")
Back to Journals » The Application of Clinical Genetics » Volume 9
Role of elosulfase alfa in mucopolysaccharidosis IVA
Authors Regier D, Tanpaiboon P
Received 28 July 2015
Accepted for publication 22 April 2016
Published 14 June 2016 Volume 2016:9 Pages 67—74
DOI https://doi.org/10.2147/TACG.S69080
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Debra S Regier, Pranoot Tanpaiboon
Division of Genetics and Metabolism, Children’s National Medical Center, Washington, DC, USA
Abstract: Mucopolysaccharidosis type IVA (MPS IVA or Morquio A) is an autosomal recessive lysosomal storage disease which results in a striking skeletal phenotype, but does not negatively impact the intellect of the patient. MPS IVA has a phenotypic continuum that ranges from a severe and rapidly progressing form to a slowly progressive form. The clinical diagnosis is often made in the preschool years based on abnormal bone findings on physical examination and dysplasia on radiographic imaging. Supportive care has been the mainstay in caring for patients. Orthopedic physicians often form the core of the care team due to the early and severe skeletal abnormalities; however, systemic disease is common and requires aggressive monitoring and management. Interdisciplinary care teams often consist of medical geneticists, cardiologists, pulmonary specialists, gastroenterologists, otolaryngologists, audiologists, and ophthalmologists. With the US Food and Drug Administration’s approval of elosulfase alfa, patients >5 years of age now have access to this medication from the time of diagnosis. The clinical trial with once weekly intravenous dosing (2.0 mg/kg per week) showed improvement in the 6-minute walk test. The composite end point analysis to evaluate the combining changes from baseline in 6-minute walk test, 3-minute stair climb test, and respiratory function showed that at a dose of 2.0 mg/kg per week, subjects performed better when compared to placebo. This indication was clinically meaningful in the treatment group. The treatment was generally well tolerated, and the uncommon infusion reactions responded well to traditional enzyme replacement therapy infusion reaction management algorithms. Currently, clinical trials are underway to determine the efficacy and safety in MPS IVA patients <5 years of age.
Keywords: mucopolysaccharidosis type IVA, elosulfase alfa, enzyme replacement therapy, Morquio syndrome, lysosomal storage disease, keratan sulfate
Introduction
Lysosomal storage disorders (LSDs) are a group of over 50 inherited disorders that affect the function of lysosomes. These organelles are responsible for metabolizing and recycling several macromolecule cellular components. Thus, dysfunction of these organelles leads to lysosome damage and often abnormalities of the lysosome anatomy. LSDs are classified based on the major accumulated substrate, underlying mechanism, or defective enzyme. Subgroups of each disorder are usually further subdivided based on their age of symptom onset (reviewed in Hendriksz et al1 and Regier et al2).
The mucopolysaccharidoses are a group of LSDs characterized by the accumulation of glycosaminoglycans, namely mucopolysaccharides. Mucopolysaccharidoses are classified into seven types (type I, II, III, IV, VI, VII, and IX) based on the accumulation of storage products and the underlying enzymatic deficiency. Mucopolysaccharidosis type IVA (MPS IVA) is an autosomal recessive LSD caused by genetic changes in the gene GALNS, leading to deficient or ineffective activity of the lysosomal enzyme N-acetylgalactosamine-6-sulfatase (GALNS). This rare disease has an incidence of 1 in 71,000 to 1,179,000.3 GALNS hydrolyzes the sulfate located on the ends of keratan sulfate (KS) and chondroitin-6-sulfate (C6S). Therefore, deficiency of GALNS leads to accumulation of KS and C6S.
MPS IVA caused by GALNS is clinically identical to MPS IVB, which causes the deficiency of β-galacotosidase (reviewed in Regier and Tifft4). Thus, it is important that enzymatic and/or genetic testing be performed to determine the underlying enzymatic defect in MPS IV since enzyme replacement is available in MPS IVA but not in MPS IVB.
The natural history of MPS IVA
MPS IVA was originally described by orthopedic experts due to the severity of the skeletal dysplasia; however, as skeletal management has improved, the natural history of this disorder has expanded to include respiratory, heart, hearing, eye, dental, and liver involvement. Thus, patients with MPS IV are followed by multidisciplinary teams to optimize their outcomes, often with orthopedics at the center of the care group.2
At birth, children with MPS IV have no unique identifiers. However, within the first few years of life, the most severe patients present with physical examination findings consistent with bony abnormalities. Kyphoscoliosis, genu valgum, and pectus carinatum are the most common presenting symptoms.5 The milder forms of MPS IV often present with hip problems such as pain, stiffness, and Legg-Calve-Perthes disease in late childhood or adolescence. In both cases, progressive bone and join involvement over time leads to short stature, pain, and arthritis. The bone progression can become disfiguring and lead to pain as well as impaired mobility and ability to perform activities of daily living (ADLs). Often, patients notice difficulty with hand strength and dexterity due to bony changes in the hands and lower arms. The more devastating skeletal concern, odontoid hypoplasia with cervical instability can lead to neurological damage and devastating neurological outcomes.5 Spinal canal stenosis could occur at any level. Upper and lower extremities malalignment are universal, but the severity varies. Common skeletal abnormalities include ulnar deviation of the wrist joint, hip dysplasia, hip dislocation, and genu valgum. Growth abnormalities are universal findings and usually severe. Height velocity is normal until 1–2 years old and then falls below normal.1,6
Extraskeletal manifestations of MPS IVA are similar to other MPS and include impaired respiratory function, valvular heart disease, obstructive sleep apnea (OSA), hearing impairment, corneal clouding, dental abnormalities in both shape and structure, and, less commonly, hepatomegaly.2
The longitudinal, prospective Morquio A clinical assessment program reviewed a decline of endurance tests, 6-minute walk test (6MWT), and 3-minute stair climb test (3MSCT), suggesting decreased functional ability over time.7 Both endurance tests have been used to monitor overall disease progression in the clinic and in elosulfase alfa clinical trials. The 6MWT assesses the walking distance in 6 minutes, and the 3MSCT assesses the number of stairs that subjects can climb within 3 minutes. These functional endurance tests are used as an indirect assessment of determining the overall function and integrated responses of multiple systems working together, including musculoskeletal, neurological, cardiovascular, and pulmonary systems.
Besides the progression of bone pathology and decline of endurance, natural history studies have revealed that other organ systems also show progressive symptoms. Significant morbidity is observed due to nonskeletal involvement.8 In a natural history study over 2 years, changes in the forced vital capacity (FVC) and maximum voluntary ventilation (MVV) were observed. For patients <14 years, the values increased, as would be expected for growth; however, the values decreased in older patients, which raised concerns about disease progression.7 The compromised respiratory function and decreased FVC and MVV are related to multiple factors, including progression of bone abnormalities and airway obstruction.
MPS IVA is a progressive condition and affects multiple organ systems. Life expectancy is shortened in the severe form, and general well-being is impaired. Bone abnormalities leading to joint and bone deformities and spinal cord compression are the major cause of mobility limitation, pain, and difficulties in ADLs. Several factors affect respiratory function such as short stature, chest wall abnormalities (pectus and kyphoscoliosis), and chronic OSA.
Current management strategies for symptoms of MPS IVA
A multidisciplinary approach is the key to management. Careful management with orthopedic intervention when necessary is crucial for MPS IV patients.2 In cases of difficult airway secondary to glycosaminoglycans accumulation in both upper and lower airway, odontoid hypoplasia, impaired respiratory function, and cardiac problems, it is crucial that anesthesia care be administered by teams that understand the needs of the MPS IV population.9 Due to the severe growth abnormalities, specific growth charts have been developed to allow for normalization of children’s growth.10 Abnormalities of the spine and malalignment of both upper and lower extremities are progressive and could lead to disability. Patients should be evaluated by orthopedics and neurosurgery closely, at least annually. Neurological examination should be performed at least every 6 months due to the risk of spinal cord compression. This would likely include radiography of spine and lower extremities, as well as whole-spine MRI annually and when clinically indicated.
Pulmonary specialty involvement is essential to manage respiratory compromise and sleep apnea.2 Close follow-up by a cardiology team is needed to monitor for and treat valvular heart disease.6 An audiology team follows and treats hearing impairment aggressively. Eye examinations and intervention for corneal clouding is important to optimize visual outcomes in patients.8 While the hepatomegaly noted in patients is not acutely dangerous, the liver size can affect other organs due to the short stature of patients. Finally, dental abnormalities can lead to pain and cosmetic difficulties if appropriate dental care is not identified early.8 Use of multidisciplinary clinics often improves patient satisfaction and quality of life by reducing the total number of hospital visits and facilitation of care coordination.
Within the context of the numerous systemic complications, it is crucial that the care delivery team optimize learning and social environments for families with MPS IV. Baseline intelligence is normal; thus, optimization of the environment and neurologic protection to prevent damage is crucial for outcomes.2
In 2014, the “International guidelines for the management and treatment of MPS IVA syndrome” was released.1 These guidelines were a summary of two meetings of international experts with extensive experience in managing MPS IVA. As summarized in Table 1, the recommendations include an extensive baseline evaluation, with reevaluation at intervals to identify systemic changes early to allow for monitoring and intervention.
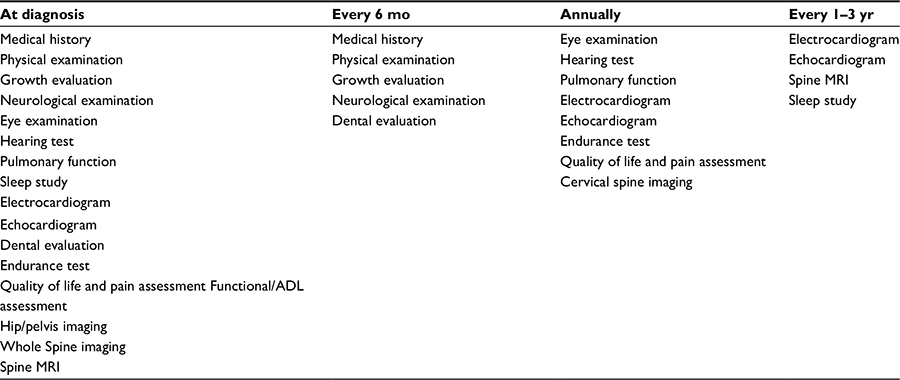 | Table 1 Monitoring recommendations for mucopolysaccharidosis type IVA (MPS IV) patients Note: Data from Regier et al.2 aMultiple evaluations performed at the time of diagnosis for baseline measurements should be repeated, as clinically indicated. Abbreviation: mo, months; yr, years; ADL, activities of daily living. |
Treatments for MPS IV have been limited to supportive care until recently. For example, adenotonsillectomy for OSA, hearing aids for sensorineural hearing loss, and keratoplasty for corneal clouding have been recommended. Aggressive supportive and symptomatic surgical and nonsurgical interventions for skeletal abnormalities have been used. While other MPS were shown to have benefited from hematopoietic stem cell transplantation, the experience with MPS IV has been limited and benefits have not been well defined.2
Pharmacology, mode of action, pharmacokinetics of elosulfase alfa
Enzyme replacement therapy (ERT) has become a standard of care for many forms of LSDs. The first ERT was approved for Gaucher disease in 1996. The limitations of ERT are infusion reaction, high cost, and low penetration of the central nervous system. With more clinical experience, the algorithm to prevent and manage infusion reactions has improved. The cost continues to be a major hurdle for treatment and an important conversation in the treatment of rare diseases. The low penetration of enzymes through the blood–brain barrier, leading to low central nervous system penetration, limits the efficacy of all ERTs. For example, ERT for Gaucher disease shows improvements in systemic symptoms of type I patients; however, the most severe forms – type II and III – with neurological regression and early death do not show improvement with peripheral administration of ERT. Thus, the efficacy of ERT in these cases is limited and does not change the end outcome – death from neurological complications. Since MPS IVA does not have effects on the intellect of patients, it makes it a perfect candidate for ERT, providing beneficial outcomes. It is important to note that the enzyme treatment can prevent disease progression to a certain extent but cannot reverse most of the pathology present from before treatment. Currently ERT has been approved for four types of MPS: MPS I (laronidase; Aldurazyme®, Genzyme, Boston, MA, USA), II (idursulfase; Elaprase®, Shire, Dublin, Republic of Ireland), IVA (elosulfase alfa; Vimizim®, Biomarin, Novato, CA, USA), and VI (galsulfase; Naglazyme®, Biomarin).
Elosulfase alfa mechanism of action
ERT is available only as an intravenous infusion formulation. The mechanism of action is to provide the enzymatic activity deficient due to MPS IVA.
Elosulfase alfa is a purified human enzyme produced using recombinant DNA technology (recombinant human enzyme or rhGALNS) in the Chinese hamster ovary cell line. The sequence and enzymatic activity of elosulfase alfa is identical to the human form. Elosulfase alfa is identical to the natural human enzyme in terms of the amino acid sequence and N-linked glycosylation.11,12 One of the oligosaccharide chains contains bis-mannose 6 phosphate, which binds a receptor at the cell surface (cation-independent mannose-6-phosphate receptor). Elosulfase alfa replaces the endogenous enzyme GALN and leads to catabolism of glycosaminoglycans C6S and KS.13,14 The enzyme is active in low pH environments and therefore has low activity outside of lysosomes.
Pharmacokinetics and pharmacodynamics
In humans, pharmacokinetics (PKs) and pharmacodynamics of elsosulfase alfa were studied in Phase I/II (MOR-002), Phase II (MOR-008), and Phase III (MOR-004) clinical trials. MOR-002is a completed Phase I/II, multicenter, open-label, dose escalation study. The primary objectives were to evaluate the safety, tolerability, and efficacy of weekly infusion of elosulfase alfa (BMN110). The study enrolled 20 subjects aged 5–18 years with MPS IVA. Subjects received elosulfase alfa over a 36-week dose escalation period of 12 weeks each of 0.1, 1.0, and 2.0 mg/kg per week, followed by 36–48 weeks of additional treatment at 1.0 mg/kg per week. MOR-004 is a multinational, multicenter, double-blind, randomized, placebo-controlled, 24-week Phase III study in patients with Morquio A syndrome. The objective of this study was to determine two different dose regimens (2.0 mg/kg per week and 2.0 mg/kg every other week [QOW]) infused intravenously compared to placebo for a total of 24 weeks. MOR-008 is the ongoing Phase II, randomized, double-blind, multicenter, pilot study of BMN110 dose 2.0 and 4.0 mg/kg per week administered for an initial treatment period of 27 weeks.
MOR-002 revealed that the PK profile of elosulfase alfa is not linear over the dose range of 0.1–2.0 mg/kg per week. The mean values for area under the plasma concentration time curve from time zero to the time of last measurable concentration (AUC0–t) and maximum plasma concentration (Cmax) increased more than the increase in dose.11 In vitro studies in human fibroblasts when using 2.0 mg/kg per week yielded plasma concentration higher than those seen with 1.0 mg/kg QOW; Cmax 2,023 and 503 ng/mL, respectively. Furthermore, the plasma concentration of elosulfase alfa at 2.0 mg/kg per week was sustained above the Kuptake (2.8–4 nM) of the M6PR for approximately 6 hours, which was around 1 hour longer than those seen in dose 1.0 mg/kg QOW.11
According to data from MOR-004 for both dosing regimens – 2.0 mg/kg per week and 2.0 mg/kg QOW – elosulfase alfa was detected in plasma at the first time point 60 minutes after starting infusion and reached maximum concentration between 120 and 240 minutes.15,16 Plasma half-life was short in both dosing regimens; at week 0, t1/2 was 7 minutes for both groups; and at week 22, t1/2 was 19 and 36 minutes in QOW and weekly dosing groups, respectively.11,19 On the other hand, the in vitro study in human Morquio fibroblast showed a half-life of 5–7 days.11 The results indicated that the enzyme’s half-life in plasma is shorter than in tissue, consistent with the uptake of the enzyme from the plasma to the tissue, as seen in other ERT regimens previously described.
The safety of a high dose (4.0 mg/kg per week) was studied in Phase II MOR-008. The results were similar to MOR-004 in that after week 22, the mean t1/2 was less than an hour, although with high dose at week 23, the mean t1/2s were 23.2 and 31.1 minutes for 2.0 and 4.0 mg/kg per week groups, respectively.17
Consistent with the findings from MOR-002, the differences between dosages in the mean AUC0–t and Cmax were higher than dose differences demonstrated in MOR-004 and MOR-008. This finding indicated that PKs of elosulfase alfa are not linear over this dosing range.11,17,19 The changes over time were attributed to neutralizing antibodies that occur in all patients.14,18 The pharmacodynamic outcome was measured by the decrease of first morning void urine KS. The level of urine KS is used to determine PKs, but the level does not correlate with clinical response. Decreases of urine KS was detected in all treatment groups, consistently across the cohorts (MOR-002, -004, and -008).11,17,19
Preclinical data
MPS IVA mice were studied to determine the safety, efficacy, and tolerability of elosulfase alfa. Animals showed marked reduction of storage in visceral organs, bone marrow, heart valves, connective tissues, and ligaments after 12 weeks of treatment.20 Further evaluation was experimented in newborn MPS IVA mice receiving ERT at birth. The bone pathology study showed that chondrocytes were vacuolated, but the column structure was organized. This evidence suggested that the enzyme entered cartilage before the cartilage cell layer becomes mature bone, leading to prevention of the disorganization of cartilage structure seen in MPS IV. These results indicated that early treatment may prevent bone pathology, the major symptom of MPS IV.21
Clinical trial data
Trial participants were enrolled in a multinational, multicenter, double-blind, randomized, placebo-controlled, 24-week Phase III study (MOR-004). The cohort was divided into two different dose regimens (2.0 mg/kg per week and 2.0 mg/kg QOW) infused intravenously compared to placebo. This study recruited only subjects with the ability to ambulate. All subjects had an average 6MWT distance ≥30 and ≤325 m during screening. A total of 176 patients (ages 5–57 years) were enrolled. One patient discontinued due to withdrawal of consent from the weekly treatment group. The primary efficacy measure was 6MWT, a measure of endurance. Secondary efficacy outcomes were 3MSCT and urine KS normalization. Tertiary measures evaluated were pulmonary function (FVC, MVV, and forced expiratory volume in 1 second). Additional tests and measures were collected, including anthropometric measures, audiogram to evaluate hearing, echocardiogram to evaluate cardiac valve function, corneal clouding and radiographic examination, MPS Health Assessment Questionnaires to evaluate quality of life, inflammatory markers (tumor necrosis factor α), and bone–cartilage metabolism (c-terminal crosslinked C-telopeptide CTX1 and type IIA collagen N-propeptide PIIANP).11,19
At week 24, elosulfase alfa administered weekly significantly improved the 6MWT distance compared to placebo (22.5 m; P=0.0174). The 3MSCT was not significantly changed in both weekly and QOW cohorts compared to placebo. Rapid and sustained urine KS was found in both treatment arms. Other tertiary improvements were detected numerically, including FVC, MVV, standing height, and growth rate in subjects with expected open growth plates (males <18 years; females <15 years).15,19 The differences in audiometry, echocardiogram, corneal clouding, or lower extremities bone length were small. Inflammatory marker was not changed in treatment group. Levels of CTX1 and PIIANP were not increased after treatment (elevation of both markers suggests bone formation). Since the tertiary end points of the Phase III study were not designed to have sufficient power, different composite end point analysis methods such as prespecified composite end point and O’Brien’s rank-sum composite end point were applied to allow for evaluation of treatment impact across multiple domains. The analyses demonstrated that the 2.0 mg/kg per week group had received benefit of treatment when the combination of changes from baseline of 6MWT, 3MSCT, and MVV were explored.15 The trial results recommended studies in younger and pre- or mildly symptomatic patients to determine the maximal benefit of treatment.15
The findings from ongoing high-dose 4.0 mg/kg per week versus recommended dose 2.0 mg/kg per week in high-functioning subjects (≥7 years and able to walk ≥200 m in 6MWT at screening) revealed numerical improvement in 3MSCT, cardiopulmonary exercise test, muscle strength, and pain in 4.0 mg/kg per week arm.
Treatment administration
The recommended dose is 2.0 mg/kg per week intravenous infusion over 3.5–4.5 hours, based on infusion volume. The final volume is 100 mL for patients <25 kg and 250 mL for patients >25 kg. The ERT is diluted in 0.9% sodium chloride injection. For patients <25 kg, the infusion rate should be 3 mL/h for the first 15 minutes and increased to 6 mL/h for the next 15 minutes. If this rate is tolerated, it can be increased by 6 mL/h every 15 minutes, not to exceed 36 mL/h.
For patients weighing ≥25 kg, initial infusion rate should be 6 mL/h for 15 minutes, and then the ramping regimen is begun at 12 mL/h for 15 minutes to a maximal rate of 72 mL/h.
Premedication with nonsedative antihistamine with or without antipyretics should be administered 30–60 minutes before infusion. A nonsedating formulation is preferred due to the risk of sleep apnea and airway difficulty in a sedating antihistamine.
Adverse events and reactions
Due to the size of the cohort and the short period of time in use, it is likely that additional adverse reactions may be identified as use of this ERT continues. Of the 235 patients enrolled in the clinical trail, 16 (6.8%) experienced signs and symptoms consistent with anaphylaxis (cough, erythema, throat tightness, urticarial, hypotension, dyspnea, gastrointestinal symptoms). The timing of these reactions was as early as 30 minutes after beginning infusion to 3 hours after infusion was completed. All but two subjects were able to receive subsequent elosulfase alfa infusion with infusion rate adjustments and/or medical intervention. Hypersensitivity reactions occurred in 18.7% and were observed from 30 minutes after start of infusion to as late as 6 days after infusions.11,12 Of the data from MOR-004, the most common adverse events were headache (20.3%), pyrexia (18.6%), vomiting (15.3%), nausea (13.6%), diarrhea (11.9%), fatigue (11.9%), upper abdominal pain, upper (8.5%), cough (8.5%), oropharyngeal pain (5.1%), abdominal pain (3.4%), and chills (1.7%).19
Most recently, a small study with children <5 years of age was completed. The group received 2.0 mg/kg per week for 52 weeks. Adverse events requiring infusion interruption and medical intervention occurred in 0.8% of infusions. Both growth velocity and urine KS levels were improved on treatment.22
Adverse reactions – prevention and management
Premedication with a nonsedating antihistamine with or without an antipyretic reduced the risk of adverse reactions in Phase I/II clinical trials.19 Premedication with sedating antihistamine, antipyretic, H2 blockers, leukotriene receptor antagonists, oral cromolyn sodium (personal communication), and/or steroids should be considered if the patient has a history of adverse reactions or allergies. Additionally, decreasing the infusion rate by 25%–50%, especially during the first 1–2 hours, is helpful if the reaction is early in the infusion course (personal experience). For patients with airway management or positive pressure during sleep secondary to sleep apnea, an airway patency evaluation should be performed prior to beginning ERT. Furthermore, oxygen should be available during the infusion therapy.
Postponement of ERT infusion should be highly considered in patients with acute infection, fever, or respiratory illness, due to an increased risk of hypersensitivity reactions.13
For acute hypersensitivity reaction, immediate medical care should be initiated. For mild-to-moderate reactions, slowing or stopping infusion should be considered. Additionally, administering antihistamines, antipyretics, and/or steroids should be considered.
For severe anaphylaxis reactions, infusion should be discontinued and breathing and circulation evaluated. Stabilizing the airway, giving oxygen supplementation, and administering epinephrine intramuscular injection and/or intravenous fluids for hypotension should be considered.
Patient evaluation of elosulfase alfa
Clinical trials demonstrated that treatment improved 6MWT and provided positive and meaningful changes in several clinical parameters. Preclinical studies indicated that early and presymptomatic treatment could prevent some progression of bone pathology. Further studies are necessary to demonstrate the efficacy of treatment in the relatively healthy population. Treatment should be initiated as soon as the diagnosis is made. The results of treatment may be different in each patient due to the high heterogeneity of the natural history. Expectation and goals of treatment of each family are different and should be discussed before starting the treatment. The efficacy of ERT on bone is not impressive. Patients should continue to follow closely with orthopedics. Furthermore, growth did not improve with treatment; however, the age of treatment initiation has not been clearly elucidated. The one study in children <5 years of age has shown short-term growth velocity improvement; however, long-term studies have not been published. Various parameters should be closely monitored throughout the treatment course with ERT. As with any ERT requiring weekly infusion, the loss of quality of life due to the frequent infusions must be offset by the perceived benefit of the treatment. This logistic issue could be managed but requires collaboration from other supporting systems and persons. For example, home infusion therapy could be preferably provided during the weekends or weekday evenings for the adult patients to avoid their work obligation issue, while pediatric patients can receive treatment during the weekdays since arrangement can be made more easily for their education requirement such as homework assignments or homeschooling (especially severe mobility impaired patients). Patient satisfaction has been fair based on the severity of the symptoms existing before ERT. For severe groups, the expectations are improvement of pain, ADLs, and ability to ambulate without using equipment. Even small changes such as ability to walk independently for a few feet could impact life (personal conversation). For mild-to-moderate symptom groups, the major expectations are improvement of endurance, exercise tolerance, and possible prevention of bone deformity progression. The consistency in receiving infusions was found to be higher in the home infusion group than those receiving infusions at an infusion center, due to the travel requirement.
The future of elosulfase alfa
Long-term outcome studies will be crucial to determine the role of elosulfase alfa on the bone outcomes of patients. Furthermore, based on the natural history outcomes described, long-term changes in progression of disease will be crucial to demonstrate the long-term efficacy of ERT treatment in MPS IVA.7 One measurement of initiation of elosulfase alfa treatment will be the number of patients that remain in the untreated arm of the natural history study, either by choice or due to inaccessibility of the ERT.
Donida et al described a cohort of MPS IVA patients receiving ERT therapy with elevated interleukin-6 and decreased glutathione levels, suggesting a proinflammatory and pro-oxidant state in these patients.23 The study did not include a cohort of MPS IVA patients not receiving ERT for comparison. This study supports further investigation of using antioxidants and antiinflammatory agents in combination with ERT to optimize patient outcomes.
Disclosure
The authors reports no conflicts of interest in this work.
References
Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet A. 2015;167A:11–25. | ||
Regier DS, Oetgen M, Tanpaiboon P. Mucopolysaccharidosis type IVA. In: Pagon RA, Adam MP, Ardinger HH, et al, editors, GeneReviews®. Seattle, WA. University of Washington. Available from: http://www.ncbi.nlm.nih.gov/books/NBK148668/. Accessed April 25, 2016. | ||
Leadley RM, Lang S, Misso K, et al. A systematic review of the prevalence of Morquio A syndrome: challenges for study reporting in rare diseases. Orphanet J Rare Dis. 2014;9:173. | ||
Regier DS, Tifft CJ. GLB1-related disorders. In: Pagon RA, Adam MP, Ardinger HH, et al, editors, GeneReviews®. Seattle, WA. University of Washington. Available from: http://www.ncbi.nlm.nih.gov/books/NBK164500/. Accessed April 25, 2016. | ||
Montano AM, Tomatsu S, Gottesman GS, Smith M, Orii T. International Morquio A Registry: clinical manifestation and natural course of Morquio A disease. J Inherit Metab Dis. 2007;30:165–174. | ||
Harmatz P, Mengel KE, Giugliani R, et al. The Morquio A Clinical Assessment Program: baseline results illustrating progressive, multisystemic clinical impairments in Morquio A subjects. Mol Genet Metab. 2013;109:54–61. | ||
Harmatz PR, Mengel KE, Giugliani R, et al. Longitudinal analysis of endurance and respiratory function from a natural history study of Morquio A syndrome. Mol Genet Metab. 2015;114:186–194. | ||
Hendriksz CJ, Al-Jawad M, Berger KI, et al. Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J Inherit Metab Dis. 2013;36:309–322. | ||
Theroux MC, Nerker T, Ditro C, Mackenzie WG. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr Anaesth. 2012;22:901–907. | ||
Montano AM, Tomatsu S, Brusius A, Smith M, Orii T. Growth charts for patients affected with Morquio A disease. Am J Med Genet A. 2008;146A:1286–1295. | ||
Biomarin Pharmaceuticals. Vimizim (elosulfase alfa) for the treatment of mucopolysaccharidosis type IVA (Morquio syndrome): briefing document for the endocrinologic and metabolic drugs advisory committee 2013; 2013. Available from: http://www.fda.gov/downloads/advisorycommittees/committeesmeetingmaterials/drugs/endocrinologicandmetabolicdrugsadvisorycommittee/ucm375127.pdf. Accessed July 2015. | ||
Food and Drug Administration. Vimizim (elosulfase alpha) injection, for intravenous use: US prescribing information; 2014. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125460s000lbl.pdf. Accessed January 2016. | ||
Goldenberg MM. Pharmaceutical approval update. P & T: a peer-reviewed. J Formul Manage. 2014;39:337–344. | ||
Sanford M, Lo JH. Elosulfase alfa: first global approval. Drugs. 2014;74:713–718. | ||
Hendriksz CJ, Giugliani R, Harmatz P, et al. Multi-domain impact of elosufase alfa in Morquio A syndrome in the pivotal phase III trial. Mol Genet Metab. 2015;114:178–185. | ||
Schweighardt B, Tompkins T, Lau K, et al. Immunogenicity of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome: results from MOR-004, a phase III trial. Clin Ther. 2015;37:1012–1021. e1016. | ||
Burton BK, Berger KI, Lewis GD, et al. Safety and physiological effects of two different doses of elosulfase alfa in patients with morquio a syndrome: a randomized, double-blind, pilot study. Am J Med Genet A. 2015;167:2272–2281. | ||
Qi Y, Musson DG, Schweighardt B, et al. Pharmacokinetic and pharmacodynamic evaluation of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome. Clin Pharmacokinet. 2014;53:1137–1147. | ||
Hendriksz CJ, Burton B, Fleming TR, et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014;37:979–990. | ||
Tomatsu S, Montano AM, Ohashi A, et al. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum Mol Genet. 2008;17:815–824. | ||
Tomatsu S, Montano AM, Oikawa H, et al. Enzyme replacement therapy in newborn mucopolysaccharidosis IVA mice: early treatment rescues bone lesions? Mol Genet Metab. 2015;114:195–202. | ||
Jones SA, Bialer M, Parini R, et al. Safety and clinical activity of elosulfase alfa in pediatric patients with Morquio A syndrome (mucopolysaccharidosis IVA) less than 5 y. Pediatr Res. 2015;78:717–722. | ||
Donida B, Marchetti DP, Biancini GB, et al. Oxidative stress and inflammation in mucopolysaccharidosis type IVA patients treated with enzyme replacement therapy. Biochim Biophys Acta. 2015;1852:1012–1019. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.