosolic GPX4 on the plasma membrane, and the DHODH/CoQ10 axis collaborates with mitochondrial GPX4 in the mitochondria. So far the subcellular localization of the GCH1/BH4/DHFR axis is not fully-illustrated.32
The cyst(e)ine/GSH/GPX4 axis is the first discovered and most extensively studied pathway in suppressing ferroptosis.33 GPX4 is a selenoprotein that catalyzes the reduction and detoxification of lipid ROS production in mammalian cells.34 As the major PLOOH-neutralizing enzyme, GPX4 reduces the PLOOHs to corresponding alcohols (PLOHs), with two electrons most commonly from GSH.34 As the most abundant antioxidant in mammalian cells, GSH plays a critical role in the generation of iron-sulfur clusters and catalysis of GPX4 and glutathione-S-transferases.14 After GPX4-mediated reduction of PLOOH, the oxidized GSH (GSSG) will be recycled by GSH-disulfide reductase with electrons provided by NADPH/H+.14 Cysteine is the rate-limiting substrate for the biosynthesis of reduced GSH.35 Cysteine can be (1) imported from environment: by a neutral amino acid transporter/in its oxidized form (cystine) by the system xc- cystine/glutamate antiporter (a transmembrane protein complex containing catalytic subunit solute carrier family 7 member 11 [SLC7A11] and regulatory subunit solute carrier family 3 member 2 [SLC3A2]) and further deoxidized via GSH or thioredoxin reductase 1;36 or (2) synthesized using methionine and glucose in the trans-sulfuration pathway.37
The FSP1/CoQ10 axis was identified in 2019 by two independent studies using an overexpression screen in one study and a synthetic lethal CRISPR-Cas9 knockout screen in the other.38,39 FSP1 (previously known as apoptosis-inducing factor mitochondrial 2), which is initially called p53-responsive gene 3,40 is a target of transcription factors NRF2,41 CRBP,42 and PPARα.43 Ferroptosis induced by pharmacological inhibition or genetic deletion of GPX4 can be completely suppressed by FSP1, however, the anti-ferroptotic function of FSP1 disappears with the mutation of the myristoylation site. Mechanistically, ferroptosis inhibition by FSP1 is mediated via CoQ10 in a NADPH-dependent manner. On the one hand, FSP1 functions as oxidoreductase to reduce CoQ10 to ubiquinol, which traps lipid peroxyl radicals that mediate lipid autoxidation; on the other hand, FSP1 regenerates the oxidized α-tocopherol radical (vitamin E), which is an effective natural chain-breaking antioxidant.38,39 Similarly, DHODH is a mitochondria-localized enzyme that can also reduce CoQ10 to ubiquinol, thus detoxifying mitochondrial LPO and conferring defenses to ferroptosis in parallel to mitochondrial GPX4.44
BH4 and dihydrobiopterin are metabolic products of GCH1.45 Recently it has been reported that BH4 protects phospholipids containing two PUFA tails against oxidative degradation via functioning as an endogenous radical-trapping antioxidant or participating in ubiquinone synthesis.46 In addition, endosomal sorting complexes required for transport-III–Dependent Membrane Repair systems confer resistance to oxidative damage in ferroptosis.47 These defense systems operate synergistically or complementarily in negatively regulating ferroptosis.
Role of Ferroptosis in Fibrotic Diseases
Studies on ferroptosis or fibrosis that converge on cellular metabolism have revealed an intimate relationship between ferroptosis and fibrosis and their shared metabolic pathway. Increased glycolysis, upregulation of glutaminolysis, and enhanced fatty acid oxidation are critical drivers of metabolomic reprogramming of activated fibroblasts,2 while under certain conditions tricarboxylic acid cycle fueled by glutaminolysis may play a major role in ferroptosis induction.48 Besides, the perturbation of cellular iron and redox homeostasis involves in transforming growth factor-β (TGF-β)-induced epithelial-mesenchymal transition (EMT) during fibrosis. Dramatic decline in the FHC as well as rise in intracellular free iron during TGF-β-induced EMT contribute to iron overload, which boosts ROS production.49 Conversely, ROS also takes part in TGF-β-induced EMT and modulates TGF-β’s fibrogenic effects through different pathways.50,51 Downregulated GSH in TGF-β-induced EMT of fibrosis was observed in vivo, in vitro, and in human beings.52–55 Additionally, upregulated LPO indicated by an increased level of malondialdehyde was reported in experimental animal models of pulmonary fibrosis.56 The fibrosis-related pathways are illustrated in Figure 3 and summarized in Table 2. The overlaps between drugs targeting ferroptosis in fibrotic diseases are depicted in Figure 4 and summarized in Table 3.
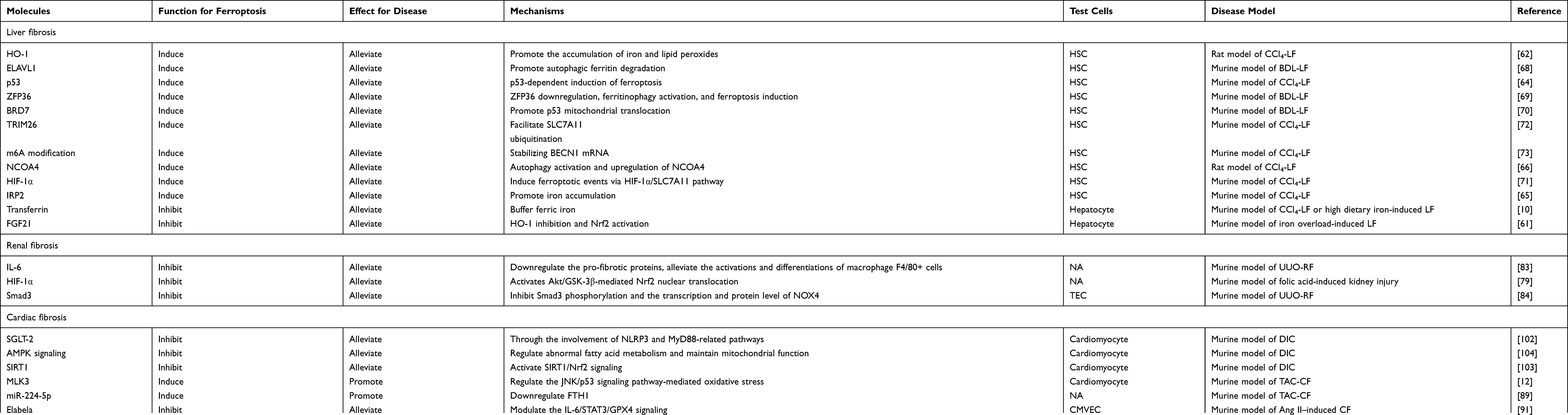 |
Table 2 Molecules Involved in Ferroptosis and Fibrosis |
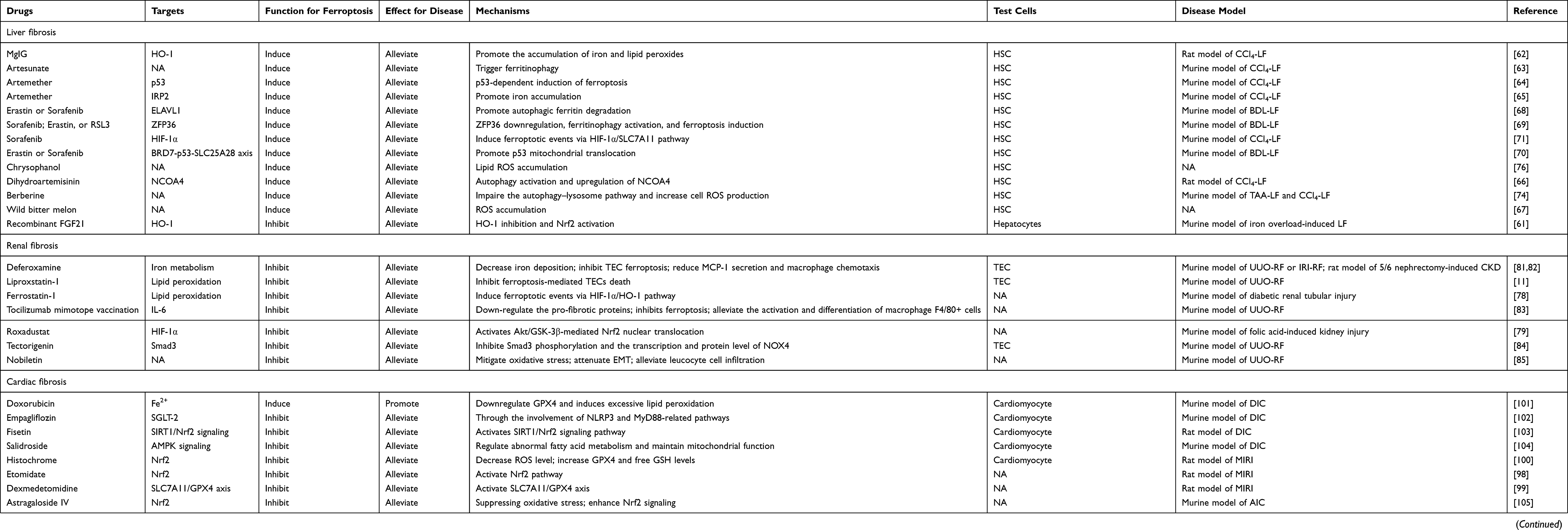 |
Table 3 Drugs Targeting Ferroptosis in Fibrosis |
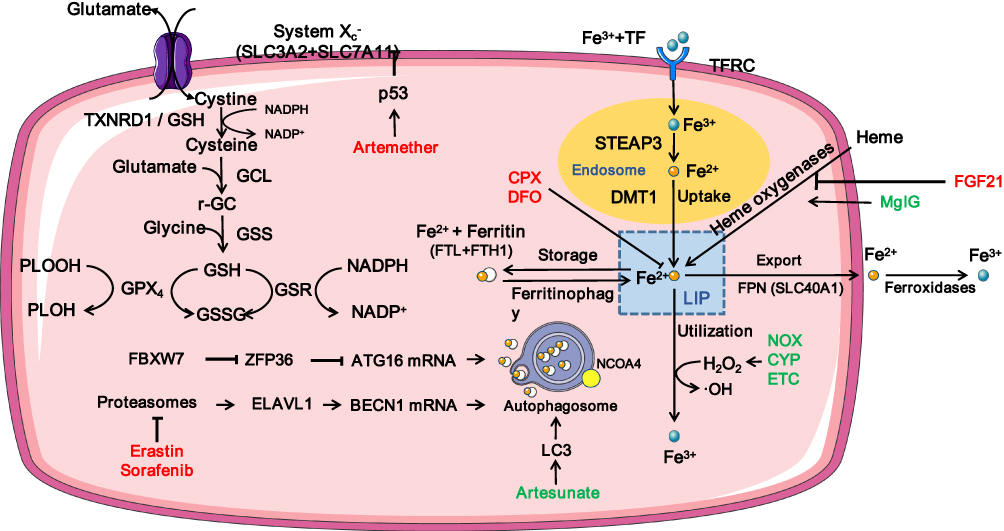 |
Figure 3 Fibrotic-related pathways in ferroptosis. Abbreviations: SXC, System Xc-; SLC7A11, solute carrier family-7 member-11; GCL, glutamate-cysteine ligase; GSS, Glutathione Synthetase; GSR, glutathione-disulfide reductase; GSH, glutathione; GSSG, oxidized glutathione; GPX4, glutathione peroxidase 4; PLOH, phospholipid alcohol; PLOOH, dihydroorotate dehydrogenase; Fe3+, ferric; TF, transferrin; TFRC, transferrin receptor; LTF, lactotransferrin; STEAP3, six-transmembrane epithelial antigen of the prostate 3; Fe2+, ferrous; DMT1, divalent metal transporter 1; FTL, ferritin light chain; FTH1, ferritin heavy chain 1; FPN, Ferroportin; LIP, labile iron pool; FGF21, Fibroblast growth factor 21; HO-1, heme oxygenase-1; MgIG, Magnesium isoglycyrrhizinate; CPX, Ciclopirox; DFO, deferoxamine; NOX, NADPH oxidase; CYP, cytochrome P450; ETC, electron transport chain; LC3, m icrotubule-associated protein light chain 3; FBXW7, F-box and WD repeat domain containing 7; ZFP36, ZFP36 ring finger protein; ATG16, autophagy related 16; ELAVL1, ELAV like RNA binding protein 1; BECN1, Beclin 1; NCOA4, nuclear receptor coactivator 4. |
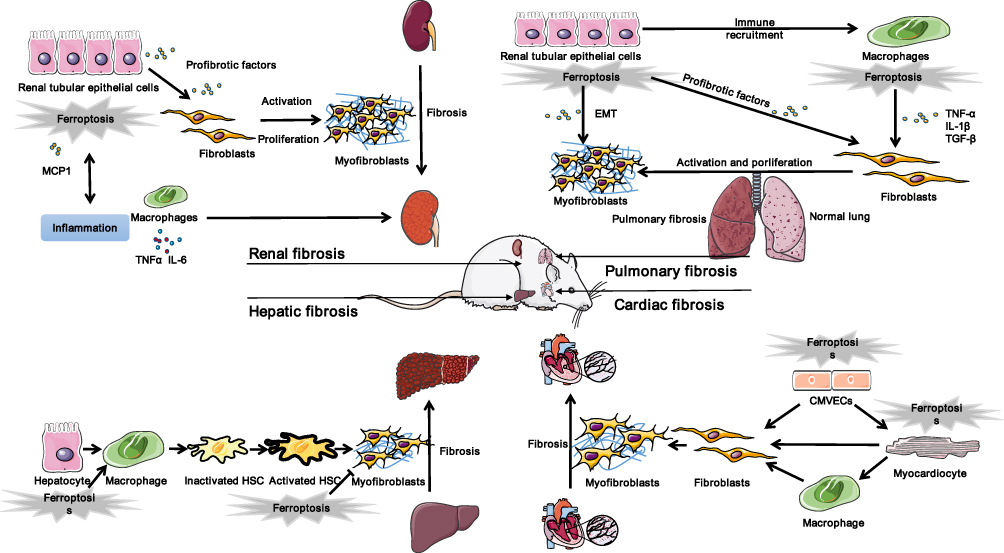 |
Figure 4 Ferroptosis in fibrotic therapy. Abbreviations: TNFα, tumor necrosis factor α; IL-6, interleukin-6; TGF-β, transforming growth factor-β; MCP1, Monocyte chemoattractant protein-1; EMT, epithelial mesenchymal transition; HSC, hepatic stellate cell; CMVEC, cardiac microvascular endothelial cell. |
Ferroptosis and Hepatic Fibrosis
Hepatic fibrosis refers to the replacement of damaged normal tissue by fibrous scar which is mainly composed of excessive deposition of ECM.57 There are two main types of chronic liver injuries that lead to liver inflammation and subsequent fibrosis: hepatotoxic injury (caused by chronic injury of hepatocytes) and cholestatic injury (caused by obstruction to bile flow). The conventional experimental animal model includes pericentral liver fibrosis (acute hepatocellular injury induced by hepatocellular carbon tetrachloride) and periportal liver fibrosis (cholestasis induced by bile duct ligation). Among the multiple progenitor cells in the liver, hepatic stellate cells (HSCs) are the primary source of myofibroblast, regardless of etiology.58 Besides, portal fibroblast, bone marrow-derived fibrocytes, mesothelial cells, and Gli+ mesenchymal stem cell-like cells can also transform into myofibroblasts.5 Cirrhosis is the ultimate result of hepatic fibrogenesis, which leads to hepatocarcinoma and liver failure.57
Intriguingly, ferroptosis has been reported to have contradictory effects in the processes of progression and regression of liver fibrosis. It has long been speculated that iron deposition may be a predisposing factor for liver fibrosis and regression. Liver biopsy samples obtained from patients with liver cirrhosis showed increased iron levels and ferroptosis markers. Ferroptosis of myofibroblast induced by inhibiting cystine/glutamate antiporter (xCT)/SLC7A11 aggravates chronic liver injury, which is strongly correlated with liver fibrosis.59 High dietary iron predisposes mice to liver fibrosis, which can be reversed by ferroptosis inhibitor ferrostatin-1 (Fer-1), suggesting that ferroptosis underlies fibrosis.10,60 Actually, ferroptosis-induced liver fibrosis is initiated by the excessive free non-heme iron in liver tissue. Using a hepatocyte-specific Trf knockout mice model, Yu et al10 found that loss of hepatic Trf, a serum-abundant metal-binding protein which plays a critical role in iron homeostasis, contributes to ferroptosis-induced liver fibrosis. Specifically, in the context of Trf insufficiency, Slc39a14 transports large amounts of non-Trf-bound iron into hepatocytes, thereby promotes ferroptosis-induced liver fibrosis under high dietary iron and carbon tetrachloride (CCI4) injections.10 Iron overload promote ferroptosis in hepatocytes via heme oxygenase-1 (HO-1) pathway, which results in liver injury and fibrosis. Fibroblast growth factor 21 (FGF21) is an endocrine member of the FGF family that plays an important role in energy balance and metabolism of glucose and lipid. Wu et al61 found that FGF21 promotes HO-1 ubiquitination and degradation as well as activates nuclear factor erythroid 2-related factor 2 (Nrf2), both of which provide a mechanistic explanation for the inhibitory effects of FGF21 on ferroptosis and liver fibrosis. Nevertheless, several other studies reported that ferroptosis has an inhibitory effect on liver fibrosis, possibly by inactivating HSCs and inducing HSCs death, two main mechanisms of removal of hepatic myofibroblasts during the regression of liver fibrosis.58 Magnesium isoglycyrrhizinate, a natural product derived from natural glycyrrhizic acid, markedly attenuated CCl4-induced hepatic fibrosis by HO-1 mediated HSCs ferroptosis.62 Artesunate, a water-soluble hemisuccinate derivative of artemisinin, remarkably promoted ferroptosis of activated HSCs in a ferritinophagy-mediated way, the anti-fibrosis effect of which can be completely abolished by deferoxamine (DFO).63 Consistently, Artemether (ART), another derivative of artemisinin which is the primary choice for the therapy of malaria, ameliorates liver fibrosis and HSCs activation by P53-dependent induction of ferroptosis.64 Another study reported that iron regulatory protein 2-iron-ROS axis is also indispensable for the anti-fibrotic effect of ART via inducing ferroptosis in HSC.65 Moreover, dihydroartemisinin, another artemisinin derivative, was also found to alleviate hepatic fibrosis through inducing ferroptosis in HSCs, which is closely related to autophagy activation.66 Recently, it is reported that wild bitter melon extract as a potential antifibrotic agent of liver fibrosis attenuates lipopolysaccharide-induced HSCs activation via the regulation of ER stress and ferroptosis.67
Activation and transdifferentiation of quiescent HSCs into matrix-producing myofibroblasts represented a milestone in liver fibrosis.57 Targeting to scavenge HSCs by activating ferroptosis is considered to be a potential strategy to treat liver fibrosis. Several molecules such as ELAV like RNA binding protein 1 (ELAVL1), ZFP36 ring finger protein (ZFP36), bromodomain-containing protein 7 (BRD7), and tripartite motif-containing protein 26 (TRIM26) have been reported to play crucial roles in regulating ferroptosis in HSCs. ELAVL1, a ubiquitous RNA-binding protein which regulates mRNA cargos containing AU-rich elements (AREs) in the 3rd-untranslated region (3ʹ-UTR), regulates ferroptosis in liver fibrosis in a posttranscriptional manner. Erastin or sorafenib treatment upregulates the expression of ELAVL1 via inhibiting the ubiquitin-proteasome pathway. The increased ELAVL1 binds to BECN1 mRNA and promotes BECN1/Beclin1 generation, thereby triggering autophagic ferritin degradation and finally inducing HSCs ferroptosis and alleviating liver fibrosis.68 Similarly, ZFP36 destabilizes autophagy related 16 like 1 (ATG16L1) mRNA through binding to the AREs within the 3ʹ-UTR, thus inhibiting macroautophagy/autophagy activation and mediating ferroptosis resistance. Ferroptosis-inducing compounds downregulate ZFP36 protein expression by the ubiquitin ligase F-box and WD repeat domain containing 7 (FBXW7/CDC4) which recognizes the SFSGLPS motif. Therefore, treatment erastin and sorafenib ameliorate liver fibrosis by inducing ZFP36 downregulation, ferritinophagy activation, and ferroptosis in HSCs.69 Another study reported that ferroptosis inducers increased BRD7 protein expression via the inhibition of the ubiquitin-proteasome pathway. BRD7 directly binds with the p53 N-terminal transactivation domain, thereby promoting p53 mitochondrial translocation, which subsequently elevated the activity of solute carrier family 25 member 28 (SLC25A28) that resulted in excessive deposition of redox-active iron and hyperfunction of electron transfer chain. Collectively, Erastin or sorafenib treatment has been shown to suppress murine liver fibrosis by inducing HSCs ferroptosis via BRD7-P53-SLC25A28 axis.70 In addition, sorafenib was also reported to trigger HSC ferroptosis via hypoxia-inducible factor (HIF)-1α/SLC7A11 signaling, which attenuates liver injury and fibrosis in a mouse model of liver fibrosis induced by CCl4.71 An E3 ubiquitin ligament TRIM26 was decreased in fibrotic liver tissues, which was originally identified as a tumor suppressor in hepatocellular carcinoma. A recent study found that TRIM26 promotes HSCs ferroptosis to mitigate liver fibrosis through mediating the ubiquitination of SLC7A11, the subunit in xCT system for lipid ROS scavenging.72 By using RNA sequencing analysis, Shen et al73 found that N6-methyladenosine (m6A) modification also enhances HSC ferroptosis, possibly by activating autophagy via stabilizing BECN1 mRNA. Berberine (BBR) has been explored as a potential anti-liver fibrosis agent, Yi et al74 reported that BBR attenuated liver fibrosis by promoting Fe2+ redox to activate ROS-mediated HSC ferroptosis. On the one hand, BBR suppressed the autophagy–lysosome pathway and increased cell ROS, which further accelerated the breakdown of the iron-storage protein ferritin and subsequent iron release from ferritin in HSCs; on the other hand, impaired autophagy enhanced BBR-mediated ferritin proteolysis to increase cellular Fe2+ overload via the ubiquitin–proteasome pathway in HSCs. Other studies discovered that exosomal miR-222 derived from hepatitis B virus-infected hepatocytes promote HSCs activation through inhibiting TFRC-induced ferroptosis.75 Chrysophanol, a natural plant composition, alleviates hepatitis B virus X protein-induced HSC activation and liver fibrosis by regulating ferroptosis.76 Collectively, these findings will provide a new perspective to understand the mechanism of ferroptosis and find effective treatment in liver fibrosis.
Ferroptosis and Renal Fibrosis
Renal fibrosis refers to excessive deposition of the fibrotic matrix within the parenchyma during chronic kidney injury, which represents the common final pathway of nearly all chronic and progressive nephropathies.77 Ferroptosis has been implicated in the development of renal fibrosis in distinct experimental animal models such as diabetic nephropathy,78 high fat diet-induced renal injury,60 folic acid-induced kidney injury,79 unilateral ureter obstruction (UUO),11 ischemia/reperfusion injury (IRI),80 and 5/6 nephrectomy-induced chronic kidney disease (CKD).81 Renal tubular epithelial cells (TECs) are one of the most vulnerable cell types to ferroptotic stress.11 A recent study reported that ureteral obstruction leads to ferroptosis in renal TECs, which are essential for UUO-induced renal fibrosis. Specifically, ferroptotic cell death in TECs triggers the secretion of profibrotic mediators such as TGF-β, CTGF, and PDGF, which subsequently regulate the proliferation and differentiation of interstitial fibroblasts in a paracrine fashion. The ferroptosis inhibitor liproxstatin-1(Lip-1) alleviated ferroptosis-induced renal fibrosis by restraining ferroptosis-mediated TECs death and lessening the activation of surrounding fibroblasts via inhibiting the paracrine activity of profibrotic factors of epithelial cells.11 Another study of a mouse model with UUO or IRI demonstrated that TECs ferroptosis facilitates monocyte chemotactic protein 1 secretion and macrophage chemotaxis, thus promoting interstitial fibrosis, which can be largely mitigated by ferroptosis inhibitor Fer-1 or DFO.82 In addition to triggering cell death of TECs, ferroptotic stress may also enhance the damage-associated state of proximal tubular cells, which leads to persistent inflammation and fibrosis during failed renal repair after severe injury and the acute kidney injury-to-CKD transition.80 Consequently, experimental reagents and traditional Chinese medicine as well as its effective components targeting ferroptosis show strong therapeutic potential for renal fibrosis in preclinical studies. In a 5/6 nephrectomy-induced CKD rat model, renal interstitial fibrosis is exacerbated by ferroptosis inducer cisplatin, while ferroptosis inhibitor showed antifibrotic effects by regulating the TGF-β1/Smad3 signaling pathway.81 Tocilizumab, a humanized monoclonal antibody targeting interleukin-6 (IL-6) signaling, ameliorates the renal fibrosis and reduces ferroptosis simultaneously in the UUO model.83 However, in this study the internal relationship of ferroptosis and fibrosis requires further exploration. Tectorigenin, a small molecule derived from the medicinal herbal Belamcanda chinensis, has proved to possess protective activities against ferroptosis and fibrosis both in vivo and in vitro, which is mediated by Smad3-induced expression of NADPH oxidase 4 (NOX4).84 Nobiletin, a flavonoid in the peel of citrus fruits, ameliorates ferroptosis-associated injury and renal fibrosis in the kidney of UUO mice, suggesting that Nob treatment has anti-ferroptosis and antifibrotic effects.85 Pretreatment of FG-4592, an inhibitor of prolyl hydroxylase of HIF, plays a protective role in the early stage of folic acid-induced kidney injury and retards the progression of renal fibrosis, mainly by alleviating ferroptosis via stabilizing HIF-1α and Akt/GSK-3β-mediated Nrf2 activation.79 Interestingly, the HIF-1α/HO-1 pathway was also reported to take part in aggravation of tubular injury and fibrosis by ferroptosis in the kidneys of diabetic mouse models.78 However, the exact mechanisms of these reagents and drugs in induction of ferroptosis and treatment of fibrosis are not fully elucidated. Therefore, in-depth exploration with a genetic model are demanding areas for future research to advance the field.
Ferroptosis and Myocardial Fibrosis
Myocardial fibrosis includes reactive fibrosis and replacement fibrosis. While the former occurs in perivascular spaces and corresponds to similar fibrogenic responses in other tissues, the latter occurs at the site of myocyte loss.86 Myofibroblasts in the injured heart are largely derived from resident fibroblasts, which are the most abundant cells in the myocardium. In response to injury, cardiac fibroblasts proliferate and differentiate into myofibroblasts under the mediation of classic factors such as TGF-β, endothelin-1, and angiotensin-1.87 Myocardial infarction (induced by permanent occlusion of the left anterior descending coronary artery) and pressure overload-induced cardiac hypertrophy (triggered by transverse aortic constriction) are two common experimental models for cardiac fibrosis. Cardiac fibrosis has deleterious effects on both myocardial contractility and myocardial electrophysiology, resulting in a growing incidence of arrhythmia and sudden cardiac death. Cardiac fibrosis is thought to play crucial roles in cardiac remodeling, which is closely associated with heart failure.88 Moreover, ferroptosis is enhanced in heart failure. Myocardium in murine model of heart failure showed apparent fibrosis as well as accumulation of iron and malondialdehyde,89 and there is evidence that cell death mediated by ferroptosis combined with activated autophagy promotes heart failure progression through the TLR4-NOX4 pathway.90 During the advanced stages of chronic heart failure, ferroptosis induced by Mixed lineage kinase 3 (MLK3) via JNK/p53 signaling pathway in cardiomyocytes mediates myocardial fibrosis. MiR-351 negatively regulated the expression of MLK3, thereby exerting a protective effect on ventricular remodeling in heart failure caused by pressure overload.12 In another study of myocardial fibrosis in response to pressure overload, elabela and Fer-1 alleviate Ang II-mediated ferroptosis of cardiac microvascular endothelial cells as well as subsequent myocardial fibrosis through modulating the IL-6/STAT3/GPX4 signaling pathway.91 SLC7A11 gene-encoded plasma membrane xCT is identified as a cardioprotective factor in cardiac hypertrophic diseases, since xCT alleviates angiotensin II–induced cardiac fibrosis and pathological cardiac remodeling by inhibiting ferroptosis.92
Many studies have shown that ferroptosis occurs in myocardial infarction, myocardial IRI, and heart failure, all of which can lead to the formation of fibrosis.87 GPX4 is significantly downregulated at the transcriptional level in the early and middle stages of myocardial infarction, which results in the accumulation of lipid peroxide and subsequently ferroptosis in cardiomyocytes.93 Similarly, the Nrf2/Hmox1 pathway involves in the iron excess which leads to ferroptotic cell death during early and middle stages of MI.94 Lip-1 protects myocardium against IRI by inhibiting ferroptosis through increasing GPX4 and decreasing ROS.95 Fer-1, another ferroptosis inhibitor, alleviates myocardial damage during hypoxia/reoxygenation by mitigating ER stress in diabetes myocardial IRI.96 Rapamycin reduces IRI by suppressing ferroptosis via regulating iron transportation and controlling iron metabolism in cardiomyocytes.97 Accordingly, drugs targeting ferroptosis showed antifibrosis effects in animal experiments of myocardial ischemia-reperfusion injury. Etomidate, a short acting anesthetic, alleviated IRI-induced myocardial fibrosis by inhibiting ferroptosis through Nrf2 pathway, which can be eliminated by erastin.98 Another anesthetic, dexmedetomidine, has also been shown to possess a protective role in IRI-induced myocardial injury and fibrosis by inhibiting ferroptosis via enhancing the expression of SLC7A11 and GPX4.99 A study of IRI also reported that intravenous injection of histochrome significantly mitigates cardiac fibrosis. Mechanistically, histochrome treatment downregulates intracellular and mitochondrial ROS levels by activating the Nrf2 pathway. In vitro experiment indicated that histochrome also inhibited erastin- and RSL3-induced ferroptosis in rat neonatal cardiomyocytes by maintaining the intracellular glutathione level and through upregulated activity of GPX4.100 In summary, ferroptosis participants in the pathogenesis of multiple fibrosis-related cardiovascular diseases, targeting ferroptosis may be an effective therapeutic strategy for these diseases, thereby reducing the formation of myocardial fibrosis.
Interstitial fibrosis also occurs in doxorubicin (DOX)-induced cardiomyopathy (DIC) and adriamycin (ADR)-induced cardiomyopathy. Ferroptosis has been proven to contribute to play a key role in progression of DIC and it has been reported that ferroptosis is the main form of RCD in DOX cardiotoxicity. Specifically, DOX induces mitochondria-dependent ferroptosis by decreasing GPX4 and promoting excessive LPO through DOX-Fe2+ complex in mitochondria.101 The selective inhibitor of the sodium glucose co-transporter 2 empagliflozin reduced ferroptosis and fibrosis in doxorubicin-treated mice through the involvement of NLRP3 and MyD88-related pathways.102 Fisetin, a natural flavonoid with cardioprotective effects, markedly ameliorates myocardial fibrosis and ferroptosis of cardiomyocytes in the DOX-induced cardiomyopathy rat and H9c2 cell models. Further studies showed that fisetin protects against DOX-induced cardiomyopathy by inhibiting ferroptosis via SIRT1/Nrf2 signaling pathway activation.103 Salidroside, an extraction from traditional Chinese medicine Rhodiola rosea, also significantly attenuated ferroptosis and fibrosis in a mice model of DIC. However, the molecular mechanism is different; the antiferroptotic effects of salidroside are mediated by activating AMPK-dependent signaling pathways including regulating abnormal fatty acid metabolism and maintaining mitochondrial function.104 In addition, Astragaloside IV, an active ingredient isolated from traditional Chinese medicine Astragalus membranaceus, exerts protective effects against adriamycin-induced myocardial fibrosis, which may partly attribute to its anti-ferroptotic effect via Nrf2 signaling.105 Overall, targeting ferroptosis may represent a novel treatment that improves cardiac fibrosis from drug-induced cardiomyopathy.
Ferroptosis and Pulmonary Fibrosis
The etiology of lung fibrosis is diverse. In addition to infections, toxics, and radiation, scleroderma and sarcoidosis can also give rise to lung fibrosis.106 Intratracheal administration of bleomycin (BLM) is the most frequently used experimental animal model of pulmonary fibrosis. Histopathologically, pulmonary fibrosis is characterized by destruction of the alveolar structure, lung fibroblast proliferation, and ECM deposition.107 Pulmonary fibrosis reduces lung compliance, disturbs gas exchange, and restricts lung function.108 As the disease continues to advance, pulmonary hypertension, right-sided heart failure, respiratory failure, and even death occur.108
Idiopathic pulmonary fibrosis (IPF), a rapidly progressive and usually fatal lung disease without clear cause and effective therapy, is the most pernicious of pulmonary fibrosis.109−111 It is now well-established that ferroptosis plays a key role in the pathogenesis of pulmonary fibrosis. Iron accumulation substantially increased both in lung tissue sections from patients with IPF and in experimental bleomycin-induced pulmonary fibrosis, which corresponded with the pulmonary fibrosis and a decline in lung function.13,112 Bioinformatics analysis also supported that ferroptosis-related genes are significantly correlated with the development of IPF.113–115 Moreover, some ferroptosis-related genes in the bronchoalveolar lavage showed prognostic value in patients with IPF.113,115 Current studies mainly focus on the role of ferroptosis in the alveolar epithelial injury, fibroblast-to-myofibroblast transition, and epithelial-mesenchymal transition (EMT), all of which are important biological processes in the development of IPF. It is now established that dysfunction of alveolar epithelial cells, especially alveolar type 2 (AT II) cells, drive the pathogenesis of IPF.13 Correspondingly, pulmonary iron overload leads to ferroptosis of AT II cells and pulmonary fibrosis, and ferroptosis inhibitor DFO completely reverses the pro-fibrosis phenotype and mitigates mitochondrial damage induced by BLM via decreasing iron deposition and ferroptosis in AT II cells.13 In another study, exogenous administration of a water-soluble iron salt, Fe3+ ammonium citrate in human lung fibroblasts can increase cellular proliferation, enhance the expression of ECM-related genes, resulting in the development of IPF.116 DFO has also been reported to inhibit TGF-β-induced EMT, thus ameliorating BLM-induced IPF.112 In line with this finding, ferroptosis inducer erastin promotes EMT in a mouse lung epithelial cell line, which is accompanied by reactive oxygen species (ROS) production and upregulated expression of heme oxygenase-1.117 Histone methyltransferase SET domain bifurcated 1(SETDB1) is decreased in TGF-β-induced EMT, which plays an antifibrotic role in pulmonary fibrosis. Knockdown of SETDB1 enhances TGF-β-induced EMT and ferroptosis in human alveolar epithelial cells, as decreased SETDB1 reduces the histone H3 lysine 9 trimethylation of Snai1 and enhances the occurrence of TGF-β-induced EMT, which further increased lipid ROS and Fe2+ ions.118 Therefore, ferroptosis occurs during EMT induced by TGF-β; however, the mechanism underlying the relationship between ferroptosis and EMT warrants further study.119 Other studies investigating the relationship of ferroptosis and fibroblast-to-myofibroblast transition in fibrosis reported that Erastin promoted fibroblast-to-myofibroblast differentiation via increasing LPO and inhibiting the expression of GPX4, which can be blocked by Fer-1.120 Upregulation of lncRNA zinc finger antisense 1 (lncRNA ZFAS1) was observed in BLM-induced PF rat lung tissue and in TGF-β1-induced human fetal lung fibroblast cells. As a competing endogenous RNA, ZFAS1 was demonstrated to promote fibroblast activation and LPO by sponging miR-150-5p and downregulating SLC38A1 expression, suggesting the role of lncRNA ZFAS1/miR-150- 5p/SLC38A1 axis in the progression of PF.121 Apart from epithelial cells and fibroblast, ferroptosis in macrophages also promotes the development of IPF. Liu et al122 demonstrated that silicosis induces ferroptosis in macrophages, which leads to the secretion of pro-fibrotic cytokines and fibrosis. In summary, these preliminary explorations provide evidences of iron deposition and ferroptosis in alveolar macrophages, AT II cells and fibroblasts contribute to the development and progression of IPF.
Radiation-induced lung fibrosis (RILF) is a category of radiation-induced lung injury (RILI), which is a common and severe complication of thoracic radiotherapy.123 It is now believed that both direct radiation damage to DNA and radiation-enhanced production of ROS cause cellular damage in RILF.124–126 In addition, ROS caused by irradiation is closely related to inflammation and fibrosis.125,126 Apart from apoptosis, several lines of evidence suggested that ferroptosis might play a critical role in the process of RILF. Firstly, the expression level of GPX4 is obviously decreased in RILF mice.127 Secondly, ROS levels are significantly upregulated in acute RILI.128 Thirdly, ferroptosis inhibitor Lip-1 inhibited the collagen deposition and reduced hydroxyproline in the RILF model.127 Fourthly, the pathological changes in RILI are substantially alleviated by ferroptosis inhibitors.128 Finally, studies of Li et al127 demonstrated that ferroptosis inhibitor Lip-1 mitigated RILF through downregulation of TGF-β1 via the activating Nrf2 pathway.
Paraquat (PQ), an efficacious and widely-used herbicide, is considered as one of the most frequently-used and incurable poisoning substances for suicide.129 Exposure to PQ causes severe damage in the lungs and eventually leads to pulmonary fibrosis.129 Generally, the toxicity of PQ is the result of formation of high energy oxygen free radicals and peroxidation of unsaturated lipids in the cell.129 Ferroptosis inhibitors with antioxidant properties appear to be one of the main mechanisms in PQ detoxification.130 For example, iron chelators DFO prevent PQ toxicity via inhibition of hydroxyl radical production and inhibition of PQ uptake by AT II cells.129,131 While vitamin E serves as a lipophilic antioxidant to inhibit LPO in ferroptosis, vitamin E deficiency increases acute PQ toxicity and deterioration of histological lung damage in animals, with a decreased survival rate.129 Edaravone protects the lung against PQ toxicity by alleviating oxidative stress and inflammatory responses.132 In the same year, it was reported that edaravone suppresses ferroptosis by scavenging free radicals and lessening oxidative stress.133 Ebselen is an inhibitor of apoptosis and ferroptosis which also has anti-inflammatory and cytoprotective effects.133 As ebselen efficiently scavenges ROS and RNS, it also exhibits antioxidant activity.134,135 More than 30 years ago, it was found that ebselen combined with N-acetylcysteine can be used to treat PQ poisoning. Mechanistically, ebselen might confer resistance to ferroptosis, possibly by inhibiting the activity of glutaminase and NADPH-oxidase and suppressing inflammasome mediated inflammatory pathways.91 Thiazolidinedione alleviates the PQ toxicity by inhibiting ferroptosis via decreasing ACSL4 activity and increasing ROS scavenging enzymatic activity.136 Together these findings raise the possibility of targeting ferroptosis for the treatment of PQ poisoning.
Concluding Remarks
Novel therapeutic strategies are in urgent demand for fibrotic diseases. A growing number of studies have provided insights into the mechanisms and factors associated with the regulation of fibrosis by ferroptosis. New techniques like single-cell multi-omics and genetic approaches makes it possible to explore mechanisms of ferroptosis and fibrosis at the single-cell level with unexampled resolution.80 In this context, the development of antibodies and drugs that modulate ferroptosis via both its induction and its inhibition holds great potential for the treatment of patients with a broad spectrum of fibrotic diseases, as the ultimate goal of understanding the role of ferroptosis in fibrosis is to leverage this relationship for the therapeutic benefit of patients suffering from fibrotic diseases. Research in this field is still in its infancy, and much work is needed to elucidate the detailed molecular mechanisms and regulatory networks through which ferroptosis participates in fibrotic diseases. Besides, a translational gap remains where we can convert the putative targets into effective clinical drugs for ferroptosis-based disease therapy.137 Targeting ferroptosis may unleash unprecedented opportunities for pharmacological intervention. Future work is expected to provide novel therapeutic strategies for preventing, controlling, and treating fibrosis.
Abbreviations
ECM, extracellular matrix; RCD, regulated cell death; GPX4, glutathione peroxidase 4; LPO, lipid peroxidation; PLOOHs, phospholipid hydroperoxides; LOXs, lipoxygenases; POR, cytochrome P450 oxidoreductase; ROS, reactive oxygen species; LIP, labile iron pool; Fe3+, ferric; Trf, transferrin; TFRC, transferrin receptor; Fe2+, ferrous; FHC, ferritin heavy chain; PUFA-PLs, phospholipids containing polyunsaturated fatty acid chains; PUFAs, polyunsaturated fatty acids; ACSL4, Acyl-CoA synthetase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; GSH, glutathione; FSP1, ferroptosis suppressor protein 1; CoQ10, coenzyme Q10, ubiquinol; DHODH, dihydroorotate dehydrogenase; GCH1, GTP cyclohydrolase 1; BH4, tetrahydrobiopterin; DHFR, dihydrofolate reductase; SLC7A11, solute carrier family-7 member-11; TGF-β, transforming growth factor-β; EMT, epithelial-mesenchymal transition; HSCs, hepatic stellate cells; Fer-1, ferrostatin-1; CCI4, tetrachloride; HO-1, heme oxygenase-1; FGF21, Fibroblast growth factor 21; Nrf2, nuclear factor erythroid 2-related factor 2; DFO, deferoxamine; ART, Artemether; ELAVL1, ELAV like RNA binding protein 1; ZFP36, ZFP36 ring finger protein; BRD7, bromodomain-containing protein 7; TRIM26, tripartite motif-containing protein 26; AREs, AU-rich elements; 3ʹ-UTR, 3rd-untranslated region; ATG16L1, autophagy related 16 like 1; FBXW7/CDC4, F-box and WD repeat domain containing 7; SLC25A28, solute carrier family 25 member 28; HIF, hypoxia-inducible factor; BBR, Berberine; UUO, unilateral ureteral obstruction; IRI, ischemia/reperfusion injury; CKD, chronic kidney disease; TECs, tubular epithelial cells; Lip-1, liproxstatin-1; IL-6, interleukin-6; NOX4, NADPH oxidase 4; MLK3, Mixed lineage kinase 3; DOX, doxorubicin; DIC, doxorubicin-induced cardiomyopathy; ADR, adriamycin; BLM, bleomycin; AT II, alveolar type 2; ZFAS1, zinc finger antisense 1; RILF, radiation-induced lung fibrosis; RILI, radiation-induced lung injury; PQ, paraquat.
Funding
This work was funded by the National Natural Science Foundation of China (Grant Nos. 81900064 and 82170079).
Disclosure
The authors have declared conflicts of interest in relation to this work.
References
1. Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol. 2019;15(12):705–730. doi:10.1038/s41584-019-0322-7
2. Henderson NC, Rieder F, Wynn TA. Fibrosis: from mechanisms to medicines. Nature. 2020;587(7835):555–566. doi:10.1038/s41586-020-2938-9
3. Jun JI, Lau LF. Resolution of organ fibrosis. J Clin Invest. 2018;128(1):97–107. doi:10.1172/JCI93563
4. Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov. 2020;19(1):57–75. doi:10.1038/s41573-019-0040-5
5. Weiskirchen R, Weiskirchen S, Tacke F. Organ and tissue fibrosis: molecular signals, cellular mechanisms and translational implications. Mol Aspects Med. 2019;65:2–15. doi:10.1016/j.mam.2018.06.003
6. Rockey DC, Bell PD, Hill JA. Fibrosis–a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138–1149. doi:10.1056/NEJMra1300575
7. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
8. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
9. Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8:586578. doi:10.3389/fcell.2020.586578
10. Yu Y, Jiang L, Wang H, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739. doi:10.1182/blood.2019002907
11. Zhang B, Chen X, Ru F, et al. Liproxstatin-1 attenuates unilateral ureteral obstruction-induced renal fibrosis by inhibiting renal tubular epithelial cells ferroptosis. Cell Death Dis. 2021;12(9):843. doi:10.1038/s41419-021-04137-1
12. Wang J, Deng B, Liu Q, et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 2020;11(7):574. doi:10.1038/s41419-020-02777-3
13. Cheng H, Feng D, Li X, et al. Iron deposition-induced ferroptosis in alveolar type II cells promotes the development of pulmonary fibrosis. Biochim Biophys Acta Mol Basis Dis. 2021;1867(12):166204. doi:10.1016/j.bbadis.2021.166204
14. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
15. Zhang DL, Ghosh MC, Rouault TA. The physiological functions of iron regulatory proteins in iron homeostasis - an update. Front Pharmacol. 2014;5:124. doi:10.3389/fphar.2014.00124
16. Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226. doi:10.3389/fcell.2020.590226
17. Wang Y, Liu Y, Liu J, Kang R, Tang D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. 2020;531(4):581–587. doi:10.1016/j.bbrc.2020.07.032
18. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–245. doi:10.1016/j.chembiol.2008.02.010
19. Geng N, Shi BJ, Li SL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. 2018;22(12):3826–3836. doi:10.26355/eurrev_201806_15267
20. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–4975. doi:10.1073/pnas.1603244113
21. Dixon SJ, Stockwell BR. The hallmarks of ferroptosis. Ann Rev Cancer Biol. 2019;3(1):35–54. doi:10.1146/annurev-cancerbio-030518-055844
22. Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. 2018;4(3):387–396. doi:10.1021/acscentsci.7b00589
23. Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302–309. doi:10.1038/s41589-020-0472-6
24. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15(12):1137–1147. doi:10.1038/s41589-019-0408-1
25. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi:10.1038/nchembio.2238
26. Dixon SJ, Winter GE, Musavi LS, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10(7):1604–1609. doi:10.1021/acschembio.5b00245
27. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi:10.1038/nchembio.2239
28. Zou Y, Henry WS, Ricq EL, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585(7826):603–608. doi:10.1038/s41586-020-2732-8
29. Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2018;508:997–1003.
30. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5(1):108. doi:10.1038/s41392-020-00216-5
31. Lee JY, Kim WK, Bae KH, Lee SC, Lee EW. Lipid metabolism and ferroptosis. Biology. 2021;10(3):184.
32. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;4:1–6.
33. Seiler A, Schneider M, F?Rster H, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8(3):237–248. doi:10.1016/j.cmet.2008.07.005
34. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
35. Meister A. Glutathione metabolism. Methods Enzymol. 1995;251:3–7.
36. Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018;38(1):12. doi:10.1186/s40880-018-0288-x
37. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23(2):270–278. doi:10.1038/cdd.2015.93
38. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi:10.1038/s41586-019-1705-2
39. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi:10.1038/s41586-019-1707-0
40. Horikoshi N, Cong J, Kley N, Shenk T. Isolation of differentially expressed cDNAs from p53-dependent apoptotic cells: activation of the human homologue of the Drosophila peroxidasin gene. Biochem Biophys Res Commun. 1999;261(3):864–869. doi:10.1006/bbrc.1999.1123
41. Chorley BN, Campbell MR, Wang X, et al. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40(15):7416–7429. doi:10.1093/nar/gks409
42. Nguyen HP, Yi D, Lin F, et al. Aifm2, a NADH oxidase, supports robust glycolysis and is required for cold- and diet-induced thermogenesis. Mol Cell. 2020;77(3):600–617.e604. doi:10.1016/j.molcel.2019.12.002
43. Venkatesh D, O’Brien NA, Zandkarimi F, et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev. 2020;34(7–8):526–543. doi:10.1101/gad.334219.119
44. Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590. doi:10.1038/s41586-021-03539-7
45. Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347(Pt 1):1–16. doi:10.1042/bj3470001
46. Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41–53. doi:10.1021/acscentsci.9b01063
47. Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522(2):415–421. doi:10.1016/j.bbrc.2019.11.110
48. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32(6):920–937. doi:10.1016/j.cmet.2020.10.011
49. Zhang KH, Tian HY, Gao X, et al. Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Res. 2009;69(13):5340–5348. doi:10.1158/0008-5472.CAN-09-0112
50. Gorowiec MR, Borthwick LA, Parker SM, Kirby JA, Saretzki GC, Fisher AJ. Free radical generation induces epithelial-to-mesenchymal transition in lung epithelium via a TGF-beta1-dependent mechanism. Free Radic Biol Med. 2012;52(6):1024–1032. doi:10.1016/j.freeradbiomed.2011.12.020
51. Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 2015;6:565–577. doi:10.1016/j.redox.2015.09.009
52. Felton VM, Borok Z, Willis BC. N-acetylcysteine inhibits alveolar epithelial-mesenchymal transition. Am J Physiol Lung Cell Mol Physiol. 2009;297(5):L805–812. doi:10.1152/ajplung.00009.2009
53. Liu RM, Vayalil PK, Ballinger C, et al. Transforming growth factor beta suppresses glutamate-cysteine ligase gene expression and induces oxidative stress in a lung fibrosis model. Free Radic Biol Med. 2012;53(3):554–563. doi:10.1016/j.freeradbiomed.2012.05.016
54. Beeh KM, Beier J, Haas IC, Kornmann O, Micke P, Buhl R. Glutathione deficiency of the lower respiratory tract in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2002;19(6):1119–1123. doi:10.1183/09031936.02.00262402
55. Muramatsu Y, Sugino K, Ishida F, Tatebe J, Morita T, Homma S. Effect of inhaled N-acetylcysteine monotherapy on lung function and redox balance in idiopathic pulmonary fibrosis. Respir Investig. 2016;54(3):170–178. doi:10.1016/j.resinv.2015.11.004
56. Deger Y, Yur F, Ertekin A, Mert N, Dede S, Mert H. Protective effect of α-tocopherol on oxidative stress in experimental pulmonary fibrosis in rats. Cell Biochem Funct. 2007;25(6):633–637. doi:10.1002/cbf.1362
57. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2020;18(Suppl. 1):151–166.
58. Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Delivery Rev. 2019;121:27–42.
59. Du K, Oh SH, Dutta RK, et al. Inhibiting xCT/SLC7A11 induces ferroptosis of myofibroblastic hepatic stellate cells but exacerbates chronic liver injury. Liver Int. 2021;41(9):2214–2227. doi:10.1111/liv.14945
60. Luo Y, Chen H, Liu H, et al. Protective effects of ferroptosis inhibition on high fat diet-induced liver and renal injury in mice. Int J Clin Exp Pathol. 2020;13(8):2041–2049.
61. Wu A, Feng B, Yu J, et al. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021;46:102131. doi:10.1016/j.redox.2021.102131
62. Sui M, Jiang X, Chen J, Yang H, Zhu Y. Magnesium isoglycyrrhizinate ameliorates liver fibrosis and hepatic stellate cell activation by regulating ferroptosis signaling pathway. Biomed Pharmacother. 2018;106:125–133. doi:10.1016/j.biopha.2018.06.060
63. Kong Z, Liu R, Cheng Y. Artesunate alleviates liver fibrosis by regulating ferroptosis signaling pathway. Biomed Pharmacother. 2019;109:2043–2053. doi:10.1016/j.biopha.2018.11.030
64. Wang L, Zhang Z, Li M, et al. P53-dependent induction of ferroptosis is required for artemether to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. Iubmb Life. 2019;71:45–56.
65. Li Y, Jin C, Shen M, et al. Iron regulatory protein 2 is required for artemether -mediated anti-hepatic fibrosis through ferroptosis pathway. Free Radic Biol Med. 2020;160:845–859. doi:10.1016/j.freeradbiomed.2020.09.008
66. Zhang Z, Wang X, Wang Z, et al. Dihydroartemisinin alleviates hepatic fibrosis through inducing ferroptosis in hepatic stellate cells. Biofactors. 2021;47(5):801–818. doi:10.1002/biof.1764
67. Ho CH, Huang JH, Sun MS, Tzeng IS, Hsu YC, Kuo CY. Wild bitter melon extract regulates LPS-induced hepatic stellate cell activation, inflammation, endoplasmic reticulum stress, and ferroptosis. Evid Based Complement Alternat Med. 2021;2021:6671129. doi:10.1155/2021/6671129
68. Zhang Z, Yao Z, Wang L, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14(12):2083–2103. doi:10.1080/15548627.2018.1503146
69. Zhang Z, Guo M, Li Y, et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy. 2020;16(8):1482–1505. doi:10.1080/15548627.2019.1687985
70. Zhang Z, Guo M, Shen M, et al. The BRD7-P53-SLC25A28 axis regulates ferroptosis in hepatic stellate cells. Redox Biol. 2020;36:101619. doi:10.1016/j.redox.2020.101619
71. Yuan S, Wei C, Liu G, et al. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1alpha/SLC7A11 pathway. Cell Prolif. 2022;55(1):e13158. doi:10.1111/cpr.13158
72. Zhu Y, Zhang C, Huang M, Lin J, Fan X, Ni T. TRIM26 induces ferroptosis to inhibit hepatic stellate cell activation and mitigate liver fibrosis through mediating SLC7A11 ubiquitination. Front Cell Dev Biol. 2021;9:644901. doi:10.3389/fcell.2021.644901
73. Shen M, Li Y, Wang Y, et al. N(6)-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 2021;47:102151. doi:10.1016/j.redox.2021.102151
74. Yi J, Wu S, Tan S, et al. Berberine alleviates liver fibrosis through inducing ferrous redox to activate ROS-mediated hepatic stellate cells ferroptosis. Cell Death Discov. 2021;7(1):374. doi:10.1038/s41420-021-00768-7
75. Zhang Q, Qu Y, Zhang Q, et al. Exosomes derived from hepatitis B virus-infected hepatocytes promote liver fibrosis via miR-222/TFRC axis. Cell Biol Toxicol. 2022. doi:10.1007/s10565-021-09684-z
76. Kuo CY, Chiu V, Hsieh PC, et al. Chrysophanol attenuates hepatitis B virus X protein-induced hepatic stellate cell fibrosis by regulating endoplasmic reticulum stress and ferroptosis. J Pharmacol Sci. 2020;144(3):172–182. doi:10.1016/j.jphs.2020.07.014
77. Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol. 2018;80:309–326. doi:10.1146/annurev-physiol-022516-034227
78. Feng X, Wang S, Sun Z, et al. Ferroptosis enhanced diabetic renal tubular injury via HIF-1alpha/HO-1 pathway in db/db mice. Front Endocrinol (Lausanne). 2021;12:626390. doi:10.3389/fendo.2021.626390
79. Li X, Zou Y, Xing J, et al. Pretreatment with roxadustat (FG-4592) attenuates folic acid-induced kidney injury through antiferroptosis via Akt/GSK-3β/Nrf2 pathway. Oxid Med Cell Longev. 2020;2020:6286984. doi:10.1155/2020/6286984
80. Ide S, Kobayashi Y, Ide K, et al. Ferroptotic stress promotes the accumulation of pro-inflammatory proximal tubular cells in maladaptive renal repair. Elife. 2021;10. doi:10.7554/eLife.68603
81. Wang J, Wang Y, Liu Y, et al. Ferroptosis, a new target for treatment of renal injury and fibrosis in a 5/6 nephrectomy-induced CKD rat model. Cell Death Discov. 2022;8(1):127. doi:10.1038/s41420-022-00931-8
82. Zhou L, Xue X, Hou Q, Dai C. Targeting ferroptosis attenuates interstitial inflammation and kidney fibrosis. Kidney Dis. 2022;8(1):57–71. doi:10.1159/000517723
83. Yang L, Guo J, Yu N, et al. Tocilizumab mimotope alleviates kidney injury and fibrosis by inhibiting IL-6 signaling and ferroptosis in UUO model. Life Sci. 2020;261:118487. doi:10.1016/j.lfs.2020.118487
84. Li J, Yang J, Zhu B, Fan J, Hu Q, Wang L. Tectorigenin protects against unilateral ureteral obstruction by inhibiting Smad3-mediated ferroptosis and fibrosis. Phytother Res. 2022;36(1):475–487. doi:10.1002/ptr.7353
85. Lo YH, Yang SF, Cheng CC, et al. Nobiletin alleviates ferroptosis-associated renal injury, inflammation, and fibrosis in a unilateral ureteral obstruction mouse model. Biomedicines. 2022;10(3):595. doi:10.3390/biomedicines10030595
86. Gyöngyösi M, Winkler J, Ramos I, et al. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. 2017;19(2):177–191. doi:10.1002/ejhf.696
87. Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. 2019;65:70–99. doi:10.1016/j.mam.2018.07.001
88. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2021;117(6):1450–1488. doi:10.1093/cvr/cvaa324
89. Zheng H, Shi L, Tong C, Liu Y, Hou M. circSnx12 is involved in ferroptosis during heart failure by targeting miR-224-5p. Front Cardiovasc Med. 2021;8:656093. doi:10.3389/fcvm.2021.656093
90. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. 2019;516(1):37–43. doi:10.1016/j.bbrc.2019.06.015
91. Zhang Z, Tang J, Song J, et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic Biol Med. 2022;181:130–142. doi:10.1016/j.freeradbiomed.2022.01.020
92. Zhang X, Zheng C, Gao Z, et al. SLC7A11/xCT prevents cardiac hypertrophy by inhibiting ferroptosis. Cardiovasc Drugs Ther. 2021;36:437–447.
93. Park TJ, Park JH, Lee GS, et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019;10(11):835. doi:10.1038/s41419-019-2061-8
94. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107. doi:10.1016/j.redox.2019.101107
95. Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. 2019;520(3):606–611. doi:10.1016/j.bbrc.2019.10.006
96. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial Ischemia/Reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020;39(2):210–225. doi:10.1089/dna.2019.5097
97. Baba Y, Higa JK, Shimada BK, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314(3):H659–h668. doi:10.1152/ajpheart.00452.2017
98. Lv Z, Wang F, Zhang X, Zhang X, Zhang J, Liu R. Etomidate attenuates the ferroptosis in myocardial ischemia/reperfusion rat model via Nrf2/HO-1 pathway. Shock. 2021;56(3):440–449. doi:10.1097/SHK.0000000000001751
99. Yu P, Zhang J, Ding Y, et al. Dexmedetomidine post-conditioning alleviates myocardial ischemia-reperfusion injury in rats by ferroptosis inhibition via SLC7A11/GPX4 axis activation. Hum Cell. 2022;35(3):836–848. doi:10.1007/s13577-022-00682-9
100. Hwang JW, Park JH, Park BW, et al. Histochrome attenuates myocardial ischemia-reperfusion injury by inhibiting ferroptosis-induced cardiomyocyte death. Antioxidants. 2021;10(10):1624.
101. Tadokoro T, Ikeda M, Ide T, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5(9). doi:10.1172/jci.insight.132747
102. Quagliariello V, De Laurentiis M, Rea D, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. 2021;20(1):150. doi:10.1186/s12933-021-01346-y
103. Li D, Liu X, Pi W, et al. Fisetin attenuates doxorubicin-induced cardiomyopathy in vivo and in vitro by inhibiting ferroptosis through SIRT1/Nrf2 signaling pathway activation. Front Pharmacol. 2021;12:808480. doi:10.3389/fphar.2021.808480
104. Chen H, Zhu J, Le Y, et al. Salidroside inhibits doxorubicin-induced cardiomyopathy by modulating a ferroptosis-dependent pathway. Phytomedicine. 2022;99:153964. doi:10.1016/j.phymed.2022.153964
105. Luo LF, Guan P, Qin LY, Wang JX, Wang N, Ji ES. Astragaloside IV inhibits Adriamycin-induced cardiac ferroptosis by enhancing Nrf2 signaling. Mol Cell Biochem. 2021;476(7):2603–2611. doi:10.1007/s11010-021-04112-6
106. Wijsenbeek M, Cottin V, Drazen JM. Spectrum of fibrotic lung diseases. N Engl J Med. 2020;383(10):958–968. doi:10.1056/NEJMra2005230
107. Spagnolo P, Kropski JA, Jones MG, et al. Idiopathic pulmonary fibrosis: disease mechanisms and drug development. Pharmacol Ther. 2021;222:107798. doi:10.1016/j.pharmthera.2020.107798
108. Kropski JA, Blackwell TS. Progress in understanding and treating idiopathic pulmonary fibrosis. Annu Rev Med. 2019;70:211–224. doi:10.1146/annurev-med-041317-102715
109. Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389(10082):1941–1952. doi:10.1016/S0140-6736(17)30866-8
110. Lederer DJ, Martinez FJ, Longo DL. Idiopathic pulmonary fibrosis. N Engl J Med. 2018;378(19):1811–1823. doi:10.1056/NEJMra1705751
111. Martinez FJ, Collard HR, Pardo A, et al. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers. 2017;3:17074. doi:10.1038/nrdp.2017.74
112. Takahashi M, Mizumura K, Gon Y, et al. Iron-dependent mitochondrial dysfunction contributes to the pathogenesis of pulmonary fibrosis. Front Pharmacol. 2021;12:643980. doi:10.3389/fphar.2021.643980
113. Li M, Wang K, Zhang Y, et al. Ferroptosis-related genes in bronchoalveolar lavage fluid serves as prognostic biomarkers for idiopathic pulmonary fibrosis. Front Med. 2021;8:693959.
114. He J, Li X, Yu M. Bioinformatics analysis identifies potential ferroptosis key genes in the pathogenesis of pulmonary fibrosis. Front Genet. 2021;12:788417. doi:10.3389/fgene.2021.788417
115. He Y, Shang Y, Li Y, et al. An 8-ferroptosis-related genes signature from bronchoalveolar lavage fluid for prognosis in patients with idiopathic pulmonary fibrosis. BMC Pulm Med. 2022;22(1):15. doi:10.1186/s12890-021-01799-7
116. Ali MK, Kim RY, Brown AC, et al. Critical role for iron accumulation in the pathogenesis of fibrotic lung disease. J Pathol. 2020;251(1):49–62. doi:10.1002/path.5401
117. Han Y, Ye L, Du F, et al. Iron metabolism regulation of epithelial-mesenchymal transition in idiopathic pulmonary fibrosis. Ann Transl Med. 2021;9(24):1755. doi:10.21037/atm-21-5404
118. Liu T, Xu P, Ke S, et al. Histone methyltransferase SETDB1 inhibits TGF-β-induced epithelial-mesenchymal transition in pulmonary fibrosis by regulating SNAI1 expression and the ferroptosis signaling pathway. Arch Biochem Biophys. 2022;715:109087. doi:10.1016/j.abb.2021.109087
119. Sun L, Dong H, Zhang W, et al. Lipid peroxidation, GSH depletion, and SLC7A11 inhibition are common causes of EMT and ferroptosis in A549 cells, but different in specific mechanisms. DNA Cell Biol. 2021;40(2):172–183. doi:10.1089/dna.2020.5730
120. Gong Y, Wang N, Liu N, Dong H. Lipid peroxidation and GPX4 inhibition are common causes for myofibroblast differentiation and ferroptosis. DNA Cell Biol. 2019;38(7):725–733. doi:10.1089/dna.2018.4541
121. Yang Y, Tai W, Lu N, et al. lncRNA ZFAS1 promotes lung fibroblast-to-myofibroblast transition and ferroptosis via functioning as a ceRNA through miR-150-5p/SLC38A1 axis. Aging. 2020;12(10):9085–9102. doi:10.18632/aging.103176
122. Liu T, Bao R, Wang Q, et al. SiO(2)-induced ferroptosis in macrophages promotes the development of pulmonary fibrosis in silicosis models. Toxicol Res (Camb). 2022;11(1):42–51. doi:10.1093/toxres/tfab105
123. Hanania AN, Mainwaring W, Ghebre YT, Hanania NA, Ludwig M. Radiation-induced lung injury: assessment and management. Chest. 2019;156(1):150–162. doi:10.1016/j.chest.2019.03.033
124. Ogura A, Oowada S, Kon Y, et al. Redox regulation in radiation-induced cytochrome c release from mitochondria of human lung carcinoma A549 cells. Cancer Lett. 2009;277(1):64–71. doi:10.1016/j.canlet.2008.11.021
125. Lee JC, Krochak R, Blouin A, et al. Dietary flaxseed prevents radiation-induced oxidative lung damage, inflammation and fibrosis in a mouse model of thoracic radiation injury. Cancer Biol Ther. 2009;8(1):47–53. doi:10.4161/cbt.8.1.7092
126. Terasaki Y, Ohsawa I, Terasaki M, et al. Hydrogen therapy attenuates irradiation-induced lung damage by reducing oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2011;301(4):L415–426. doi:10.1152/ajplung.00008.2011
127. Li X, Duan L, Yuan S, Zhuang X, Qiao T, He J. Ferroptosis inhibitor alleviates Radiation-induced lung fibrosis (RILF) via down-regulation of TGF-beta1. J Inflamm. 2019;16:11. doi:10.1186/s12950-019-0216-0
128. Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun. 2019;519(2):240–245. doi:10.1016/j.bbrc.2019.08.165
129. Dinis-Oliveira RJ, Duarte JA, Sanchez-Navarro A, Remiao F, Bastos ML, Carvalho F. Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment. Crit Rev Toxicol. 2008;38(1):13–71. doi:10.1080/10408440701669959
130. Rashidipour N, Karami-Mohajeri S, Mandegary A, et al. Where ferroptosis inhibitors and paraquat detoxification mechanisms intersect, exploring possible treatment strategies. Toxicology. 2020;433–434:152407. doi:10.1016/j.tox.2020.152407
131. Van der Wal NA, Smith LL, van Oirschot JF, van Asbeck BS. Effect of iron chelators on paraquat toxicity in rats and alveolar type II cells. Am Rev Respir Dis. 1992;145(1):180–186. doi:10.1164/ajrccm/145.1.180
132. Yi R, Zhizhou Y, Zhaorui S, Wei Z, Xin C, Shinan N. Retrospective study of clinical features and prognosis of edaravone in the treatment of paraquat poisoning. Medicine. 2019;98(19):e15441. doi:10.1097/MD.0000000000015441
133. Homma T, Kobayashi S, Sato H, Fujii J. Edaravone, a free radical scavenger, protects against ferroptotic cell death in vitro. Exp Cell Res. 2019;384(1):111592. doi:10.1016/j.yexcr.2019.111592
134. Latunde-Dada GO. Ferroptosis: role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017;1861(8):1893–1900. doi:10.1016/j.bbagen.2017.05.019
135. Ren X, Zou L, Lu J, Holmgren A. Selenocysteine in mammalian thioredoxin reductase and application of ebselen as a therapeutic. Free Radic Biol Med. 2018;127:238–247. doi:10.1016/j.freeradbiomed.2018.05.081
136. Angeli JPF, Shah R, Pratt DA, Conrad M. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol Sci. 2017;38(5):489–498. doi:10.1016/j.tips.2017.02.005
137. Conrad M, Lorenz SM, Proneth B. Targeting ferroptosis: new hope for as-yet-incurable diseases. Trends Mol Med. 2021;27(2):113–122. doi:10.1016/j.molmed.2020.08.010
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.