")
Back to Journals » ImmunoTargets and Therapy » Volume 9
Role of Chemokines and Chemokine Receptors in Rheumatoid Arthritis
Authors Elemam NM , Hannawi S, Maghazachi AA
Received 24 December 2019
Accepted for publication 28 February 2020
Published 9 March 2020 Volume 2020:9 Pages 43—56
DOI https://doi.org/10.2147/ITT.S243636
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Michael Shurin
Noha Mousaad Elemam,1 Suad Hannawi,2 Azzam A Maghazachi1
1College of Medicine and Sharjah Institute for Medical Research, University of Sharjah, Sharjah, United Arab Emirates; 2Ministry of Health and Prevention, Department of Rheumatology, Dubai, United Arab Emirates
Correspondence: Azzam A Maghazachi
College of Medicine and Sharjah Institute for Medical Research, University of Sharjah, Sharjah, United Arab Emirates
Email [email protected]
Abstract: Rheumatoid arthritis (RA) is one of the most prevalent autoimmune diseases and a prototypic inflammatory disease, affecting the small joints of the hands and feet. Chemokines and chemokine receptors play a critical role in RA pathogenesis via immune cells recruitment. Several chemokines and chemokine receptors are abundant in the peripheral blood and in the local inflamed joints of RA. Furthermore, synthetic and biologics disease modifying anti rheumatic drugs have been reported to affect chemokines expression. Thus, many studies have focused on targeting chemokines and chemokine receptors, where some have shown positive promising results. However, most of the chemokine blockers in human trials of RA treatment displayed some failures that can be attributed to several reasons in their structures and binding affinities. Nevertheless, targeting chemokines will continue to be under development, in order to improve their therapeutic potentials in RA and other autoimmune diseases. In this review we provide an up-to-date knowledge regarding the role of chemokines and chemokine receptors in RA with an emphasis on their activities on immune cells. We also discussed the effects of drugs targeting those molecules in RA. This knowledge might provide impetus for developing new therapeutic modalities to treat this chronic disease.
Keywords: rheumatoid arthritis, chemokines, immunotherapy, immunotargets, chemokine receptors, drugs
Introduction
Rheumatoid Arthritis
Autoimmune rheumatic diseases, including systemic lupus erythematosus (SLE), Sjogren’s syndrome (SS), and rheumatoid arthritis (RA), can be difficult to diagnose as they share multiple symptoms and are of complex nature. It would take years before clinical manifestations become apparent and that will probably happen after organ/tissue damage has occurred. Hence, early diagnosis and treatment would be crucial to preventing further damage.1 Autoimmune diseases are manifestations of immune cells attacking normal tissues; however, the etiology of autoimmune diseases is not clearly defined.
Rheumatoid arthritis (RA) is one of the most prevalent autoimmune diseases (1–3% of the world’s population). RA is a prototypic inflammatory disease, being characterized by an altered state of homeostasis, in which immunological stimulation and unwanted inflammation prevail. The disordered inflammation has painful and debilitating immediate effects while causing cumulative tissue damage, which could progress into symmetric polyarthritis thus leading to lifelong discomfort, disability and shortened life expectancy.2–4 It has been reported that almost 50% of RA patients become disabled within 10 years of disease onset, and hence, their survival is lessened.5–7 RA starts with a painful inflammation in the small joints of the hands and feet, especially in the metacarpophalangeal, metatarsophalangeal, and proximal interphalangeal joints. Also, large joints can be involved such as the elbows, ankles, knees and shoulders.4,8 Being a systemic autoimmune disease, RA also affects other organs and processes such as osteoclastogensis, angiogenesis and cardiovascular, pulmonary, and skeletal disorders. In clinical setting, RA can be diagnosed by the presence of physical knee inflammation (as per the ACR/EULAR 2010 criteria) along with the presence of a high-titer of rheumatoid factor and/or anticitrullinated peptide antibodies (ACPAs).9
The standard golden therapy for RA Patients is the disease-modifying anti-rheumatic drugs (DMARDs). These drugs act by ameliorating the signs of RA in order to inhibit further progression and damage of the joints.10 The most commonly used DMARD is methotrexate. However, due to inefficacy, intolerance and side effects, there have been emerging therapeutic agents that can act on specific molecules associated with RA pathogenesis. Biologics DMARDs are prescribed only when treatment with DMARDs and/or NSAIDs failed. Currently, there are many specific biological DMARDs such as TNF-α inhibitors, IL-6R antibodies and JAK inhibitors, that are considered to be the most efficient therapeutic agents in RA.11 The known anti-TNF therapies include etanercept, infliximab, adalimumab, certolizumab, and golimumab, while other cytokine receptor blockers include anakinra (IL-1R blocker) and tocilizumab (IL-6R blocker). Nevertheless, the therapeutic strategy for RA has to be monitored by continuous assessment of the disease activity in order to reach the clinical remission phase.12,13
RA is influenced by both genetic and environmental factors, where smoking, diet, obesity, microbiota and infections have been suggested to induce the disease in genetically susceptible individuals. The clinical representation of RA is the result of a cascade of responses and close interactions between immune and non-immune cells (e.g. endothelial and fibroblast-like synoviocytes), autoantibodies, soluble mediators such as cytokines and chemokines, as well as signal transduction pathways of the innate and adaptive immune system.14 Various players of the immune system include neutrophils, macrophages, B cells, natural killer (NK) cells and T cells migrate to the synovial membrane and accumulate in the synovial fluid, leading to the release of mediators such as cytokines, chemokines, adhesion molecules, matrix metalloproteinases (MMPs) and reactive oxidative species (ROS) which consequently cause joint destruction.8
Each immune cell player can contribute to the pathogenesis of RA. For instance, M1 macrophages play a critical role in the production of several proinflammatory cytokines such as TNF-α, IL-6, IL-12, IL-23, IL-1β and IL-18,15 which promote the production of other mediators from different cell types including endothelial cells and fibroblast-like synoviocytes.16 Other innate immune cell players are neutrophils which release high levels of ROS, TNF-α, proteases, and defensins in RA joints. Various subtypes of T cells, including Th1, Th2 and Th17, take parts in immune-mediated inflammation of RA, where they become activated and then accumulate in the inflamed joints.17–19 On the other hand, regulatory T cells (Tregs) have been described to suppress disease severity in collagen induced arthritis (CIA) animal models, and this could explain the finding that Tregs are decreased in the peripheral blood of RA patients.20,21 One important arm in immune-mediated pathogenesis of RA includes B cells that react against citrullinated antigens and release antibodies that contribute to the initiation and persistence of the inflammatory process. The crosstalk among various immune cells whether via cell to cell contact or the release of mediators, is a critical aspect of the inflammatory process observed in RA. For example, activated Th17 cells are involved in the induction of inflammation by stimulating neutrophils, causing their chemoattraction into the joints.22–24 Furthermore, neutrophils play a crucial role in the activation of NK cells, as their depletion has led to impairment in the function and homeostasis of NK cells.25
Chemokines
Chemokines are chemotactic cytokines that regulate the migration of immune cells in various physiological and pathological processes. They play a crucial role in homeostasis, generation of cellular and humoral immune responses, as well as pathologic immune contribution in various diseases. Chemokines consist of a large family of more than 50 chemokine ligands and receptors, that are classified based on the assembly of cysteine residues in their primary amino acid sequence.26 Their nomenclature is based on the arrangement of the two cysteine residues dividing them into four subfamilies: CC, CXC, CX3C, and XC.27,28 In CC chemokines, the cysteine residues are next to each other, while CXC chemokines have one varying amino acid between them. On the other hand, the CX3C chemokines have three variable amino acids between these two cysteine residues, and the XC chemokines have only one cysteine amino acids.26,29 Almost all chemokine ligands are secreted from the cells with the exception of CX3CL1 and CXCL16, that have a transmembrane domain to keep the chemokines at the surface, that can be later cleaved to release the chemokine portion into the extracellular space.30,31
Chemokine receptors are expressed on all leukocytes and can be classified into two groups: 1. serpentine G protein–coupled chemokine receptors (GPCRs), and 2. atypical chemokine receptors.32 Many chemokine ligands can bind to multiple receptors, while some receptors have many ligands, especially with chemokines involved in inflammatory processes. Atypical chemokine receptors (ACKRs) are involved in regulating chemokine distribution and localization.33 These receptors play a vital role in the regulation of hemopoietic stem and progenitor cells in addition to acting as chemokine scavengers that internalize and degrade chemokines.34,35
Chemokines have several functions primarily leukocyte migration, cell proliferation, survival, differentiation, degranulation, and cytokine production. Additionally, many chemokines were shown to possess angiogenic or anti-angiogenic activities.36 Leukocyte migration is required and necessary for rapid employment of innate immune cells in order to kill pathogens, prevent microbial infection, and drive inflammation as an attempt to repair the damage.37 Furthermore, chemokines help in the lymphoid organization, regulation of the adaptive immune response, and the consequent immune memory development.38,39 Any imbalance in the chemokine system could lead to failure of immunosurveillance that can trigger diseases including autoimmunity, chronic inflammatory disease, allergy, cancer, and atherosclerosis.40,41
Chemokines in Rheumatoid Arthritis
Chronic inflammation represented in RA synovium is due to the release of a variety of mediators, including chemokines, cytokines, matrix metalloproteinases (MMPs), and growth factors, thus causing continuous activation of innate and adaptive immune systems, as illustrated in Figure 1. Extravasation of inflammatory T cells into the synovium is a crucial event in the pathogenesis of RA. Furthermore, chemokine receptors and ligands have been implicated in different processes of RA development including inflammation and angiogenesis.42,43 Numerous studies have confirmed the critical role of chemokines for Th1 cell migration into the synovium where chemokine ligands are abundantly present.39,44 Blocking these chemokine receptors has led to inhibition of inflammatory Th1 cells resulting in decreased synovitis. Neutrophils produce chemokines such as CXCL2 and CCL3 as well as trigger the production of chemokines including CXCL1, CXCL5 and CCL9 from fibroblast-like synoviocytes, endothelial cells, and macrophages.45
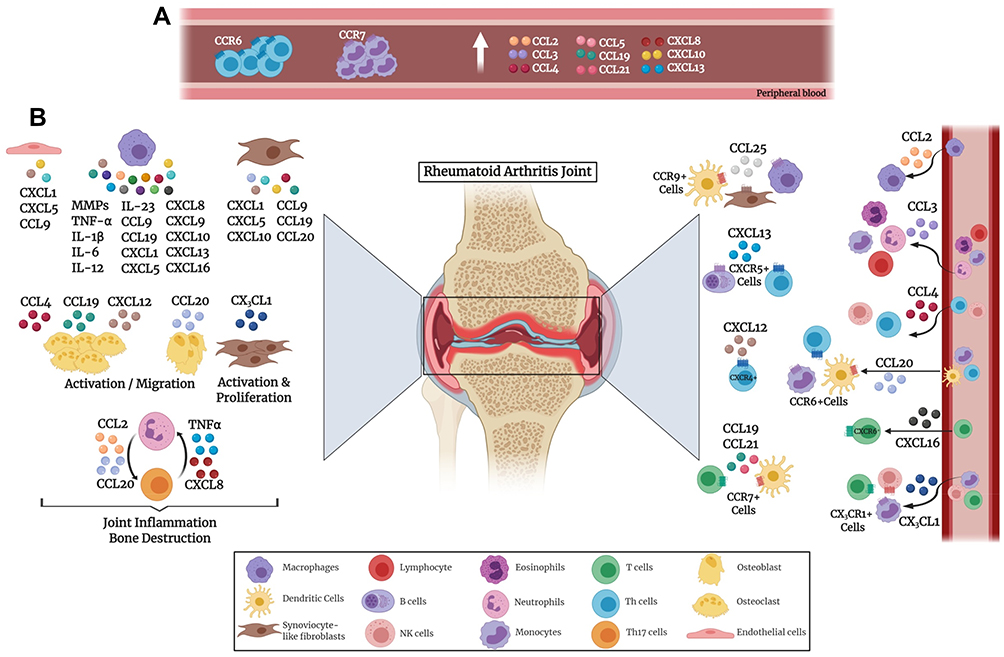 |
Figure 1 Chemokine involvement in the pathogenesis of rheumatoid arthritis. Various immune cells secrete chemokines that affect the joints. (A) Chemokines secreted in the peripheral blood by immune cells. (B) Chemokines secreted inside the joints by various immune and non-immune cells. |
Chemokine production has been reported to vary at different stages of RA. At an early phase, CCL4, CXCL4, CXCL7 and CXCL13 were expressed, whereas CCL3 and CCL9 were released at later stages.46,47 Other chemokines, including CXCL1 and CXCL5, promote inflammation and hence their levels keep escalating by persistent inflammation.45 It is worth mentioning that citrullination of chemokines (e.g. CXCL5 and CCL2) occurs in RA and has been observed in synovial fluid of RA patients. Citrullinated chemokines possess an alteration in their activities which leads to reduction in chemotaxis.48
RA patients exhibit increased levels of CCL2, CCL3, CCL4, and CXCL10 in plasma as well as synovial fluid.49–53 On the other hand, CCL5 showed an increased level in the plasma but a reduction in synovial fluid of RA patients.49 CCL2 is a potent chemoattractant for macrophages, while CCL3 recruits various lymphocytes, monocytes, and eosinophils.54 Elevated CCL2 level in RA has been strongly associated with an increase in joint infiltration by immune cells, specifically macrophages.55 Moreover, high levels of CCL3 were found in neutrophils isolated from synovial fluid.56 CCL4 has been found to be an important regulator for osteoclast migration indicating that it is a potential therapy target for bone resorptive diseases.57 A study reported that CXCL10, CCL5 and CXCL8 chemokines were elevated in the plasma of patients with active RA similar to Th1 associated proinflammatory cytokines TNFα, and IL-6.58 CXCL10 is primarily secreted by fibroblast-like synoviocytes and infiltrating macrophages in the synovium. Another study reported that there is a strong association between serum CXCL10 and disease activity scores (DAS) indicating that this chemokine can be a possible biomarker and diagnostic aid in monitoring disease progression in RA patients.59 CXCR3 is expressed primarily on NK cells60 and activated T lymphocytes especially the inflammatory Th1 cells, that secrete high levels of IFN-gamma.60,61 It has been stated that CXCR3 knockout mice would be more resistant to inflammatory autoimmune diseases.62 The interaction between CXCR3 and its ligands CXCL9, CXCL10 and CXCL11, would lead to migration of these Th1 cells such as those present in the synovial tissues of RA patients.63–65
As mentioned earlier, the interplay between immune cells consists of key players involved in the pathogenesis of RA, including neutrophils, B cells, T cells and macrophages. Th17 cells are known to generate cytokines and chemokines such as TNF-α and CXCL8 that attract neutrophils.22,24 Mutually, neutrophils further activate Th17 cells through the secretion of CCL2 and CCL20 chemokines.66 M1 macrophages secrete proinflammatory cytokines and chemokines such as TNFα, IL-1β, IL-6, IL-12, IL-23, CXCL5, CXCL8, CXCL9, CXCL10, and CXCL13 which recruit more leukocytes thus promoting RA and leading to joint destruction (Figure 1).22,67
It is well known that leukocyte migration to the joints is one of the main causes of RA pathogenesis. CCR7 was found to aid in the guidance of antigen presenting dendritic cells and T cells to the inflamed synovium and thus contributing to RA pathogenesis. This infiltration in the synovial tissues is partially attributed to CCL21 and its receptor CCR7, where their blockage led to prevention of the migration.68 It has been reported that plasma CCL19 level and monocyte CCR7 surface expression were higher in RA patients.69,70 Likewise, CCL19 and CCL21 were reported to be higher in RA patients in comparison to osteoarthritis individuals.69,71 Also, CCL19 has been found at high concentrations in the synovium where it was reported to be expressed by fibroblasts and macrophages.69,70 In addition, inflammatory molecules such as lipopolysaccharide, TNF-α and IL-1β were found to promote CCR7 expression.71 Moreover, CCL19 and CCL21 stimulated osteoclast migration as well as bone resorption by osteoclasts in an animal model of RA,71 and CCL21 drives osteoclastogensis in RA through M1 macrophage polarization of Th17 cells as well as neovascularization.68 CCL25 and its receptor CCR9 were both detected in the RA synovium. CCR9 was reported to be expressed on dendritic cells, macrophages, and fibroblast-like synoviocytes. In addition, stimulation with CCL25 led to the secretion of proinflammatory cytokines IL-6 and TNF-α from peripheral blood monocytes.72
A subgroup of T helper cells expressing CCR6 was detected in the peripheral blood, synovial fluid and inflamed synovial tissue of RA patients.73 These CCR6+ Th cells can be further classified to Th17, Th22, and Th17.1 which were identified to be pathogenic as they lead to the progression of chronic arthritis.74–78 A proportion of peripheral T memory cells were reported to be CCR6+, highlighting the significance of CCR6-CCL20 axis in cell migration to synovium in RA.79,80 It has been reported that the presence of the rheumatoid arthritis risk variant: dinucleotide polymorphism in the CCR6 gene (CCR6DNP genotype) is linked to RA susceptibility.81,82 CCL20 has been reported to be secreted by chondrocytes, synoviocytes and Th17 cells in the joints, as well as being able to activate osteoblasts.73,83–85 Also, CCL20 works in synergy with RANKL leading to bone resorption and destruction.83 Additionally, CCL20 contributes to T cells, monocytes and CD1a+ dendritic cells chemotaxis towards the joints.79,80,84,86
Regarding its pathogenic role in RA, CXCL12 was described to provoke osteoclastogensis by upregulating RANKL expression in synovial fibroblasts and CD4+ T cells, via TNF-α.87 CXCL12 receptor CXCR4 was correlated with the presence of synovial CD4+ T cells and thus could be associated with T cell migration and joint destruction.88,89 CXCL13 activates CXCR5 that is present on B cells and T helper cells attracting them to the follicles. CXCL13 which is known to play a critical role in RA pathogenesis, has been reported to be a novel biomarker for RA disease severity.46,90 It was found to be significantly higher in early compared to established RA. Moreover, its serum level significantly correlated with DAS28 score as well as RF and ACPAs.91 Therefore, CXCL13 can aid in the diagnosis of early RA with an enhanced diagnostic performance compared to rheumatoid factor (RF) and anti-cyclic citrullinated peptide (CCP).91 Serum baseline levels of both CXCL10 and CXCL13 were found to be elevated in RA patients, especially those that are RF and anti-CCP positive individuals.46,70,90,92,93 Additionally, serum CXCL13 was associated with inflammation, synovitis, RF and DAS28, which could be a predictor of high RA severity. Several studies have shown that CXCL16 is highly expressed in RA synovia, causing the recruitment and accumulation of CXCR6+ T cells in RA joints, which is highly associated with RA pathogenesis.94,95 It has been suggested that CXCL16 is released and expressed by synovial macrophages, where the expression was elevated by inflammatory TNF-α.95
CX3CL1 “Fractalkine”, that is present in RA synovium,96 was found to play a crucial role in monocyte chemotaxis and angiogenesis in the rheumatoid synovium. The interaction of CX3CL1 and its receptor CX3CR1 contributes to the recruitment of various immune cells including T cells, monocytes and NK cells causing inflammatory autoimmune diseases such as RA and SLE.96–99 Moreover, stimulation by CX3CL1 was found to trigger the proliferation of fibroblasts and atherosclerosis as well as leading to an increased MMPs and further inflammation in RA joints.100–104
Multiple studies in RA have been carried out with antagonists and/or neutralizing antibodies against chemokines including CCR1, CCR5, CXCR4, CXCR7, CCL19, CXCL10, CXCL12, and CXCL13, where they revealed potential to be future targets.105,106 However, further understanding of the role of these chemokines in RA is quite essential.
Effect of Chemokines in RA Therapy
It has been noted that chemical and biologic DMARDs affect chemokine expression in RA. Many studies have shown that NSAIDs, glucocorticoids and DMARDs (sulfasalazine, sulfa pyridine, methotrexate, and leflunomide) hinder the production of numerous chemokines in various clinical setups,107–111 as summarized in Figure 2. RA patients treated with biological DMARDs such as infliximab, etanercept and tocilizumab exhibited a significant reduction in serum CCL20 levels compared to before treatment.112 Furthermore, the expression of CCR7 and CCL19 chemokines reversed to normal baseline levels after 1 year of DMARD methotrexate and cyclosporine A therapy.69 Similarly, levels of CCL5 and CCL19 were reported to be decreased by rituximab and anti-TNF therapies, respectively.69,70,113 Hence, blocking CCR7 and its ligands CCL19 and CCL21 could be a therapeutic approach targeting inflammation and bone destruction in RA. Besides, serum CCL19 was reported to be a promising predictive diagnostic tool for effective response to rituximab therapy.70
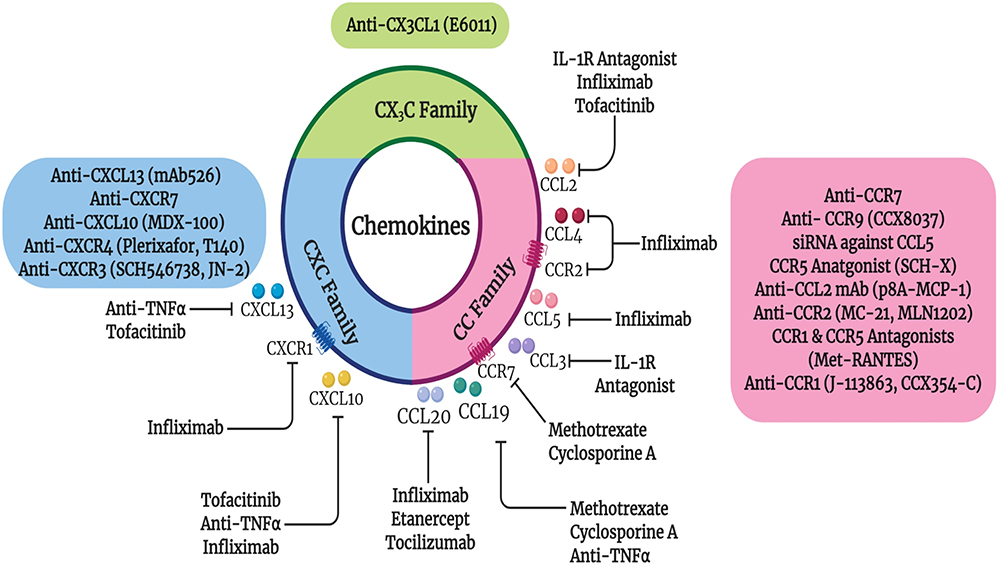 |
Figure 2 Effect of current RA therapies on chemokines as well as the current chemokine targeted therapies in RA. Closed arrows indicate antagonism. |
Additionally, RA patients that responded to IL-1 receptor antagonist (IL-1Ra) therapy had significantly lower mean changes in the serum CCL2 and CCL3 levels compared to non-responders or placebo. This indicates that CCL2 and CCL3 may be convenient markers for IL-1Ra efficient treatment.51 Likewise, chemokine ligands such as CXCL10, CCL2, and CCL4 levels decreased significantly upon treatment with infliximab along with a reduction of chemokine receptors CCR2 and CXCR1 on T cells.52 Furthermore, RA patients that responded to TNF inhibitors had higher baseline serum levels of CXCL10 and CXCL13, that were reduced after therapy.46,92,106 Another class of therapy, Janus kinase (JAK) inhibitor Tofacitinib caused a significant reduction of synovial mRNA chemokine expression of CCL2, CXCL10 and CXCL13.114 Therefore, levels of CXCL10 and CXCL13 could be used as prognostic tools for biological TNF and JAK inhibitor therapy.46 Also, serum baseline levels of CCL2 and CXCL8 were found to be higher in the tocilizumab responders compared to non-responder RA patients. A closely related chemoattractant, macrophage migration inhibitory factor (MIF) level was reduced after tocilizumab treatment in RA responders, highlighting the importance of MIF in RA.115
Numerous chemokine inhibitors targeting CC or CXC receptors have been investigated, with controversial outcomes, especially in autoimmune diseases.116–120 For instance, CXCR4 antagonist Plerixafor and the CCR4 monoclonal antibody Mogamulizumab, have been utilized in the mobilization of stem cells in the treatment of non-Hodgkin lymphomas and T cell leukemias, respectively.105 Additionally, CCR5 antagonists Aplaviroc, Maraviroc and Vicriviroc have been utilized for the treatment of HIV infection.121 Another CCR9 antagonist Vercirnon, displayed promising results in Crohn’s disease.72 Interestingly, antibodies against CXCL12 and CXCL13 showed beneficial effects in vitro and in animal models of cancer and inflammatory diseases including collagen induced arthritis (CIA).122,123 Currently, many studies and clinical trials have investigated chemokine targeting in autoimmune diseases including RA (Figure 2).118,124–131
Another autoimmune disease, Sjögren’s Syndrome (SS), occurs when the immune system attacks exocrine glands. Similar to RA, SS is a debilitating progressive disease. CXCL13 expression was found to be increased in salivary tissue and associated with disease progression in SS mouse model.132 In addition, CXCL13 was described to be elevated in the serum and saliva of SS patients, highlighting its importance in SS.132 Studies have reported that blockage of CXCL13 reduced glandular inflammation and hence could be an effective therapeutic strategy in SS mouse model as well as in SS patients.132,133 Besides, mAb 526, an anti-CXCL13 antibody, has shown therapeutic efficacy in various autoimmune diseases including mice models of RA or CIA as well as multiple sclerosis model (experimental autoimmune encephalomyelitis or EAE).134 In addition, inhibition of CXCR7, another receptor for CXCL12, decreased inflammation in the joints and reduced angiogenesis in CIA model.135
A recent study reported engineered nanobodies that target CCR7 which can be used for therapeutic and diagnostic purposes.136 Another study indicated that both prophylactic and therapeutic treatments of CIA-humanized CCR7 mice with anti-human CCR7 monoclonal antibody led to complete resistance to CIA and arrested progression, thus highlighting that CCR7 could be a potential therapeutic target in RA.136
One potential therapeutic target in RA is CX3CL1 which has been blocked in a clinical trial using a monoclonal antibody as a treatment for RA.137 The humanized anti-CX3CL1 monoclonal antibody E6011 was found to be safe, well tolerated, and exhibited efficacy for 52 weeks in active RA patients who were non-responders or intolerant to MTX or TNF inhibitor therapies.138,139 Moreover, E6011 was found to hinder the migration of CX3CR1+ macrophages as well as blocking joint destruction via reducing inflammatory cytokines and suppressing osteoclastogenesis.139,140 All of these data support the utilization of anti-CX3CL1 monoclonal antibody in Phase 2 clinical trials for further assessment.
Different CXCR4 antagonists have shown positive results in tempering synovitis in animal models of arthritis including Plerixafor and T140.141,142 Previously, SCH546738, a synthetic compound targeting CXCR3, was found to weaken the development of CIA in mice.143 Furthermore, a CXCR3 antagonist JN-2, was reported to improve arthritis symptoms in a CIA animal model.144 This was suggested to be due to inhibiting cell migration and pro-inflammatory cytokine expression from macrophages and CD4+ T cells.144 A study by Broeren et al reported that CXCR3 ligand, CXCL10 increases inflammatory mediators which are present in the serum of patients with RA.145 The CXCL10 promoter-regulated IL-10 overexpression was described to lead to a reduction in inflammatory cytokine production. For that reason, this vector was suggested to provide a possible gene therapy approach for RA.145 Additionally, the use of blocking monoclonal antibody against CXCL10 as therapy for arthritis led to halting its progression.146,147 Clinical trials using anti-CXCL10 monoclonal antibody (MDX-1100) for RA patients with an ineffective response to methotrexate showed that blocking CXCL10 significantly improved the response rate, suggesting a possible therapeutic use in humans.148 This has been supported by a decrease in the levels of C-reactive protein and disease activity score (DAS) as well as an improvement in the ACR20 (i.e. 20% improvement in RA symptoms) and physical function.148
CCR9 antagonist, CCX8037 or knockdown of CCR9 repressed arthritis symptoms in mice.72 A neutralizing mAb against CCL2 was shown to reduce ankle swelling along with a decrease in the number of monocytes/macrophages recruited to the joints.149 While the treatment with a small-molecule inhibitor of endogenous CCL2 (p8A-MCP-1) displayed a positive clinical efficacy on adjuvant-induced arthritis (AIA),150 its receptor CCR2 is expressed by CD14+ monocytes, demonstrating a vital role in monocyte recruitment during CIA.149 Consequently, low doses of a monoclonal antibody against CCR2, the MC-21 showed some improvement in CIA.151 On the other hand, administration of an anti-CCR2 antibody (MLN1202) was implemented in humans with no observed reduction in the numbers of inflammatory cells.126,152 Another possible chemokine target was a highly abundant chemokine in RA synovium, i.e. CCR5 that is expressed by inflammatory Th1 cells and tissue-resident macrophages.107,153,154 Local injection of small interfering RNA (siRNA) against CCR5 in a rat model of adjuvant-induced arthritis was found to repress joint inflammation and swelling, highlighting that CCR5 inhibition may be a promising target for therapy.155 Additionally, CCR5 antagonist SCH-X was found to hinder the development of CIA.156 A single nucleotide polymorphism of CCR5 influenced RA severity and immune infiltration to joints where individuals bearing the unfunctional Δ32-CCR5 variant, were more likely to develop less severe RA symptoms.157 However, CCR5 antagonists such as Maraviroc and AZD5672 did not exhibit any clinical efficacy in RA patients in terms of ACR20 response.158 Dual antagonists have been under development, where dual targeting of CCR1 and CCR5 via Met-RANTES caused prevention of arthritis in CIA and AIA animal models.159,160
Several studies suggested that CCR2 and CCR5 are not as critical as CCR1 for the migration of monocytes towards the synovial compartment in RA.127,131 As mentioned earlier, CCR1 levels are elevated on several leukocytes present in the RA synovium, along with its multiple ligands.130,161 This has been supported by immunohistochemistry findings reporting the presence of CCR1+ cells promoting inflammatory monocyte infiltration into RA synovial tissue.162 The CCR1 antagonist J-113863 displayed a positive clinical efficacy in murine CIA.163 However, in the past, Phase II clinical trials using oral CCR1 antagonists, such as CP-481,715 (Pfizer), MLN3897 (Millennium), BMS-817399 (Bristol-Myers Squibb) and c-4462 failed to induce noticeable activities in RA patients.118,124,164–166 This could be attributed to persistent high level of receptor blockage which may be necessary to inhibit monocyte migration in the synovium. Another possibility could be that CCR1 ligands can interact with multiple receptors including CCR2 and CCR5,164 mediating their effects in RA. However, a recent clinical trial using an oral CCR1 antagonist, CCX354-C was performed to evaluate its safety and efficacy in RA patients.167 The clinical trial so called CARAT-2 reported that CCX354-C revealed good safety and tolerability profiles that can be implemented in clinical aspects in RA.167 A slight elevation in the ACR20 was observed in RA patients using CCX354-C but the difference was not significant. Nevertheless, treatment with CCX354-C revealed more than 90% receptor occupancy that was required for effective blockade of infiltration of inflammatory cells.131,168
Targeting chemokines as a therapeutic approach has shown several obstacles that might hinder their development and approval in various diseases. One of the main reason for the failure is that chemokine receptors are not cell-specific and can be shared by several inflammatory and anti–inflammatory cells, making their blockage difficult to reach reasonable therapeutic effects.169,170 Furthermore, many chemokines are engaged in physiological and developmental processes where any intervention could lead to undesirable adverse effects.54,94,107,171,172 It is worth mentioning that single targeting is not quite effective as the binding affinities are much limited in vivo compared to in vitro studies.173–175 Additionally, the use of animal models could create difficulties, as the affinity of a compound for a rodent chemokine receptor can differ noticeably from its affinity for the equivalent human chemokine. Also, as mentioned earlier, chemokines could become citrullinated in RA and hence, they might not be inhibited by chemokine blockers that are designed for unmodified chemokines.48 Therefore, it is vital to find the appropriate chemokine targets in order to be able to succeed in blocking pathogenic lymphocyte recruitment to the RA synovium.
Conclusions
Chemokine signaling has shown to be critical in RA pathogenesis, as several chemokines and their respective receptors contribute to immune cell recruitment in arthritic joints. Therefore, targeting chemokines could be a suitable therapeutic approach in the treatment of RA. Nevertheless, many studies with antagonists and antibodies directed against chemokines and chemokine receptors failed upon translation into clinical trials. Currently, the development of more effective chemokine therapies is underway, where it would provide new opportunities for clinical trials for the treatment of RA and similar autoimmune diseases. We aim from this article to shed some lights on the importance of chemokines and chemokine receptors in RA disease. This information should provide a solid background for developing new drugs or other therapeutic modalities to target chemokine or their receptors due to their vital importance in disease initiation, progression and development.
Abbreviations
ACKRs, Atypical chemokine receptors; ACPAs, Anti–citrullinated protein antibodies; ACR20, American College of Rheumatology 20; CCP, Cyclic citrullinated peptide; CD, Cluster of Differentiation; CIA, Collagen-induced arthritis; DAS, Disease Activity Score; DMARDs, Disease Modifying Anti Rheumatic Drugs; DNP, Dinucleotide polymorphism; EAE, Experimental Autoimmune Encephalomyelitis; GPCRs, G-protein Coupled Receptor; IL, Interleukin; JAK, Janus Kinase; MIF, Migration inhibitory factor; MMPs, Matrix Metalloproteinases; MTX, Methotrexate; NK, Natural Killer; RA, Rheumatoid Arthritis; RANKL, Receptor activator of nuclear factor kappa-Β ligand/ tumor necrosis factor ligand superfamily member 11; RF, Rheumatoid Factor; ROS, Reactive Oxygen Species; siRNA, Small interfering RNA; SS, Sjogren’s Syndrome; SLE, Systemic Lupus Erythematosus; Th, T helper; TNF-α, Tumor Necrosis Factor alpha; Tregs, Regulatory T cells.
Acknowledgments
Work done in the authors’ laboratory is supported by University of Sharjah grant numbers 1701090222-P and 1701090223-P, and by Terry-Fox Foundation.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The authors report no conflicts of interest in this work.
References
1. Barturen G, Beretta L, Cervera R, Van Vollenhoven R, Alarcón-Riquelme ME. Moving towards a molecular taxonomy of autoimmune rheumatic diseases. Nat Rev Rheumatol. 2018;14:75. doi:10.1038/nrrheum.2017.220
2. Pincus T, Brooks RH, Callahan LF. Prediction of long-term mortality in patients with rheumatoid arthritis according to simple questionnaire and joint count measures. Ann Intern Med. 1994;120(1):26–34. doi:10.7326/0003-4819-120-1-199401010-00005
3. Silman AJ, Pearson JE. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002;4(Suppl 3):S265–S272. doi:10.1186/ar578
4. Aletaha D, Smolen JS. Diagnosis and management of rheumatoid arthritis: a review. JAMA. 2018;320(13):1360–1372. doi:10.1001/jama.2018.13103
5. Young A, Koduri G, Batley M, et al. Mortality in rheumatoid arthritis. Increased in the early course of disease, in ischaemic heart disease and in pulmonary fibrosis. Rheumatology. 2007;46(2):350–357. doi:10.1093/rheumatology/kel253
6. Gabriel Sherine E, Crowson Cynthia S, Kremers Hilal M, et al. Survival in rheumatoid arthritis: a population‐based analysis of trends over 40 years. Arthritis Rheum. 2003;48(1):54–58. doi:10.1002/art.10705
7. Malviya G, Salemi S, Laganà B, Diamanti AP, D’Amelio R, Signore A. Biological therapies for rheumatoid arthritis: progress to date. BioDrugs. 2013;27(4):329–345. doi:10.1007/s40259-013-0021-x
8. Chen S-J, Lin G-J, Chen J-W, et al. Immunopathogenic mechanisms and novel immune-modulated therapies in rheumatoid arthritis. Int J Mol Sci. 2019;20(6):1332. doi:10.3390/ijms20061332
9. Scherer HU, Huizinga TWJ, Krönke G, Schett G, Toes REM. The B cell response to citrullinated antigens in the development of rheumatoid arthritis. Nat Rev Rheumatol. 2018;14:157. doi:10.1038/nrrheum.2018.10
10. Smolen JS, van der Heijde D, Machold KP, Aletaha D, Landewé R. Proposal for a new nomenclature of disease-modifying antirheumatic drugs. Ann Rheum Dis. 2014;73(1):3. doi:10.1136/annrheumdis-2013-204317
11. Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960. doi:10.1136/annrheumdis-2016-210715
12. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. doi:10.1038/nature01661
13. Alam J, Jantan I, Bukhari SNA. Rheumatoid arthritis: recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed Pharmacother. 2017;92:615–633. doi:10.1016/j.biopha.2017.05.055
14. Holmdahl R, Malmström V, Burkhardt H. Autoimmune priming, tissue attack and chronic inflammation — the three stages of rheumatoid arthritis. Eur J Immunol. 2014;44(6):1593–1599. doi:10.1002/eji.v44.6
15. Sidiropoulos PI, Goulielmos G, Voloudakis GK, Petraki E, Boumpas DT. Inflammasomes and rheumatic diseases: evolving concepts. Ann Rheum Dis. 2008;67(10):1382. doi:10.1136/ard.2007.078014
16. Romano M, Sironi M, Toniatti C, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6(3):315–325. doi:10.1016/S1074-7613(00)80334-9
17. Jung MS, Lee J, Baek YS, et al. Fraxinellone attenuates rheumatoid inflammation in mice. Int J Mol Sci. 2018;19(3):829. doi:10.3390/ijms19030829
18. Myers LK, Stuart JM, Kang AH. A CD4 cell is capable of transferring suppression of collagen-induced arthritis. J Immunol. 1989;143(12):3976–3980.
19. Pöllinger B. IL-17 producing T cells in mouse models of multiple sclerosis and rheumatoid arthritis. J Mol Med (Berl). 2012;90(6):613–624. doi:10.1007/s00109-011-0841-4
20. Kelchtermans H, Geboes L, Mitera T, Huskens D, Leclercq G, Matthys P. Activated CD4+CD25+ regulatory T cells inhibit osteoclastogenesis and collagen-induced arthritis. Ann Rheum Dis. 2009;68(5):744. doi:10.1136/ard.2007.086066
21. Dulic S, Vásárhelyi Z, Sava F, et al. T-Cell subsets in rheumatoid arthritis patients on long-term anti-TNF or IL-6 receptor blocker therapy. Mediators Inflamm. 2017;2017:6894374. doi:10.1155/2017/6894374
22. Navegantes KC, de Souza Gomes R, Pereira PAT, Czaikoski PG, Azevedo CHM, Monteiro MC. Immune modulation of some autoimmune diseases: the critical role of macrophages and neutrophils in the innate and adaptive immunity. J Transl Med. 2017;15(1):36. doi:10.1186/s12967-017-1141-8
23. Milanova V, Ivanovska N, Dimitrova P. TLR2 elicits IL-17-mediated RANKL expression, IL-17, and OPG production in neutrophils from arthritic mice. Mediators Inflamm. 2014;2014:14. doi:10.1155/2014/643406
24. Pelletier M, Maggi L, Micheletti A, et al. Evidence for a cross-talk between human neutrophils and Th17 cells. Blood. 2010;115(2):335–343. doi:10.1182/blood-2009-04-216085
25. Jaeger BN, Donadieu J, Cognet C, et al. Neutrophil depletion impairs natural killer cell maturation, function, and homeostasis. J Exp Med. 2012;209(3):565–580. doi:10.1084/jem.20111908
26. Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J. 2018;285(16):2944–2971. doi:10.1111/febs.14466
27. Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12(2):121–127. doi:10.1016/S1074-7613(00)80165-X
28. Murphy PM, Baggiolini M, Charo IF, et al. International union of pharmacology. XXII. Nomenclature for chemokine receptors. Pharmacol Rev. 2000;52(1):145.
29. Miller MC, Mayo KH. Chemokines from a structural perspective. Int J Mol Sci. 2017;18(10):2088. doi:10.3390/ijms18102088
30. Bazan JF, Bacon KB, Hardiman G, et al. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385(6617):640–644. doi:10.1038/385640a0
31. Matloubian M, David A, Engel S, Ryan JE, Cyster JG. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol. 2000;1(4):298–304. doi:10.1038/79738
32. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32(1):659–702. doi:10.1146/annurev-immunol-032713-120145
33. Nibbs RJ, Graham GJ. Immune regulation by atypical chemokine receptors. Nat Rev Immunol. 2013;13(11):815–829. doi:10.1038/nri3544
34. Weber M, Blair E, Simpson CV, et al. The chemokine receptor D6 constitutively traffics to and from the cell surface to internalize and degrade chemokines. Mol Biol Cell. 2004;15(5):2492–2508. doi:10.1091/mbc.e03-09-0634
35. Fra AM, Locati M, Otero K, et al. Cutting edge: scavenging of inflammatory CC chemokines by the promiscuous putatively silent chemokine receptor D6. J Immunol. 2003;170(5):2279. doi:10.4049/jimmunol.170.5.2279
36. Ridiandries A, Tan JTM, Bursill CA. The role of CC-chemokines in the regulation of angiogenesis. Int J Mol Sci. 2016;17(11):1856. doi:10.3390/ijms17111856
37. Sokol CL, Luster AD. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7(5):a016303. doi:10.1101/cshperspect.a016303
38. Lian J, Luster AD. Chemokine-guided cell positioning in the lymph node orchestrates the generation of adaptive immune responses. Curr Opin Cell Biol. 2015;36:1–6. doi:10.1016/j.ceb.2015.05.003
39. Comerford I, Kara EE, McKenzie DR, McColl SR. Advances in understanding the pathogenesis of autoimmune disorders: focus on chemokines and lymphocyte trafficking. Br J Haematol. 2014;164(3):329–341. doi:10.1111/bjh.2014.164.issue-3
40. Rolin J, Maghazachi AA. Implications of chemokines, chemokine receptors, and inflammatory lipids in atherosclerosis. J Leukoc Biol. 2014;95(4):575–585. doi:10.1189/jlb.1113571
41. Rolin J, Maghazachi AA. Implications of chemokine receptors and inflammatory lipids in cancer. Immunotargets Ther. 2013;3:9–18. doi:10.2147/ITT.S32049
42. Szekanecz Z, Kim J, Koch AE. Chemokines and chemokine receptors in rheumatoid arthritis. Semin Immunol. 2003;15(1):15–21. doi:10.1016/S1044-5323(02)00124-0
43. Koch AE. Chemokines and their receptors in rheumatoid arthritis: future targets? Arthritis Rheum. 2005;52(3):710–721. doi:10.1002/(ISSN)1529-0131
44. White GE, Iqbal AJ, Greaves DR. CC chemokine receptors and chronic inflammation—therapeutic opportunities and pharmacological challenges. Pharmacol Rev. 2013;65(1):47. doi:10.1124/pr.111.005074
45. Chou RC, Kim ND, Sadik CD, et al. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33(2):266–278. doi:10.1016/j.immuni.2010.07.018
46. Greisen SR, Schelde KK, Rasmussen TK, et al. CXCL13 predicts disease activity in early rheumatoid arthritis and could be an indicator of the therapeutic `window of opportunity’. Arthritis Res Ther. 2014;16(5):434. doi:10.1186/s13075-014-0434-z
47. Yeo L, Adlard N, Biehl M, et al. Expression of chemokines CXCL4 and CXCL7 by synovial macrophages defines an early stage of rheumatoid arthritis. Ann Rheum Dis. 2016;75(4):763. doi:10.1136/annrheumdis-2014-206921
48. Yoshida K, Korchynskyi O, Tak PP, et al. Citrullination of epithelial neutrophil–activating peptide 78/CXCL5 results in conversion from a non–monocyte-recruiting chemokine to a monocyte-recruiting chemokine. Arthritis Rheumatol. 2014;66(10):2716–2727. doi:10.1002/art.v66.10
49. Sucur A, Jajic Z, Artukovic M, et al. Chemokine signals are crucial for enhanced homing and differentiation of circulating osteoclast progenitor cells. Arthritis Res Ther. 2017;19(1):142. doi:10.1186/s13075-017-1337-6
50. Chen C-Y, Fuh L-J, Huang -C-C, et al. Enhancement of CCL2 expression and monocyte migration by CCN1 in osteoblasts through inhibiting miR-518a-5p: implication of rheumatoid arthritis therapy. Sci Rep. 2017;7(1):421. doi:10.1038/s41598-017-00513-0
51. Bao J, Liu W, Bao Y-X. Recombinant human interleukin receptor antagonist influences serum chemokines in patients with rheumatoid arthritis. Cent Eur J Immunol. 2014;39(2):170–173. doi:10.5114/ceji.2014.43717
52. Eriksson C, Rantapää-Dahlqvist S, Sundqvist KG. Changes in chemokines and their receptors in blood during treatment with the TNF inhibitor infliximab in patients with rheumatoid arthritis. Scand J Rheumatol. 2013;42(4):260–265. doi:10.3109/03009742.2012.754937
53. Katrib A, Tak PP, Bertouch JV, et al. Expression of chemokines and matrix metalloproteinases in early rheumatoid arthritis. Rheumatology. 2001;40(9):988–994. doi:10.1093/rheumatology/40.9.988
54. Szekanecz Z, Vegvari A, Szabo Z, Koch AE. Chemokines and chemokine receptors in arthritis. Front Biosci (Schol Ed). 2010;2:153–167. doi:10.2741/s53
55. Koch AE, Kunkel SL, Harlow LA, et al. Enhanced production of monocyte chemoattractant protein-1 in rheumatoid arthritis. J Clin Invest. 1992;90(3):772–779. doi:10.1172/JCI115950
56. Hatano Y, Kasama T, Iwabuchi H, et al. Macrophage inflammatory protein 1 alpha expression by synovial fluid neutrophils in rheumatoid arthritis. Ann Rheum Dis. 1999;58(5):297–302. doi:10.1136/ard.58.5.297
57. Xuan W, Feng X, Qian C, et al. Osteoclast differentiation gene expression profiling reveals chemokine CCL4 mediates RANKL-induced osteoclast migration and invasion via PI3K pathway. Cell Biochem Funct. 2017;35(3):171–177. doi:10.1002/cbf.3260
58. Bahlas S, Damiati L, Dandachi N, Sait H, Alsefri M, Pushparaj PN. Rapid immunoprofiling of cytokines, chemokines and growth factors in patients with active rheumatoid arthritis using luminex multiple analyte profiling technology for precision medicine. Clin Exp Rheumatol. 2019;37(1):112–119.
59. van Hooij A, Boeters DM, Tjon Kon Fat EM, et al. Longitudinal IP-10 serum levels are associated with the course of disease activity and remission in patients with rheumatoid arthritis. Clin Vaccine Immunol. 2017;24(8):e00060–e00017. doi:10.1128/CVI.00060-17
60. Loetscher M, Loetscher P, Brass N, Meese E, Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28(11):3696–3705. doi:10.1002/(ISSN)1521-4141
61. Má G-L, Sánchez-Madrid F, Rodríguez-Frade JM, et al. CXCR3 chemokine receptor distribution in normal and inflamed tissues: expression on activated lymphocytes, endothelial cells, and dendritic cells. Lab Invest. 2001;81(3):409–418.
62. Karin N, Razon H. Chemokines beyond chemo-attraction: CXCL10 and its significant role in cancer and autoimmunity. Cytokine. 2018;109:24–28. doi:10.1016/j.cyto.2018.02.012
63. Di Domenicantonio A. Rheumatoid arthritis and the alpha-chemokine IP-10. Clin Ter. 2014;165(6):e447–e451. doi:10.7417/CT.2014.1791
64. Qin S, Rottman JB, Myers P, et al. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J Clin Invest. 1998;101(4):746–754. doi:10.1172/JCI1422
65. Mohan K, Issekutz TB. blockade of chemokine receptor CXCR3 inhibits T cell recruitment to inflamed joints and decreases the severity of adjuvant arthritis. J Immunol. 2007;179(12):8463. doi:10.4049/jimmunol.179.12.8463
66. Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10(7):479–489. doi:10.1038/nri2800
67. Morand EF, Leech M. Macrophage migration inhibitory factor in rheumatoid arthritis. Front Biosci. 2005;10:12–22. doi:10.2741/1501
68. Van Raemdonck K, Umar S, Palasiewicz K, et al. CCL21/CCR7 signaling in macrophages promotes joint inflammation and Th17-mediated osteoclast formation in rheumatoid arthritis. Cell Mol Life Sci. 2019. doi:10.1007/s00018-019-03235-w
69. Ellingsen T, Hansen I, Thorsen J, et al. Upregulated baseline plasma CCL19 and CCR7 cell-surface expression on monocytes in early rheumatoid arthritis normalized during treatment and CCL19 correlated with radiographic progression. Scand J Rheumatol. 2014;43(2):91–100. doi:10.3109/03009742.2013.803149
70. Sellam J, Rouanet S, Hendel-Chavez H, et al. CCL19, a B Cell chemokine, is related to the decrease of blood memory B Cells and predicts the clinical response to rituximab in patients with rheumatoid arthritis. Arthritis Rheum. 2013;65(9):2253–2261. doi:10.1002/art.38023
71. Lee J, Park C, Kim HJ, et al. Stimulation of osteoclast migration and bone resorption by C-C chemokine ligands 19 and 21. Exp Mol Med. 2017;49(7):e358. doi:10.1038/emm.2017.100
72. Yokoyama W, Kohsaka H, Kaneko K, et al. Abrogation of CC chemokine receptor 9 ameliorates collagen-induced arthritis of mice. Arthritis Res Ther. 2014;16(5):445. doi:10.1186/s13075-014-0445-9
73. Hirota K, Yoshitomi H, Hashimoto M, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med. 2007;204(12):2803. doi:10.1084/jem.20071397
74. Paulissen SMJ, van Hamburg JP, Dankers W, Lubberts E. The role and modulation of CCR6+ Th17 cell populations in rheumatoid arthritis. Cytokine. 2015;74(1):43–53. doi:10.1016/j.cyto.2015.02.002
75. Maggi L, Santarlasci V, Capone M, et al. Distinctive features of classic and nonclassic (Th17 derived) human Th1 cells. Eur J Immunol. 2012;42(12):3180–3188. doi:10.1002/eji.201242648
76. Leipe J, Grunke M, Dechant C, et al. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010;62(10):2876–2885. doi:10.1002/art.v62:10
77. Nistala K, Moncrieffe H, Newton KR, Varsani H, Hunter P, Wedderburn LR. Interleukin-17–producing T cells are enriched in the joints of children with arthritis, but have a reciprocal relationship to regulatory T cell numbers. Arthritis Rheum. 2008;58(3):875–887. doi:10.1002/(ISSN)1529-0131
78. Li X, Yuan F-L, Lu W-G, et al. The role of interleukin-17 in mediating joint destruction in rheumatoid arthritis. Biochem Biophys Res Commun. 2010;397(2):131–135. doi:10.1016/j.bbrc.2010.05.111
79. Ruth JH, Shahrara S, Park CC, et al. Role of macrophage inflammatory protein-3α and its ligand CCR6 in rheumatoid arthritis. Lab Invest. 2003;83(4):579–588. doi:10.1097/01.LAB.0000062854.30195.52
80. Lee AYS, Körner H. CCR6 and CCL20: emerging players in the pathogenesis of rheumatoid arthritis. Immunol Cell Biol. 2014;92(4):354–358. doi:10.1038/icb.2013.97
81. Kochi Y, Okada Y, Suzuki A, et al. A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat Genet. 2010;42(6):515–519. doi:10.1038/ng.583
82. Stahl EA, Raychaudhuri S, Remmers EF, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet. 2010;42(6):508–514. doi:10.1038/ng.582
83. Lisignoli G, Piacentini A, Cristino S, et al. CCL20 chemokine induces both osteoblast proliferation and osteoclast differentiation: increased levels of CCL20 are expressed in subchondral bone tissue of rheumatoid arthritis patients. J Cell Physiol. 2007;210(3):798–806. doi:10.1002/(ISSN)1097-4652
84. Matsui T, Akahoshi T, Namai R, et al. Selective recruitment of CCR6-expressing cells by increased production of MIP-3 alpha in rheumatoid arthritis. Clin Exp Immunol. 2001;125(1):155–161. doi:10.1046/j.1365-2249.2001.01542.x
85. Haudenschild DR, Nguyen B, Chen J, D’Lima DD, Lotz MK. Rho kinase–dependent CCL20 induced by dynamic compression of human chondrocytes. Arthritis Rheum. 2008;58(9):2735–2742. doi:10.1002/art.v58:9
86. Page G, Lebecque S, Miossec P. Anatomic localization of immature and mature dendritic cells in an ectopic lymphoid organ: correlation with selective chemokine expression in rheumatoid synovium. J Immunol. 2002;168(10):5333–5341. doi:10.4049/jimmunol.168.10.5333
87. Kim H-R, Kim K-W, Kim B-M, Jung H-G, Cho M-L, Lee S-H. Reciprocal activation of CD4+ T cells and synovial fibroblasts by stromal cell–derived factor 1 promotes RANKL expression and osteoclastogenesis in rheumatoid arthritis. Arthritis Rheumatol. 2014;66(3):538–548. doi:10.1002/art.v66.3
88. Kanbe K, Chiba J, Inoue Y, Taguchi M, Yabuki A. SDF-1 and CXCR4 in synovium are associated with disease activity and bone and joint destruction in patients with rheumatoid arthritis treated with golimumab. Mod Rheumatol. 2016;26(1):46–50. doi:10.3109/14397595.2015.1054088
89. Buckley CD, Amft N, Bradfield PF, et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol. 2000;165(6):3423–3429. doi:10.4049/jimmunol.165.6.3423
90. Bugatti S, Manzo A, Vitolo B, et al. High expression levels of the B cell chemoattractant CXCL13 in rheumatoid synovium are a marker of severe disease. Rheumatology (Oxford). 2014;53(10):1886–1895. doi:10.1093/rheumatology/keu163
91. Allam SI, Sallam RA, Elghannam DM, El-Ghaweet AI. Clinical significance of serum B cell chemokine (CXCL13) in early rheumatoid arthritis patients. Egypt Rheumatol. 2019;41(1):11–14. doi:10.1016/j.ejr.2018.04.003
92. Han BK, Kuzin I, Gaughan JP, Olsen NJ, Bottaro A. Baseline CXCL10 and CXCL13 levels are predictive biomarkers for tumor necrosis factor inhibitor therapy in patients with moderate to severe rheumatoid arthritis: a pilot, prospective study. Arthritis Res Ther. 2016;18:93. doi:10.1186/s13075-016-0995-0
93. Jones JD, Hamilton BJ, Challener GJ, et al. Serum C-X-C motif chemokine 13 is elevated in early and established rheumatoid arthritis and correlates with rheumatoid factor levels. Arthritis Res Ther. 2014;16(2):R103–R103. doi:10.1186/ar4552
94. Nanki T, Shimaoka T, Hayashida K, Taniguchi K, Yonehara S, Miyasaka N. Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum. 2005;52(10):3004–3014. doi:10.1002/(ISSN)1529-0131
95. van der Voort R, van Lieshout AWT, Toonen LWJ, et al. Elevated CXCL16 expression by synovial macrophages recruits memory T cells into rheumatoid joints. Arthritis Rheum. 2005;52(5):1381–1391. doi:10.1002/art.21004
96. Ruth JH, Volin MV, Haines GK
97. Yoneda O, Imai T, Goda S, et al. Fractalkine-mediated endothelial cell injury by NK cells. J Immunol. 2000;164(8):4055–4062. doi:10.4049/jimmunol.164.8.4055
98. Choi J, Selmi C, Leung PSC, Kenny TP, Roskams T, Gershwin ME. Chemokine and chemokine receptors in autoimmunity: the case of primary biliary cholangitis. Expert Rev Clin Immunol. 2016;12(6):661–672. doi:10.1586/1744666X.2016.1147956
99. Yajima N, Kasama T, Isozaki T, et al. Elevated levels of soluble fractalkine in active systemic lupus erythematosus: potential involvement in neuropsychiatric manifestations. Arthritis Rheum. 2005;52(6):1670–1675. doi:10.1002/(ISSN)1529-0131
100. Blaschke S, Koziolek M, Schwarz A, et al. Proinflammatory role of fractalkine (CX3CL1) in rheumatoid arthritis. J Rheumatol. 2003;30(9):1918.
101. Wang X, Xia S, Fu B. RNAseq analysis of synovial fibroblasts in human rheumatoid arthritis. Mol Med Rep. 2014;10(1):241–247. doi:10.3892/mmr.2014.2182
102. Sawai H, Park YW, He X, Goronzy JJ, Weyand CM. Fractalkine mediates T cell–dependent proliferation of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2007;56(10):3215–3225. doi:10.1002/(ISSN)1529-0131
103. Volin MV, Huynh N, Klosowska K, Chong KK, Woods JM. Fractalkine is a novel chemoattractant for rheumatoid arthritis fibroblast-like synoviocyte signaling through MAP kinases and Akt. Arthritis Rheum. 2007;56(8):2512–2522. doi:10.1002/(ISSN)1529-0131
104. Pingiotti E, Cipriani P, Marrelli A, et al. Surface expression of fractalkine receptor (CX3CR1) on CD4+/CD28− T Cells in RA patients and correlation with atherosclerotic damage. Ann N Y Acad Sci. 2007;1107(1):32–41. doi:10.1196/annals.1381.004
105. Asquith DL, Bryce SA, Nibbs RJB. Targeting cell migration in rheumatoid arthritis. Curr Opin Rheumatol. 2015;27(2):204–211. doi:10.1097/BOR.0000000000000150
106. Dennis G
107. Szekanecz Z, Koch AE, Tak PP. Chemokine and chemokine receptor blockade in arthritis, a prototype of immune-mediated inflammatory diseases. Neth J Med. 2011;69(9):356–366.
108. Loetscher P, Dewald B, Baggiolini M, Seitz M. Monocyte chemoattractant protein 1 and interleukin 8 production by rheumatoid synoviocytes. Effects of anti-rheumatic drugs. Cytokine. 1994;6(2):162–170. doi:10.1016/1043-4666(94)90038-8
109. Barsig J, Yam G, Lehner MD, Beume R. Methotrexate treatment suppresses local cytokine and chemokine production in rat adjuvant arthritis. Drugs Exp Clin Res. 2005;31(1):7–11.
110. Volin MV, Campbell PL, Connors MA, Woodruff DC, Koch AE. The effect of sulfasalazine on rheumatoid arthritic synovial tissue chemokine production. Exp Mol Pathol. 2002;73(2):84–92. doi:10.1006/exmp.2002.2460
111. Ho CY, Wong CK, Li EK, Tam LS, Lam CWK. Suppressive effect of combination treatment of leflunomide and methotrexate on chemokine expression in patients with rheumatoid arthritis. Clin Exp Immunol. 2003;133(1):132–138. doi:10.1046/j.1365-2249.2003.02192.x
112. Kawashiri S-Y, Kawakami A, Iwamoto N, et al. Proinflammatory cytokines synergistically enhance the production of chemokine ligand 20 (CCL20) from rheumatoid fibroblast-like synovial cells in vitro and serum CCL20 is reduced in vivo by biologic disease-modifying antirheumatic drugs. J Rheumatol. 2009;36(11):2397–2402. doi:10.3899/jrheum.090132
113. Portalès P, Fabre S, Vincent T, et al. Peripheral blood T4 cell surface CCR5 density as a marker of activity in rheumatoid arthritis treated with anti-CD20 monoclonal antibody. Immunology. 2009;128(1 Suppl):e738–e745. doi:10.1111/j.1365-2567.2009.03076.x
114. Boyle DL, Soma K, Hodge J, et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann Rheum Dis. 2015;74(6):1311–1316. doi:10.1136/annrheumdis-2014-206028
115. Kasama T, Isojima S, Umemura M, et al. Serum macrophage migration inhibitory factor levels are correlated with response to tocilizumab therapy in patients with rheumatoid arthritis. Rheumatol Int. 2014;34(3):429–433. doi:10.1007/s00296-013-2778-0
116. Allegretti M, Cesta MC, Garin A, Proudfoot AEI. Current status of chemokine receptor inhibitors in development. Immunol Lett. 2012;145(1):68–78. doi:10.1016/j.imlet.2012.04.003
117. Striz I, Brabcova E, Kolesar L, Sekerkova A. Cytokine networking of innate immunity cells: a potential target of therapy. Clin Sci (Lond). 2014;126(9):593–612. doi:10.1042/CS20130497
118. Haringman JJ, Kraan MC, Smeets TJM, Zwinderman KH, Tak PP. Chemokine blockade and chronic inflammatory disease: proof of concept in patients with rheumatoid arthritis. Ann Rheum Dis. 2003;62(8):715–721. doi:10.1136/ard.62.8.715
119. Reynolds G, Cooles FAH, Isaacs JD, Hilkens CMU. Emerging immunotherapies for rheumatoid arthritis. Hum Vaccin Immunother. 2014;10(4):822–837. doi:10.4161/hv.27910
120. Szekanecz Z, Koch AE. Successes and failures of chemokine-pathway targeting in rheumatoid arthritis. Nat Rev Rheumatol. 2016;12(1):5–13. doi:10.1038/nrrheum.2015.157
121. Henrich TJ, Kuritzkes DR. HIV-1 entry inhibitors: recent development and clinical use. Curr Opin Virol. 2013;3(1):51–57. doi:10.1016/j.coviro.2012.12.002
122. Zhong C, Wang J, Li B, et al. Development and preclinical characterization of a humanized antibody targeting CXCL12. Clin Cancer Res. 2013;19(16):4433. doi:10.1158/1078-0432.CCR-13-0943
123. Finch DK, Ettinger R, Karnell JL, Herbst R, Sleeman MA. Effects of CXCL13 inhibition on lymphoid follicles in models of autoimmune disease. Eur J Clin Invest. 2013;43(5):501–509. doi:10.1111/eci.2013.43.issue-5
124. Vergunst CE, Gerlag DM, von Moltke L, et al. MLN3897 plus methotrexate in patients with rheumatoid arthritis: safety, efficacy, pharmacokinetics, and pharmacodynamics of an oral CCR1 antagonist in a phase IIa, double-blind, placebo-controlled, randomized, proof-of-concept study. Arthritis Rheum. 2009;60(12):3572–3581. doi:10.1002/art.v60:12
125. Clucas AT, Shah A, Zhang Y, Chow VF, Gladue RP. Phase I evaluation of the safety, pharmacokinetics and pharmacodynamics of CP-481,715. Clin Pharmacokinet. 2007;46(9):757–766. doi:10.2165/00003088-200746090-00003
126. Vergunst CE, Gerlag DM, Lopatinskaya L, et al. Modulation of CCR2 in rheumatoid arthritis: a double-blind, randomized, placebo-controlled clinical trial. Arthritis Rheum. 2008;58(7):1931–1939. doi:10.1002/art.v58:7
127. Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8(1):23–33. doi:10.1038/nrd2734
128. van Kuijk AWR, Vergunst CE, Gerlag DM, et al. CCR5 blockade in rheumatoid arthritis: a randomised, double-blind, placebo-controlled clinical trial. Ann Rheum Dis. 2010;69(11):2013. doi:10.1136/ard.2010.131235
129. Haringman JJ, Gerlag DM, Smeets TJM, et al. A randomized controlled trial with an anti-CCL2 (anti–monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54(8):2387–2392. doi:10.1002/art.21975
130. Berahovich RD, Miao Z, Wang Y, Premack B, Howard MC, Schall TJ. Proteolytic activation of alternative CCR1 ligands in inflammation. J Immunol. 2005;174(11):7341. doi:10.4049/jimmunol.174.11.7341
131. Lebre MC, Vergunst CE, Choi IYK, et al. Why CCR2 and CCR5 blockade failed and why CCR1 blockade might still be effective in the treatment of rheumatoid arthritis. PLoS One. 2011;6(7):e21772. doi:10.1371/journal.pone.0021772
132. Kramer JM, Klimatcheva E, Rothstein TL. CXCL13 is elevated in Sjögren’s syndrome in mice and humans and is implicated in disease pathogenesis. J Leukoc Biol. 2013;94(5):1079–1089. doi:10.1189/jlb.0113036
133. Sharma A, Kiripolsky J, Klimatcheva E, et al. Early BAFF receptor blockade mitigates murine Sjögren’s syndrome: concomitant targeting of CXCL13 and the BAFF receptor prevents salivary hypofunction. Clin Immunol. 2016;164:85–94. doi:10.1016/j.clim.2016.01.015
134. Klimatcheva E, Pandina T, Reilly C, et al. CXCL13 antibody for the treatment of autoimmune disorders. BMC Immunol. 2015;16(1):6. doi:10.1186/s12865-015-0068-1
135. Watanabe K, Penfold MET, Matsuda A, et al. Pathogenic role of CXCR7 in rheumatoid arthritis. Arthritis Rheum. 2010;62(11):3211–3220. doi:10.1002/art.27650
136. Jakobs BD, Spannagel L, Purvanov V, Uetz-von Allmen E, Matti C, Legler DF. Engineering of nanobodies recognizing the human chemokine receptor CCR7. Int J Mol Sci. 2019;20(10):2597. doi:10.3390/ijms20102597
137. Nanki T, Imai T, Kawai S. Fractalkine/CX3CL1 in rheumatoid arthritis. Mod Rheumatol. 2017;27(3):392–397. doi:10.1080/14397595.2016.1213481
138. Tanaka Y, Takeuchi T, Umehara H, et al. Safety, pharmacokinetics, and efficacy of E6011, an antifractalkine monoclonal antibody, in a first-in-patient Phase 1/2 study on rheumatoid arthritis. Mod Rheumatol. 2018;28(1):58–65. doi:10.1080/14397595.2017.1337056
139. Tanaka Y, Takeuchi T, Umehara H, et al. Safety, pharmacokinetics, and efficacy of E6011, an anti-fractalkine monoclonal antibody, in a first-in-patient phase 1/2 study on rheumatoid arthritis: 52-week results [abstract]. Arthritis Rheumatol. 2017;69(suppl 10).
140. Imai T, Yasuda N. Therapeutic intervention of inflammatory/immune diseases by inhibition of the fractalkine (CX3CL1)-CX3CR1 pathway. Inflamm Regen. 2016;36(1):9. doi:10.1186/s41232-016-0017-2
141. Matthys P, Hatse S, Vermeire K, et al. AMD3100, a potent and specific antagonist of the stromal cell-derived factor-1 chemokine receptor CXCR4, inhibits autoimmune joint inflammation in IFN-gamma receptor-deficient mice. J Immunol. 2001;167(8):4686–4692. doi:10.4049/jimmunol.167.8.4686
142. Tamamura H, Fujisawa M, Hiramatsu K, et al. Identification of a CXCR4 antagonist, a T140 analog, as an anti-rheumatoid arthritis agent. FEBS Lett. 2004;569(1):99–104. doi:10.1016/j.febslet.2004.05.056
143. Jenh C-H, Cox MA, Cui L, et al. A selective and potent CXCR3 antagonist SCH 546738 attenuates the development of autoimmune diseases and delays graft rejection. BMC Immunol. 2012;13:2. doi:10.1186/1471-2172-13-2
144. Kim B, Lee J-H, Jin WJ, Kim H-H, Ha H, Lee ZH. JN-2, a C-X-C motif chemokine receptor 3 antagonist, ameliorates arthritis progression in an animal model. Eur J Pharmacol. 2018;823:1–10. doi:10.1016/j.ejphar.2018.01.037
145. Broeren MGA, de Vries M, Bennink MB, et al. Disease-regulated gene therapy with anti-inflammatory interleukin-10 under the control of the CXCL10 promoter for the treatment of rheumatoid arthritis. Hum Gene Ther. 2015;27(3):244–254. doi:10.1089/hum.2015.127
146. Salomon I, Netzer N, Wildbaum G, Schif-Zuck S, Maor G, Karin N. Targeting the function of IFN-γ-inducible protein 10 suppresses ongoing adjuvant arthritis. J Immunol. 2002;169(5):2685. doi:10.4049/jimmunol.169.5.2685
147. Wildbaum G, Netzer N, Karin N. Plasmid DNA encoding IFN-γ-inducible protein 10 redirects antigen-specific T cell polarization and suppresses experimental autoimmune encephalomyelitis. J Immunol. 2002;168(11):5885. doi:10.4049/jimmunol.168.11.5885
148. Yellin M, Paliienko I, Balanescu A, et al. A Phase II, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of MDX-1100, a fully human anti-CXCL10 monoclonal antibody, in combination with methotrexate in patients with rheumatoid arthritis. Arthritis Rheum. 2012;64(6):1730–1739. doi:10.1002/art.v64.6
149. Ogata H, Takeya M, Yoshimura T, Takagi K, Takahashi K. The role of monocyte chemoattractant protein-1 (MCP-1) in the pathogenesis of collagen-induced arthritis in rats. J Pathol. 1997;182(1):106–114. doi:10.1002/(ISSN)1096-9896
150. Shahrara S, Proudfoot AEI, Park CC, et al. Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J Immunol. 2008;180(5):3447. doi:10.4049/jimmunol.180.5.3447
151. Brühl H, Cihak J, Plachý J, et al. Targeting of Gr-1+, CCR2+ monocytes in collagen-induced arthritis. Arthritis Rheum. 2007;56(9):2975–2985. doi:10.1002/(ISSN)1529-0131
152. Kurihara T, Warr G, Loy J, Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med. 1997;186(10):1757–1762. doi:10.1084/jem.186.10.1757
153. Takeuchi T, Kameda H. What is the future of CCR5 antagonists in rheumatoid arthritis? Arthritis Res Ther. 2012;14(2):114. doi:10.1186/ar3775
154. Tuttle DL, Harrison JK, Anders C, Sleasman JW, Goodenow MM. Expression of CCR5 increases during monocyte differentiation and directly mediates macrophage susceptibility to infection by human immunodeficiency virus type 1. J Virol. 1998;72(6):4962–4969. doi:10.1128/JVI.72.6.4962-4969.1998
155. Duan H, Yang P, Fang F, Ding S, Xiao W. CCR5 small interfering RNA ameliorated joint inflammation in rats with adjuvant-induced arthritis. Immunol Lett. 2014;162(2, Part B):258–263. doi:10.1016/j.imlet.2014.09.018
156. Vierboom MPM, Zavodny PJ, Chou C-C, et al. Inhibition of the development of collagen-induced arthritis in rhesus monkeys by a small molecular weight antagonist of CCR5. Arthritis Rheum. 2005;52(2):627–636. doi:10.1002/art.20850
157. Zapico I, Coto E, Rodríguez A, Alvarez C, Torre JC, Alvarez V. CCR5 (chemokine receptor-5) DNA-polymorphism influences the severity of rheumatoid arthritis. Genes Immun. 2000;1(4):288–289. doi:10.1038/sj.gene.6363673
158. Gerlag DM, Hollis S, Layton M, et al. Preclinical and clinical investigation of a CCR5 antagonist, AZD5672, in patients with rheumatoid arthritis receiving methotrexate. Arthritis Rheum. 2010;62(11):3154–3160. doi:10.1002/art.27652
159. Shahrara S, Proudfoot AEI, Woods JM, et al. Amelioration of rat adjuvant-induced arthritis by Met-RANTES. Arthritis Rheum. 2005;52(6):1907–1919. doi:10.1002/art.21033
160. Plater-Zyberk C, Hoogewerf AJ, Proudfoot AE, Power CA, Wells TN. Effect of a CC chemokine receptor antagonist on collagen induced arthritis in DBA/1 mice. Immunol Lett. 1997;57(1–3):117–120. doi:10.1016/S0165-2478(97)00075-8
161. Booth G, Newham P, Barlow R, Raines S, Zheng B, Han S. Gene expression profiles at different stages of collagen-induced arthritis. Autoimmunity. 2008;41(7):512–521. doi:10.1080/08916930802095210
162. Brennan FM, Maini RN, Feldmann M. Role of pro-inflammatory cytokines in rheumatoid arthritis. Springer Semin Immunopathol. 1998;20(1):133–147. doi:10.1007/BF00832003
163. Amat M, Benjamim CF, Williams LM, et al. Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br J Pharmacol. 2006;149(6):666–675. doi:10.1038/sj.bjp.0706912
164. Ronald PG, Matthew FB, Samuel HZ. CCR1 antagonists: what have we learned from clinical trials. Curr Top Med Chem. 2010;10(13):1268–1277. doi:10.2174/156802610791561237
165. Kivitz A, Maciag P, Gulati P, et al. THU0109 Lack of efficacy of CCR1 antagonist BMS-817399 in patients with moderate to severe rheumatoid arthritis: results of 12-week proof-of-concept study. Ann Rheum Dis. 2014;73(Suppl 2):215. doi:10.1136/annrheumdis-2014-eular.3871
166. Naya A, Ishikawa M, Matsuda K, et al. Structure-activity relationships of xanthene carboxamides, novel CCR1 receptor antagonists. Bioorg Med Chem. 2003;11(6):875–884. doi:10.1016/S0968-0896(02)00559-X
167. Tak PP, Balanescu A, Tseluyko V, et al. Chemokine receptor CCR1 antagonist CCX354-C treatment for rheumatoid arthritis: CARAT-2, a randomised, placebo controlled clinical trial. Ann Rheum Dis. 2013;72(3):337. doi:10.1136/annrheumdis-2011-201605
168. Dairaghi DJ, Zhang P, Wang Y, et al. Pharmacokinetic and pharmacodynamic evaluation of the novel CCR1 antagonist CCX354 in healthy human subjects: implications for selection of clinical dose. Clin Pharmacol Ther. 2011;89(5):726–734. doi:10.1038/clpt.2011.33
169. Jiao Z, Wang W, Jia R, et al. Accumulation of FoxP3-expressing CD4+CD25+ T cells with distinct chemokine receptors in synovial fluid of patients with active rheumatoid arthritis. Scand J Rheumatol. 2007;36(6):428–433. doi:10.1080/03009740701482800
170. Wei S, Kryczek I, Zou W. Regulatory T-cell compartmentalization and trafficking. Blood. 2006;108(2):426–431. doi:10.1182/blood-2006-01-0177
171. Pablos JL, Santiago B, Galindo M, et al. Synoviocyte-derived CXCL12 is displayed on endothelium and induces angiogenesis in rheumatoid arthritis. J Immunol. 2003;170(4):2147–2152. doi:10.4049/jimmunol.170.4.2147
172. Shi K, Hayashida K, Kaneko M, et al. Lymphoid chemokine B cell-attracting chemokine-1 (CXCL13) is expressed in germinal center of ectopic lymphoid follicles within the synovium of chronic arthritis patients. J Immunol. 2001;166(1):650. doi:10.4049/jimmunol.166.1.650
173. Gong JH, Yan R, Waterfield JD, Clark-Lewis I. Post-onset inhibition of murine arthritis using combined chemokine antagonist therapy. Rheumatology. 2003;43(1):39–42. doi:10.1093/rheumatology/keg459
174. Zhao Q. Dual targeting of CCR2 and CCR5: therapeutic potential for immunologic and cardiovascular diseases. J Leukoc Biol. 2010;88(1):41–55. doi:10.1189/jlb.1009671
175. Matsukawa A, Yoshimura T, Fujiwara K, Maeda T, Ohkawara S, Yoshinaga M. Involvement of growth-related protein in lipopolysaccharide-induced rabbit arthritis: cooperation between growth-related protein and IL-8, and interrelated regulation among TNFalpha, IL-1, IL-1 receptor antagonist, IL-8, and growth-related protein. Lab Invest. 1999;79(5):591–600.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.