")
Back to Journals » Clinical Epidemiology » Volume 11
Role of cigarette smoking in the development of ischemic stroke and its subtypes: a Mendelian randomization study
Authors Qian Y, Ye D, Wu DJH , Feng C , Zeng Z, Ye L , Zhu R, Zhang Z , Mao Y
Received 27 May 2019
Accepted for publication 23 July 2019
Published 13 August 2019 Volume 2019:11 Pages 725—731
DOI https://doi.org/10.2147/CLEP.S215933
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eyal Cohen
Yu Qian,1,* Ding Ye,1,* David JH Wu,1,2 Chen Feng,3 Zhen Zeng,1 Lihong Ye,1 Rui Zhu,4 Zhenyu Zhang,5 Yingying Mao1
1School of Public Health, Zhejiang Chinese Medical University, Hangzhou, Zhejiang 310053, People’s Republic of China; 2University of Minnesota Medical School, Minneapolis, MN 55455, USA; 3The Second Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, Zhejiang 310053, People’s Republic of China; 4College of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou, Zhejiang 310053, People’s Republic of China; 5Welch Center for Prevention, Epidemiology, and Clinical Research, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD 21205, USA
*These authors contributed equally to this work
Purpose: Numerous studies have indicated that smokers have an increased risk of developing ischemic stroke. However, less is known about the causal relationship between cigarette smoking and ischemic stroke subtypes. In the present study, we aim to determine whether genetically predicted cigarette smoking was associated with subtypes of ischemic stroke using Mendelian randomization (MR).
Patients and methods: We used summary-level genetic association data from the MEGASTROKE consortium, including 438,847 individuals of European ancestry (34,217 cases of ischemic stroke and 404,630 controls). We used 176 single nucleotide polymorphisms as instrumental variables, which were previously identified to be associated with smoking in the Study of the Social Science Genetic Association Consortium (n=518,633). MR analyses were performed using inverse-variance-weighted method, weighted-median method, and MR-Egger regression.
Results: We found that genetically predicted smoking was associated with a higher risk of ischemic stroke (odds ratio (OR): 1.24, 95% CI: 1.10–1.39) and large artery ischemic stroke (OR: 1.52, 95% CI: 1.14–2.02), but not with risk of cardioembolic ischemic stroke or small vessel ischemic stroke. Sensitivity analyses using alternative MR approaches produced similar results.
Conclusion: Genetic predisposition toward smoking is causally associated with a higher incidence of large artery ischemic stroke. Further work is warranted to clarify the underlying mechanism of smoking in the development of large artery ischemic stroke.
Keywords: ischemic stroke, Mendelian randomization, polymorphism, single nucleotide, smoking
Plain language summary
The relationship between smoking and the risk of ischemic stroke has been known about for quite some time. Traditionally, studies have only been able to explore correlations and not causality, due to the limitations of observational analysis. In this study, we used a new method called Mendelian randomization to explore a potential causal relationship between smoking and the risk of stroke. First, we looked for changes in a person’s genome, called single nucleotide polymorphisms (SNPs), which were correlated with smoking. Because a person cannot alter their genotypes, this helps us avoid problems like reverse causation which can confound traditional analyses. We then explored whether presence of these smoking-related SNPs had any relationship to the risk of ischemic stroke by looking for the presence of these SNPs in a large stroke database. We found that for certain subtypes of ischemic stroke, there was a positive relationship between smoking-related SNPs and the risk of ischemic stroke, providing support for a causal, unidirectional relationship between smoking and risk of ischemic stroke. This finding is important because it suggests that further studies are warranted to explore why this causal relationship may exist.
Introduction
Stroke is the leading cause of death worldwide, with a mortality rate of 86.5 (83.3–89.9) per 100,000 population per year.1 Large artery ischemic stroke (LV-IS), cardioembolic ischemic stroke (CE-IS) and small vessel ischemic stroke (SV-IS) are main subtypes of ischemic stroke (IS), accounting for 11%, 22% and 18% of all stroke, respectively.2 Recently, a meta-analysis of genome-wide association studies (GWASs) suggested that the pathophysiological mechanisms are different among these IS subtypes.3 Hence, the study of IS as a whole may not be adequate to assess the effects of risk factors under the influence of subtype distribution.4 Therefore, determining differences in the risk factor profiles among IS subtypes can inform future research for more targeted solutions.
In the past few decades, epidemiological studies have demonstrated that current smokers have a higher risk of IS compared with never-smokers.5,6 However, less is known about the relationship between smoking and IS subtypes. A recent case–control study proposed that smoking might increase the susceptibility of large artery atherosclerotic stroke,7 while a population-based study suggested a negative relationship between smoking and CE-IS.8 In addition, a cross-sectional study showed that current smoking was associated with the risk of small vessel disease.9 However, as the nature of the above studies is observational, associations between smoking and IS subtypes from traditional epidemiological studies may be biased by confounding and reverse causation.
One way to circumvent the bias inherent in traditional observational studies is Mendelian randomization (MR).10 Specifically, in this method, the exposure-related genetic variants are used as instrumental variables to investigate the potential causal relationship between exposure and disease.11 As the genetic variants are not influenced by the onset of disease or confounding factors, by studying the instruments rather than exposure directly, the MR approach can overcome the potential impact of confounding and reverse causation on the exposure and the outcome.11
Therefore, in the current study, we adopted a MR approach to investigate the potential causal effect of smoking on the development of IS subtypes by using summary-level statistics from GWASs.
Materials and methods
Data sources
Stroke genotyping data were obtained from the MEGASTROKE consortium, which included 34,217 cases of ischemic stroke and 404,630 controls of European ancestry.12 The details of phenotype definition for stroke and its subtypes have been reported elsewhere.12 Briefly, cases with stroke were defined as the sudden developing signs of neurological deficit, lasting more than 24 hrs with a vascular origin. According to the clinical and imaging criteria, strokes were divided into IS and intracerebral hemorrhage. IS cases were further categorized using the Trial of Org 10172 in Acute Stroke Treatment criteria.13 Specifically, there were 4373 cases of LV-IS, 5386 cases of SV-IS, and 7193 cases of CE-IS.
Studies participating in the MEGASTROKE consortium have received ethical approval from relevant institutional review boards. As we used publicly available summary-level data from the published genome-wide meta-analysis, no additional ethical approval was required.
Selection of SNPs
The GWAS performed by the Social Science Genetic Association Consortium was used to identify association estimates between single-nucleotide polymorphisms (SNPs) and smoking (ever smoker vs never smoker).14 The details of the study protocol were available elsewhere.14 Briefly, data from the UK Biobank and the Tobacco and Genetics consortium were used in this study, which included 518,633 individuals. The phenotype of “ever smoker” was defined as whether one has ever smoked regularly. We used a total of 176 independent SNPs associated with smoking at the genome-wide significance threshold (P<5×10−8) as the instrumental variables for the following MR analysis,15 which did not overlap with any known stroke risk loci.
Statistical analysis
Statistical analyses were performed using the Mendelian Randomization package in R software v3.5.2 (https://www.r-project.org/).16 The primary analysis assessing the causal associations of smoking and IS subtypes was performed using the inverse-variance-weighted (IVW) method. The method can estimate the causal effect of smoking on IS and its subtypes when each of the genetic variant is a valid instrumental variable.17 Although the inclusion of multiple genetic variants in the MR analysis can increase statistical power, it may contain some genetic variants that do not meet the assumption of an instrumental variable in the MR analysis.18 Violation of this assumption is most likely due to the presence of pleiotropy (SNPs associated with multiple related traits),19 which may bias the causal association.20 Therefore, to improve the reliability of causal effect estimates, we alternatively used the weighted-median method which has a different assumption about the estimation of causal effects. This method can provide a more reliable estimate of the causal effect than the IVW method, when some SNPs were invalid instruments.17
To further assess the impact of potential pleiotropy on causal estimates, we performed sensitivity analyses using several other methods. First, we used the MR-Egger regression to assess the presence of average pleiotropy.21 A statistically significant intercept term from the MR-Egger regression suggests the possibility that genetic variants may not affect the outcome via the exposure of interest.21 Second, Cochran’s Q test was used to explore the heterogeneity among the SNPs used in the primary analyses.22 The presence of heterogeneity indicates that some genetic variants are invalid instruments.22 Third, we excluded those SNPs associated with at least one secondary trait at the genome-wide significance levels (P<5×10−8) by searching the GWAS Catalog (https://www.ebi.ac.uk/gwas, accessed on March 31, 2019),23 and repeated the MR analysis using the IVW, the MR-Egger regression, and the weighted-median methods.
Patient and public involvement
We adopted a MR approach by using publicly accessible summary-level statistics of published GWASs from the MEGASTROKE consortium. Thus, no additional patients were involved.
Results
Primary MR analysis of smoking with the risk of IS subtypes
As listed in Table S1, 176 SNPs were used as the instrumental variables for smoking. We found that genetically predicted ever-smokers had a higher risk of developing IS (odds ratio (OR) 1.24, 95% CI 1.10–1.39, P=3.63×10−4) and LV-IS (OR 1.52, 95% CI 1.14–2.02, P=0.004) compared with never-smokers using the IVW method (Figure 1). Similar results were obtained from the weighted-median analyses, with an odds ratio of 1.26 (1.06–1.50) for IS and 1.81 (1.17–2.80) for LV-IS. In contrast, we did not find a statistically significant causal association of smoking with the risk of CE-IS using the IVW method (OR 1.18, 95% CI 0.95–1.47, P=0.143) or the weighted-median method (OR 1.21, 95% CI 0.88–1.67, P=0.230) (Figure 1).
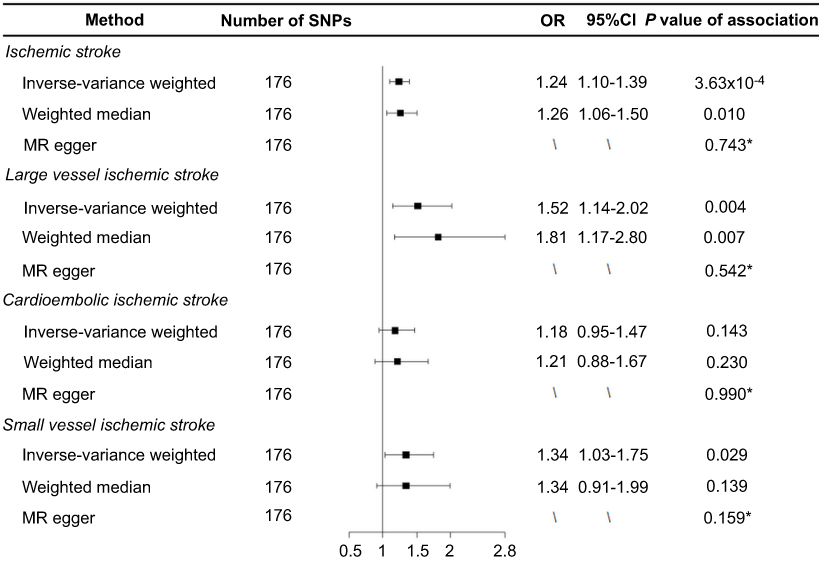 |
Figure 1 Forest plot of Mendelian randomization effect estimates for the associations of smoking and risk of ischemic stroke, large artery atherosclerotic ischemic stroke, cardioembolic ischemic stroke, and small vessel ischemic stroke. Effect estimates were derived from the inverse-variance-weighted method, weighted-median method, and MR-Egger regression. *Indicates the P-values of the intercept from the MR-Egger regression.Abbreviations: MR, Mendelian randomization; OR, odds ratio; SNP, single-nucleotide polymorphism. |
Sensitivity analysis of smoking with the risk of IS subtypes
To assess the robustness of the causal effect of smoking on IS and its subtypes, we performed MR-Egger regression and Cochran’s Q test. The intercept term from the MR-Egger regression analyses did not suggest evidence of directional pleiotropy in the MR analysis of smoking with IS (P=0.743), LV-IS (P=0.542), or SV-IS (P=0.159), respectively. Cochran’s Q test did not suggest evidence of potential heterogeneity for the causal association of smoking with LV-IS (P=0.078); however, potential heterogeneity for the associations of smoking with IS (P<0.001), CE-IS (P=0.020) and SV-IS (P=0.002) were observed, suggesting the possibility that the obtained effect estimates of these associations from the IVW method may be biased by outlier SNPs. As expected, after excluding those outlier SNPs that have strong effects on outcomes, we found that the effect estimates for the causal association of smoking with IS were attenuated but remained statistically significant (OR 1.19, 95% CI 1.06–1.34, P=0.003 for IS). We did not find a statistically significant association of smoking with CE-IS (OR 1.13, 95% CI 0.90–1.41, P=0.289) or SV-IS (OR 1.24, 95% CI 0.95–1.62, P=0.115) after excluding those outlier SNPs (Table S2).
We further assessed whether the smoking-associated SNPs used as instrumental variables were associated with secondary phenotypes by searching the GWAS Catalog. Details of the excluded SNPs and their secondary phenotypes are shown in Table S3. In total, we excluded 15 SNPs that were potentially associated with at least one secondary phenotype and repeated the MR analyses. As expected, the causal associations of smoking with IS (OR 1.20, 95% CI 1.06–1.35, P=0.004) and LV-IS (OR 1.42, 95% CI 1.05–1.91, P=0.021) remained statistically significant (Table 1). We did not detect evidence for causal associations of smoking with CE-IS (OR 1.14, 95% CI 0.90–1.43, P=0.280) or SV-IS (OR 1.26, 95% CI 0.96–1.66, P=0.105) using the IVW method or other MR approaches after excluding the potential pleiotropic SNPs.
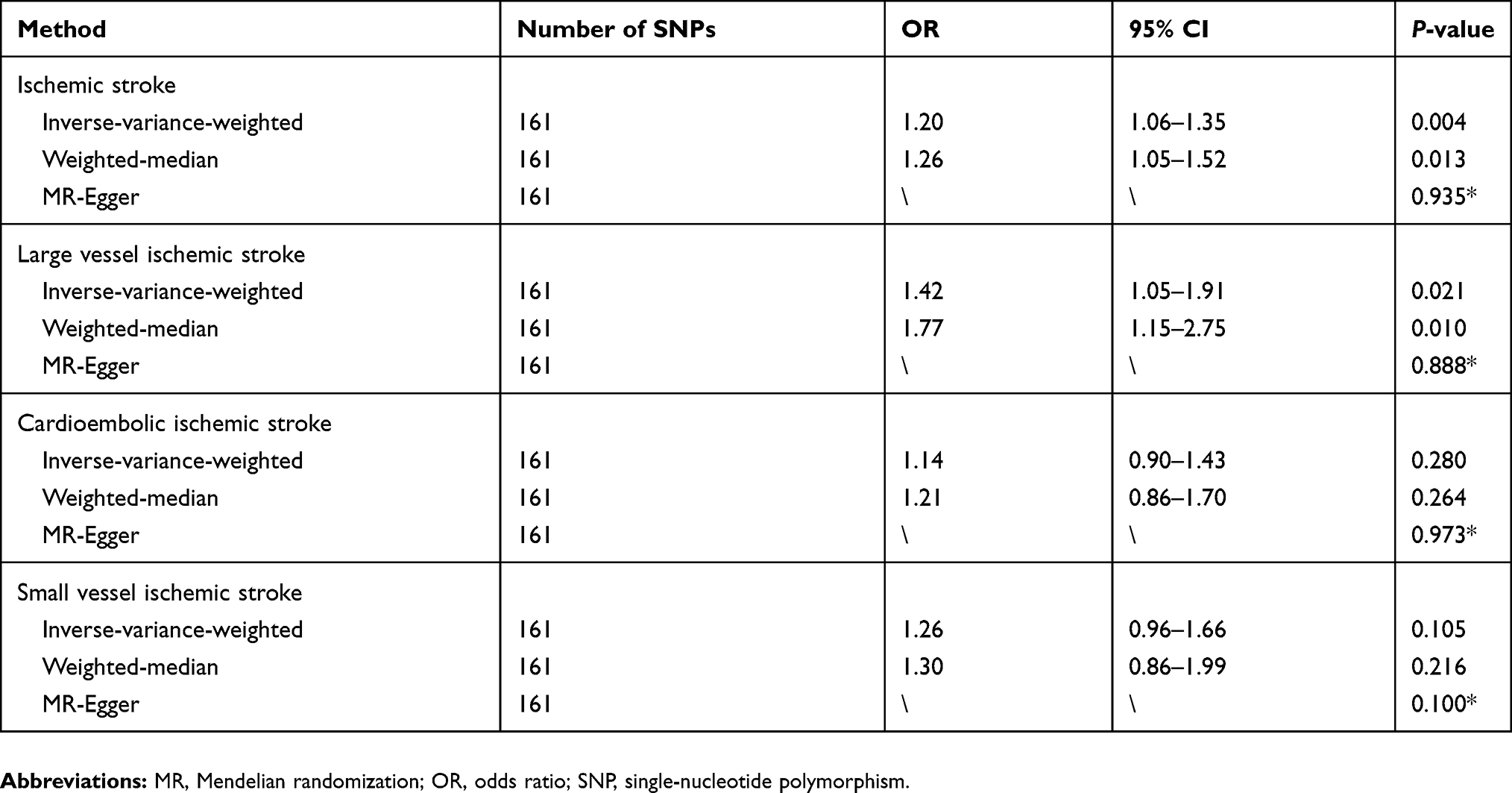 |
Table 1 MR analyses effect estimates for associations between smoking and stroke after excluding potential pleiotropic SNPs |
Discussion
This MR study provides evidence for a causal association of genetically predicted smoking with the risk of LV-IS. However, we did not find evidence of a causal association between smoking and the risk of SV-IS or CE-IS. The results were consistent in the sensitivity analyses, providing further support for the causal association between smoking and LV-IS.
Numerous studies have documented a link between smoking and the risk of IS. For instance, in a cohort study of 118,539 women aged from 30 to 55 years, the age-adjusted relative risk of IS was 2.2 (95% CI 1.5–3.3) for 1–14 cigarettes/day and 3.7 (95% CI 2.7–5.1) for 25 or more cigarettes per day.24 Recently, a case–control study containing 615 IS cases and 530 controls indicated a dose–response relationship between the number of cigarettes smoked and risk of IS, ranging from 1.46 for individuals who smoke less than 11 cigarettes per day to 5.66 for those who smoke more than 40 cigarettes per day.5 Our study suggested that genetically predicted smokers had a 1.24-fold increased risk of IS compared with nonsmokers, which was in line with these observational epidemiological studies.
In the present study, we found that genetic predisposition to smoking is associated with a 1.52-fold higher risk of LV-IS, which is consistent with some recent observational studies. For instance, a cohort study including 2656 patients with acute IS and 208 patients with transient ischemic attack found that smoking was significantly associated with the risk of extracranial atherosclerotic stenosis, which is one of the most common causes of LV-IS (OR 1.47, 95% CI 1.09–1.99).25 However, the underlying mechanisms between smoking and LV-IS remain uncertain. One possible mechanism is that smoking may induce oxidative DNA damage, which further contributes to the susceptibility of LV-IS.7 In addition, smoking may affect the development of atherosclerotic stenosis and thrombus generation in atherosclerotic arteries, leading to an increased risk of IS.25 Further studies are needed to investigate the underlying mechanisms of smoking in the development of LV-IS.
In contrast, our analysis did not provide statistically significant evidence for the relationship between smoking and CE-IS. An epidemiological study including a larger sample size of CE-IS cases reported a finding that consistent with our results. This study combining the data of Oxford Vascular Study and Oxfordshire Community Stroke Project found that smoking was not associated with CE-IS (OR 0.68, 95% CI 0.43–1.09).8 Similarly, the relationship between smoking and SV-IS remains unclear. A population-based analysis involving about 196,000 people suggested that smoking was not associated with SV-IS (OR 1.16, 95% CI 0.75–1.79).8 However, it has been proposed that tobacco smoke may increase the risk of SV-IS by inducing a strong inflammatory response.26 In our primary MR analysis, the effect estimate from the IVW method suggested a causal association between smoking and risk of SV-IS. However, it was not statistically significant after correction for multiple comparison. Moreover, the weighted-median method did not provide evidence for this relationship, and no statistically significant association was observed after excluding the outlier SNPs. Due to the possibility of inappropriate selection of instrumental variables and insufficient statistical power, studies with larger sample size and prospective design are warranted to further assess the effect of smoking on the risk of SV-IS.
The main strength of our study was to overcome the potential impact of confounding and reverse causation on the associations between smoking and IS subtypes.27,28 In addition, we used summary-level data from a large number of IS subtypes cases and controls, and used a large number of smoking-associated SNPs as instrumental variables reported from the most up-to-date GWAS of populations with European ancestry, which increased the statistical power of causal estimates. Finally, we used several alternative methods to assess possible pleiotropic biases, and obtained similar effect estimates, which indicated the robustness of our findings.
Our study has several limitations. First, the effect estimates of SNPs on smoking and stroke were obtained only from populations of European ancestry, which reduced the bias from population stratification. The reliability of these results may be reduced when extrapolating to other populations. Therefore, further studies are warranted to investigate the causal nature of smoking and IS in other populations of additional ethnic origins. Additionally, the data of smoking traits were collected through interviewer- or self-administered questionnaires, which may reduce the precision of causal estimation between smoking and the risk of IS subtypes. Due to the limited sample size of patients with IS subtypes such as SV-IS and CE-IS, the possibility of a weak causal association of smoking with SV-IS and CE-IS could not be totally excluded.
Conclusion
Genetic predisposition toward smoking is causally associated with higher incidence of large artery ischemic stroke. Further work is warranted to clarify the underlying mechanism of smoking in the development of large artery ischemic stroke.
Abbreviations
CE-IS, cardioembolic ischemic stroke; CI, confidence interval; GWAS, genome-wide association study; IS, ischemic stroke; LV-IS, large artery ischemic stroke; MR, Mendelian randomization; OR, odds ratio; SNP, single-nucleotide polymorphism; SV-IS, small vessel ischemic stroke.
Ethics approval and informed consent
We adopted a MR approach by using publicly accessible summary-level statistics of published GWASs from the MEGASTROKE consortium. Thus, no additional ethical approval was required.
Data sharing statement
Summary-level data for ischemic stroke used in the study is publicly available from the MEGASTROKE consortium (http://megastroke.org/download.html).
Acknowledgments
We thank the MEGASTROKE Consortium for providing summary statistics data for these analyses. The full list of the MEGASTROKE authors is available in the Supplementary materials. The MEGASTROKE project received funding from sources specified at http://www.megastroke.org/acknowledgements.html.
This work was supported by grants from National Natural Science Foundation of China (81602917), The Talent Project of Zhejiang Association for Science and Technology (2018YCGC003) and Zhejiang Provincial Natural Science Foundation of China (LQ15H260001). The funders had no role in the study design, data analysis, interpretation of data, or preparation of the manuscript.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Collaborators GBDCoD. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390(10100):1151–1210. doi:10.1016/S0140-6736(17)32152-9
2. Endres M, Heuschmann PU, Laufs U, Hakim AM. Primary prevention of stroke: blood pressure, lipids, and heart failure. Eur Heart J. 2011;32(5):545–552. doi:10.1093/eurheartj/ehq472
3. Traylor M, Farrall M, Holliday EG, et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): a meta-analysis of genome-wide association studies. Lancet Neurol. 2012;11(11):951–962. doi:10.1016/S1474-4422(12)70234-X
4. Song YM, Kwon SU, Sung J, et al. Different risk factor profiles between subtypes of ischemic stroke. A case-control study in Korean men. Eur J Epidemiol. 2005;20(7):605–612.
5. Markidan J, Cole JW, Cronin CA, et al. Smoking and risk of ischemic stroke in young men. Stroke. 2018;49(5):1276–1278. doi:10.1161/STROKEAHA.117.018859
6. Xu T, Bu X, Li H, et al. Smoking, heart rate, and ischemic stroke: a population-based prospective cohort study among Inner Mongolians in China. Stroke. 2013;44(9):2457–2461. doi:10.1161/STROKEAHA.111.000648
7. Shyu HY, Shieh JC, Ji-Ho L, Wang HW, Cheng CW. Polymorphisms of DNA repair pathway genes and cigarette smoking in relation to susceptibility to large artery atherosclerotic stroke among ethnic Chinese in Taiwan. J Atheroscler Thromb. 2012;19(4):316–325. doi:10.5551/jat.10967
8. Schulz UG, Rothwell PM. Differences in vascular risk factors between etiological subtypes of ischemic stroke: importance of population-based studies. Stroke. 2003;34(8):2050–2059. doi:10.1161/01.STR.0000079818.08343.8C
9. Staals J, Makin SD, Doubal FN, Dennis MS, Wardlaw JM. Stroke subtype, vascular risk factors, and total MRI brain small-vessel disease burden. Neurology. 2014;83(14):1228–1234. doi:10.1212/WNL.0000000000000837
10. Smith GD, Ebrahim S. Mendelian randomization: prospects, potentials, and limitations. Int J Epidemiol. 2004;33(1):30–42. doi:10.1093/ije/dyh132
11. Davey Smith G, Ebrahim S. What can mendelian randomisation tell us about modifiable behavioural and environmental exposures? Bmj. 2005;330(7499):1076–1079. doi:10.1136/bmj.330.7499.1076
12. Malik R, Chauhan G, Traylor M, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524–537. doi:10.1038/s41588-018-0058-3
13. Adams HP
14. Karlsson Linner R, Biroli P, Kong E, et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 2019;51(2):245–257. doi:10.1038/s41588-018-0309-3
15. Zhan Y, Fang F. Smoking and amyotrophic lateral sclerosis: a mendelian randomization study. Ann Neurol. 2019;85(4):482–484. doi:10.1002/ana.25443
16. Yavorska OO, Burgess S. Mendelian Randomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol. 2017;46(6):1734–1739. doi:10.1093/ije/dyx034
17. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–314. doi:10.1002/gepi.21965
18. Hartwig FP, Davies NM, Hemani G, Davey Smith G. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol. 2016;45(6):1717–1726. doi:10.1093/ije/dyx028
19. Kia DA, Noyce AJ, White J, et al. Mendelian randomization study shows no causal relationship between circulating urate levels and Parkinson’s disease. Ann Neurol. 2018;84(2):191–199. doi:10.1002/ana.25294
20. Dale CE, Fatemifar G, Palmer TM, et al. Causal associations of adiposity and body fat distribution with coronary heart disease, stroke subtypes, and type 2 diabetes mellitus: a mendelian randomization analysis. Circulation. 2017;135(24):2373–2388. doi:10.1161/CIRCULATIONAHA.116.026560
21. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
22. Greco MF, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34(21):2926–2940. doi:10.1002/sim.6522
23. MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS catalog). Nucleic Acids Res. 2017;45(D1):D896–D901. doi:10.1093/nar/gkw1133
24. Colditz GA, Bonita R, Stampfer MJ, et al. Cigarette smoking and risk of stroke in middle-aged women. N Engl J Med. 1988;318(15):937–941. doi:10.1056/NEJM198804143181501
25. Ji R, Pan Y, Yan H, et al. Current smoking is associated with extracranial carotid atherosclerotic stenosis but not with intracranial large artery disease. BMC Neurol. 2017;17(1):120. doi:10.1186/s12883-017-0873-7
26. Hossain M, Sathe T, Fazio V, et al. Tobacco smoke: a critical etiological factor for vascular impairment at the blood-brain barrier. Brain Res. 2009;1287:192–205. doi:10.1016/j.brainres.2009.06.033
27. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–1163. doi:10.1002/sim.3034
28. Smith GD, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. doi:10.1093/ije/dyg070
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.