")
Back to Journals » Journal of Inflammation Research » Volume 13
Role of Caspase-1 in the Pathogenesis of Inflammatory-Associated Chronic Noncommunicable Diseases
Authors Molla MD , Akalu Y , Geto Z , Dagnew B , Ayelign B , Shibabaw T
Received 17 August 2020
Accepted for publication 21 September 2020
Published 20 October 2020 Volume 2020:13 Pages 749—764
DOI https://doi.org/10.2147/JIR.S277457
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Meseret Derbew Molla,1 Yonas Akalu,2 Zeleke Geto,3 Baye Dagnew,2 Birhanu Ayelign,4 Tewodros Shibabaw1
1Department of Biochemistry, School of Medicine, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia; 2Department of Human Physiology, School of Medicine, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia; 3Department of Biomedical Sciences, School of Medicine, College of Medicine and Health Sciences, Wollo University, Dessie, Ethiopia; 4Department of Immunology and Molecular Biology, School of Biomedical and Laboratory Sciences, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Correspondence: Meseret Derbew Molla
Department of Biochemistry, School of Medicine, College of Medicine and Health Sciences, University of Gondar, PO Box 196, Gondar, Ethiopia
, Tel +251918331617
Email [email protected]
Abstract: Caspase-1 is the first and extensively studied inflammatory caspase that is activated through inflammasome assembly. Inflammasome is a cytosolic formation of multiprotein complex that aimed to start inflammatory response against infections or cellular damages. The process leads to an auto-activation of caspase-1 and consequent maturation of caspase-1 target molecules such as interleukin (IL)-1β and IL-18. Recently, the role of caspase-1 and inflammasome in inflammatory-induced noncommunicable diseases (NCDs) like obesity, diabetes mellitus (DM), cardiovascular diseases (CVDs), cancers and chronic respiratory diseases have widely studied. However, their reports are distinct and even they have reported contrasting role of caspase-1 in the development and progression of NCDs. A few studies have reported that caspase-1/inflammasome assembley has a protective role in the initiation and progression of these diseases through the activation of the noncanonical caspase-1 target substrates like gasdermin-D and regulation of immune cells. Conversely, others have revealed that caspase-1 has a direct/indirect effect in the development and progression of several NCDs. Therefore, in this review, we systematically summarized the role of caspase-1 in the development and progression of NCDs, especially in obesity, DM, CVDs and cancers.
Keywords: caspase-1, inflammasome, IL-1β, IL-18, noncommunicable diseases
Introduction
Caspases are a cysteine protease that speed up a chemical reaction via pointing their target substrates following an aspartic acid residue.1 They are grouped into apoptotic (caspase-2, 3, 6, 7, 8, 9 and 10) and inflammatory (caspase-1, 4, 5, 11 and 12) mediated caspases.2 Caspase-1 is the first studied caspase in 1989 and clearly characterized in 1992 after performing genetic sequencing.3,4 It is one of the most common and well-studied inflammatory mediated caspases that initiate inflammatory response through the activation of pro-inflammatory cytokines such as interleukin-1β (IL-1β) and IL-18.5 However, caspase-1 is produced and stored within the tissues in the form of zymogens (inactive form of a molecule) like that of other caspases. Therefore, caspase-1 should be first activated in order to be functional, and caspase-1 is activated through the assembly of an inflammasome complex.6 Inflammasomes are a cytosolic multiprotein complex formation that are recruited by external pathogen-associated molecular patterns (PAMPs) and/or internal damage-associated molecular patterns (DAMPs) stimuli, which is usually generated by immune cells. The immune cells are able to detect these stimuli through the help of pattern recognition receptors (PRRs).6,7 The primary aim of the inflammasome complex is to resist the host innate immune cells for infection or injury, but chronic inflammasomes may rather lead to chronic inflammatory disorders such as obesity, DM and CVDs.8,9
Recently, several human and animal studies have explored that different metabolic intermediates can be initiated and activate the inflammasome in the immune cells. For example, nucleotide-binding domain and leucine-rich repeat containing nod-like receptor, pyrin domain containing 3 (NLRP3) inflammasome, the most studied inflammasome, can be activated by calcium channel affecting marine toxin maitotoxin, bacterial ribonucleic acid (RNA), uric acid crystals associated with gout, extracellular ATP, ceramides, high plasma free fatty acid, high blood glucose level and islet amyloid polypeptide.10–13 NLRP3 inflammasome is a cytosolic protein complex that expressed in monocytes, dendritic cells, neutrophils, lymphocytes, epithelial cells and osteoblasts.14 Having this understanding, researchers who are focused on clinical studies revealed that inflammasome targeting therapeutic intervention for inflammatory disorders should be encouraged. This is because of the activation of IL-1β and other inflammatory cytokines that can initiate and/or aggravate the inflammation of some vital organs, including adipose tissue and pancreatic beta cells, and in turn result in metabolic disorders.15 In particular, white adipose tissue (WAT) is one of the most affected tissues and the impairment or dysregulation of adipogenesis and adipokines are frequently encountered due to metabolic disorder-associated systemic inflammation.16 Consequently, the functional impairment of WAT increases the flux of free fatty acid (FFA) into nonadipose tissues, which will be accumulated in vital organs as ectopic fat and/or toxic FA metabolites like ceramide. This causes or increases insulin resistance, which in turn stimulates oversecretion of insulin from pancreatic beta cells for physiological compensatory activities. Following this, over time oversecretion of insulin causes exhaustion of pancreatic beta cells and then resulted in the impairment of insulin secretion. Finally, this leads to hyperglycemia and other metabolic disorders that are associated with the inability of insulin action.17,18
Furthermore, studies have confirmed that activation of IL-1β and IL-18 by caspase-1 are directly associated with several metabolic disorders and their complications such as obesity, atherosclerosis, impairment of insulin action and/or secretion, cancers and nonalcoholic fatty liver disease.19,20 These diseases collectively termed as NCDs are alarmingly increasing worldwide and become the major global public health concern. Globally, NCDs are the leading cause of morbidity and mortality with high proportion rate found in middle and low income countries.21 They accounted for about 70% of deaths in the world. Out of them, cardiovascular diseases (CVDs) are the most frequent cause of death followed by cancers, chronic respiratory diseases and DM. Thus, NCDs produce a massive health and economic burdens in the world, especially in low and middle-income countries.22 Therefore, targeting the exact molecular link between caspase-1 and these NCDs is vital to overcome the health and economic burden of the population. In support of this, NCDs share common modifiable behavioral risk factors, such as unbalanced diet, inadequate physical activity, and frequent use of alcohol and tobacco, in turn lead to the activation of inflammasome and inflammatory responses, which further lead to several metabolic disorders such as obesity, hypertension, dyslipidemia and finally chronic diseases.23–25 These risk factors could have a synergizing effect overtime time and associated with the development and progression of NCDs, and finally may lead to death.23,25 However, to the concern of our search, there are a limited number of reviews on the issue, even with several original articles having been published throughout a year. Therefore, we aimed to summarize the role of caspase-1 in inflammatory-associated chronic NCDs such as obesity, DM, CVDs and cancers.
Caspase-1 Activation and Inflammation
Caspase-1 is the well-studied inflammatory mediated caspase, which is important for pro-inflammatory cytokines (proIL-1β and IL-18) maturation into IL-1β and IL-18.5 Inflammatory mediated caspases are those caspases that take part in the process of inflammation and cell death to overcome the stimulatory materials.26 The pro-caspase-1, is activated through inflammasome assembly which is stimulated by several small molecules derived from infections, tissue damages, or metabolic dysfunctions.27 Thus, caspase-1 activation is commonly observed in macrophages, dendritic cells and epithelial cells as immune defense mechanism.28 Inflammasome are a group of cytosolic multimeric protein complexes that is assembled in response to the danger signs (PAMPs and DAMPs) which is best known for their ability to control activation of pro-caspase-1, and active caspase-1 in turn regulates its target substrates.6 Innate immune cells recognize these danger signs through PRRs such as toll-like receptors (TLRs) or intracellular node-like receptors (NLRs).29 However, the external stimuli (PAMPs, including microbial infection) should have an access of cytosolic sensing to assemble inflammasome complexes since inflammasome is a cytosolic dependent intracellular process.30 There are many inflammasomes yet to be studied that are differentiated by their distinct activators, NLR/ALR components, and effector caspases. Of these, NLR families (NLRP1, NLRP3 or NLR apoptosis inhibitory protein (NAIP), NLRP6, NLRP7, NLRP12 and NLRC4 or IPAF) are the most common inflammasome responsible for host immune responses against infection, trauma or tissue necrosis. The HIN-200 family member AIM2 can also be the other common receptors for the inflammasome complex formation.31 However, NLRP3 inflammasome is by far the most studied and important inflammasome in the development and progression of inflammatory-induced metabolic diseases.32 It is activated by several PAMPs and DAMPs, but the exact molecular mechanism by which these ligands are attached to NLRP3 is not clearly addressed yet.
Several studies have reported that NLRP3 inflammasome can be activated through the efflux of potassium ion due to the NLRP3 stimuli mediated by the purinergic P2X7 receptor and phagocytosis of the pathogens by immune cells associated with the release of lysosomal cysteine protease (cathepsin B) and lysosomal swelling and damage. NLRP3 inflammasome can also be activated through RIP2 kinase-NF-kB–dependent transcriptional activation of the NLRP3 inflammasome components. The external stimuli (PAMPs and DAMPs) are recognized by TLRs, which mediates the expression of RIP2 kinase. The expression of RIP2 kinase activates the nuclear NF-kB and MAP kinases and then induces the activation of pro-inflammatory cytokines through the assembly of NLRP3 inflammasome.33–35 RIP2 kinase is one of the RIP kinase family that has the N-terminal homologous kinase domain, an intermediate domain and a C-terminal caspase activation and recruitment domain (CARD).36 However, some other inflammasome can be directly detected and bind with the ligand of danger signals. For instance, AIM2 and NLRC4 inflammasome directly binds to double-stranded DNA and bacterial flagellin, respectively.37–39 Moreover, studies have reported that pyrin containing inflammasomes can be recognized in the microbial infections with the pyrin part of the inflammasome. Thus, the pryin recognizes and binds with the modified rho GTPases such as adenylylation, glucosylation and ADP-ribosylation by bacterial toxins.40
Although different types of inflammasome have studied, the classical inflammasome complex is composed of three components; (i) a cytosolic sensor to detect microbial products or stress signals (can be NLRs, absent in melanoma 2–like receptors (ALRs), or pyrin), (ii) an adaptor protein (apoptosis-associated speck-like protein containing a CARD; ASC), and (iii) an effector caspase (pro-caspase-1).41,42 These inflammasome are an intracellular danger sign detector that primarily aim to activate procaspase-1, and in turn lead to the maturation of the target substrates (proIL-1β, IL-18 and gasdermin-D) into their biological active form, then initiate and/or mediate the inflammatory response.6,43 Once these pro-inflammatory cytokines (IL-1β and proIL-18) are activated, they act in an autocrine or paracrine way and mediate both innate and adaptive immune response through the mediation of leukocyte survival, proliferation, differentiation, recruitment and migration.44 For instance, active IL-1β binds to the IL-1 receptor and activates nuclear factor kappaB (NF-κB) and mitogen-activated protein kinase (MAPK) pathways which bring to the generation of more inflammatory mediators that influence cell migration and immune cell infiltration.45 It can also promote and keep up interferon gamma (IFN-γ) and IL-17 producing Th1 and Th17 cells, respectively46 (Figure 1).
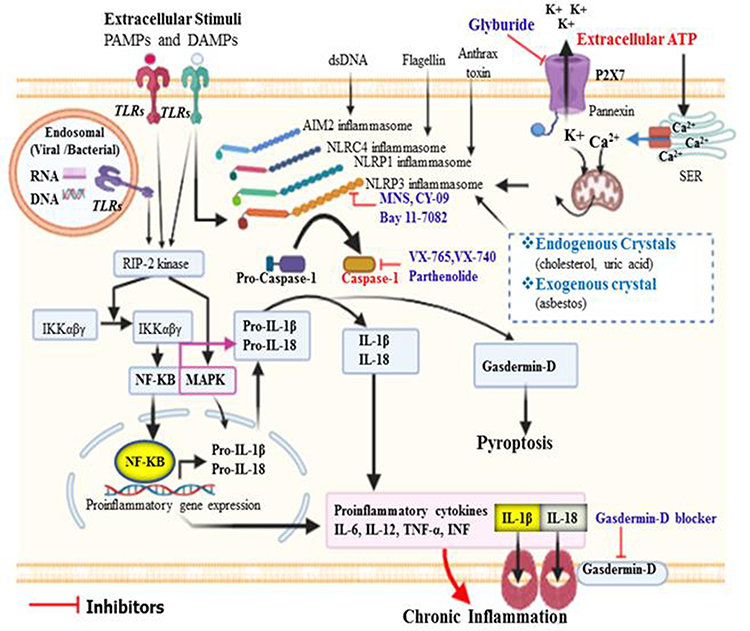 |
Figure 1 Activation of caspase-1 and its role in the initiation of inflammation. Different types of inflammasome can be activated by their distinct stimuli’s. Of these, NLRP3 can be activated by several PAMPs and DAMPs that enter into the cytosol through TLRs or phagocytosis. Moreover, in response to extracellular ATP, efflux of K+ from intracellular via P2X7 and release of Ca++ from storage site called smooth endoplasmic reticulum via Ca2+ channel leads to mitochondrial dysfunction and generation of ROS could trigger oxidative stress, which can then activate NLRP3 inflammasome. All inflammasomes finally activate procaspase-1 to caspase-1, and in turn matures pro-IL-1β and pro-IL-18 to induce inflammatory response. Caspase-1 activated gasdermin-D serves as pore-forming protein channels for IL-1β and IL-18 secretion, then high abundance of gasdermin-D at the plasma membrane results in inflammatory-induced lytic form of cell death called pyroptosis. NLRC4 inflammasome does not have ASC/adaptor protein, and the CARD domain of NLRC4 directly binds to its respective domain of procaspase-1. |
Active caspase-1 also stimulates inflammatory-induced cell death called pyroptosis through gasdermin-D activation.47,48 Therefore, inflammasome-mediated activation of caspase-1 and its target substrates are physiologically important to regulate immune cell response against microbial infections and metabolic activities.32,49 However, infection resistance or obesity-associated chronic inflammation in peripheral tissues can lead to metabolic disturbances such as obesity, type two diabetes mellitus (T2DM), insulin resistance and cardiovascular diseases.50 Chronic inflammation is usually associated with the release of oxidative metabolic intermediates like ROS, which in turn leads to organelle dysfunctions, including mitochondrial dysfunction. The disturbance of mitochondrial dynamic activity can further contribute for the development and progression of these inflammatory-induced metabolic disorders.51
Role of Caspase-1 in Obesity
Globally, the prevalence and incidence rate of obesity is becoming extremely high regardless of age and sex. Consequently, obesity is associated with many life-threatening metabolic disorders such as T2DM or insulin resistance/β-cell dysfunction, atherosclerosis, obstructive airway diseases, nonalcoholic fatty liver disease and cancers.52 So far, several human and animal experimental studies revealed that NLRP3 inflammasome and caspase-1 overexpression were found in obese subjects compared with normal, which may be directly associated with the progression and complication of obesity into further metabolic disorders.12,20,53
The activation of IL-1β and IL-18 (caspase-1 target substrates) are linked to the development of obesity, but the exact molecular mechanisms are still in research.19,20 Besides, the role of caspase-1 in the development and progression of obesity remains a controversial issue. Some experimental studies reported that development of obesity is less likely in caspase-1 deficient mice compared with the wild-type mice and other studies have released the opposite, ie the probability of developing obesity is more likely in caspase-1 deficient mice than that of the wild-type mice.54–56 In addition, one study also reported that IL-18 deficient mice have significantly increased body weight compared to wild-type at the same age.20 This may be due to the exceptional activity of IL-18. Thus, unlike IL-1β, maturation of IL-18 with active NLRP1 inflammasome activates insulin sensitivity in the peripheral tissues, and in turn stimulates activation of insulin dependent metabolic pathways, including fatty acid utilization. Consequently, accumulation of excess fat becomes reduced and will have low chance of developing obesity.57 Other studies have also reported that development of obesity-associated insulin resistance is commonly observed in IL-18 gene/receptor deficient mice than that of wild-type mice.20,58 This suggests that in healthy individuals’ caspase-1 dependent activation of IL-18 is important for metabolic homeostasis. However, in systemic inflammation the activity of IL-18 is undermined by matured IL-1β, then obesity development and progression might be prominent. In support of this, one study has also reported that the effect of inflammatory stimuli on IL-18 is slight compared to IL-1β.59
These controversies have revealed that activation of inflammatory signaling pathway had a significant effect in lipid metabolism alteration, especially in the situation of diabetes, insulin resistance, atherosclerosis, obesity and infection disorders.60 Diabetes, insulin resistance, atherosclerosis and obesity are very closely related metabolic disorders that are characterized by hyperactivation of pro-inflammatory cytokines and inflammatory signaling pathways.61 In particular, pro-inflammatory cytokines such as tumor necrosis factor (TNF), IL-1β and IL-12 over-activation during these condition attracts further inflammatory molecules and results in infiltration of inflamed organs by immune cells.59 This is supported by both human and mice studies, which revealed that macrophage infiltration of adipose tissue has been observed in obesity.62,63 This condition will be further aggravated by excessive nutrition and abnormal of production metabolic intermediate, which are the principal risk factors of obesity development and progression. Particularly, NLRP3 inflammasome is activated by several metabolic surpluses such as ceramides, saturated fatty acid and monosodium urate.10,12,64 Initially, inflammation helps to clear infection or any other noxious stimuli, but if the process persists and the noxious stimuli cannot be quickly eliminated, it will result in chronic inflammation, which lead to serious tissue damage and pathological consequences65 (Figure 2). As the stimulatory molecules are continued for a prolonged time, the adipose tissue will become hypertrophied and cause death due to the overactivation of the pro-inflammatory cytokines through hypoxia and endoplasmic reticulum (ER) stress-related mechanisms. This condition synergistically aggravates the problem and will result in the release of FFAs in the circulation.66 The circulating FFAs may further directly bind to TLR2 and TLR4, inducing NF-κB activation and production of further pro-inflammatory cytokines, including pro-IL-1β, which in turn contributes to the progression of obesity.46 Besides, active caspase-1 can also be stimulated the development and advancement of obesity through inactivation of sirtuin1 (SIRT1)67,68 (Figure 2). Sirtuin1 is a nuclear nicotinamide-adenine dinucleotide (NAD+) dependent protein deacetylase that regulates several transcriptional factors, which are very important for systemic metabolic homeostasis69 Thus, inactivation of SIRT1 by active caspase-1 results inactivation of SIRT1 target pathways.67 For instance, in adipose tissue, inactivation of SIRT1 activates the expression of peroxisome proliferator-activated receptor gamma (PPARγ), which in turn activates adipogenesis, inhibits fat mobilization70 and promotes insulin resistance by targeting NF-κB.71 In addition, in hepatic cells, inactivation of SIRT1 may lead to activation of sterol regulatory element-binding protein 1 (SREBP1), and in turn lead to the increment of de novo synthesis of both fatty acid and cholesterol.72,73
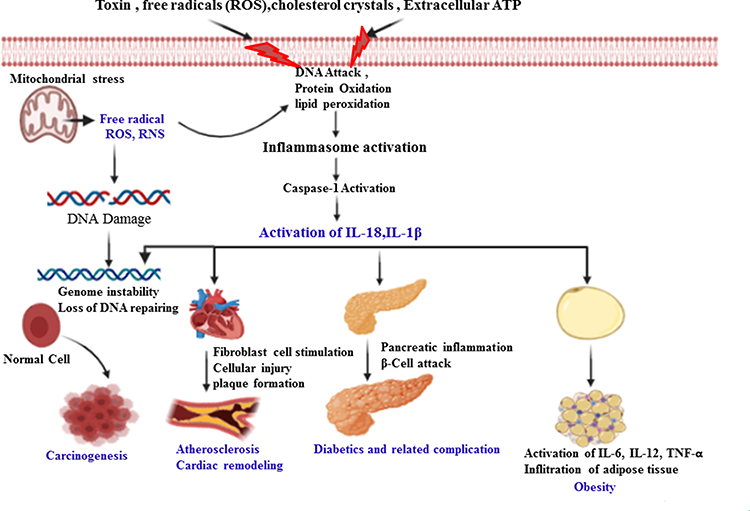 |
Figure 2 The role of caspase-1 in the pathogenesis of inflammatory-induced NCDs. Activation of caspase-1 by internal/external stimuli matures pro-inflammatory cytokines (IL-1β) and gasdermin-D to induce pyroptosis. This initiates inflammatory response and may result in cell injury and/or death, plaque formation and rupture, infiltration to several organs, in turn leading to the development and progression of obesity, atherosclerosis, cardiac remodeling and cancer formation. The release of ROS/RNS from stressed mitochondria may directly oxidize and damage cellular/extracellular proteins, carbohydrates, lipids and nucleic acids that lead to metabolic disturbance. Thus, DNA damage leads to an alteration of signaling pathways (suppression of tumor suppressor genes and promotion of tumor promoter genes) that further results in exacerbation of DNA damage, genomic instability and impairment of DNA repair system. These collectively may promote carcinogenesis. These metabolic disturbances further result in the production of NLRP3 inflammasome stimulatory molecules such as cholesterol crystals, ROS/RNS, damaged cells and ATP, which in turn activates caspase-1 in positive a loop pathway which aggravates the inflammatory process and infiltration of the inflammatory immune cells. |
Role of Caspase-1 in Diabetes Mellitus
Diabetes mellitus is a metabolic disorder characterized by the persistent elevation of blood glucose levels (hyperglycemia) due to failure of insulin production and/or insulin action.74 It is becoming one of the most common public health problems in the world and the number of affected people has increased up to fourfold in the past three decades.75 Globally, in 2019, the number of people with DM reached around 463 million, and it is estimated that by 2045, this number will reach 700 million.76 Three quarters of the disease is found in developing countries, and more than 90% of them are affected by T2DM.77 Thus, medical fees along with micro and macro vascular complication-associated death are the main concern of DM patients.75,78 For this reason, scholars have given substantial attention to the potential initiated mechanism of T2DM. The major metabolic abnormality that greatly contributed to the development of T2DM is insulin resistance and it has been addressed that obesity is the most common cause of insulin resistance.74,79 Obesity-associated insulin resistance is mainly caused by the initiation of chronic inflammations in several tissues.9,80
In line with this, researchers found that low-grade chronic inflammation have a great role in the onset and progression of T2DM.81,82 So far, studies have confirmed that the major inflammatory cytokines (IL-1 family, IL-6 and TNF-α) are highly overexpressed in patients with T2DM.83–86 The activation of these cytokines are involved in the promotion of pancreatic beta cell destruction, which in turn leads to the impairment of insulin secretion and resistance of insulin in the peripheral cells through the interference of insulin receptors.87 In fact, the exact molecular mechanism of the development of insulin resistance is not clearly understood yet, but excess accumulation of fats and inflammation of adipose tissues are believed to be a major contributing factor for it.88 While insulin resistances have been developed, it associated with the accumulation of ectopic fat which generates toxic metabolic intermediates like diacylglycerol and ceramides. Consequently, these molecules could have a potential to activate inflammasomes, especially NLRP3 inflammasome, which in turn activates IL-1β. Then, IL-1β along with other inflammatory cytokines such as IL-6, and TNF-α will further aggravate the inflammatory reaction in response to the stimuli19 (Figure 2). The maturation of these cytokines can also be directly inhibited by the activation of Akt due to the biosynthesis of ceramides and in turn affect the activity of insulin. Thus, the increment of intracellular ceramide associates with insulin resistance through the activation of phosphatase 2A to dephosphorylate Akt, which in turn inhibits insulin signaling.89,90
The major inflammatory cytokine (IL-1β) activation through caspase-1 can also be involved in DM development and progression through the interfering of the activity of insulin-signaling pathways by activating stress kinases, including the inhibitor of nuclear factor κB kinase subunit β (IKKβ) and c-Jun N-terminal kinase (JNK).15,91 The activation of these stress kinases brings the interference of the activity of insulin receptor through the phosphorylation of insulin receptor substrate 1 (IRS1), then leads to the impairment of the downstream response (PI3K activity).92–94 Cytokine activation can also directly affect the activity of insulin through the inhibition of insulin signaling molecule expression.95 As the activity of the insulin receptor is altered, the activity of insulin will be compromised and pancreatic beta cells will produce more insulin as a compensatory mechanism. However, over time pancreatic beta cells become exhausted and fail to produce enough insulin and lead to DM development.18
Furthermore, the activation of inflammatory cytokines, including IL-1β in pancreatic beta cells will directly interfere with the production of insulin via the promotion of inflammatory-induced pancreatic β cell hypertrophy and death due to pancreatic infiltrating macrophages, which in turn results hyperglycemia and associated metabolic disorders.17,18,96 This may also be provoked by the activation of the inflammasomes due to a high glucose diet leading to elevation of blood glucose levels.24 This may be due to hyperglycemia-induced upregulation of electron transport chain in β cells, which results in the generation of mitochondrial ROS, and in turn stimulates the dissociation of thioredoxin to produce thioredoxin-interacting protein (TXNIP) which binds and activates NLRP3 inflammasome. Moreover, NLRP3 inflammasome can further bind with thioredoxin-interacting protein (TXNIP); thereby promoting insulin resistance and pancreatic cell death through the inhibition of thioredoxin. Thus, the inhibition of thioredoxin results in production of more ROS which brings activation of more NLRP3 inflammasome since ROS is one of the most commonly known activators of it96,97 (Figure 2). Similarly, several studies have also reported that IL-1β deficient mice have good insulin response for athreogenic diets or high fat diets, and injection of IL-1β puts the mice in insulin resistance.54,64 The pathological role of inflammasome in T2DM is supported by other studies that revealed that the administration of insulin-secretion promoting drug (glyburide) and IL-1 receptor antagonist to treat T2DM were found to suppress the maturation of NLRP3-mediated IL-1β and improvement of β-cell function and blood glucose level.98,99
Role of Caspase-1 in Cardiovascular Diseases
CVDs are a combined name for vascular/blood vessel and heart diseases, which are life-threatening diseases. Some of these are heart failure, coronary heart disease, cardiomyopathy, peripheral vascular disease, congenital heart disease and stroke.100 The chance of developing CVDs is more common in those people having metabolic disorders than that of relatively healthy people. So far, published papers have reported that an overexpression of caspase-1 is often observed in all CVDs. However, the role of caspase-1 is not similarly reported in deferent types of CVDs. For instance, in heart failure and cardiac ischemia, it was reported that caspase-1 plays a protective role in the deterioration of the disease by inducing programed cell death to clear out the damaged cells. In their finding, caspase-1 has a pro-apoptotic activity that helps withactivation of the intrinsic apoptotic caspases such as caspase-3 and caspase-9 bringing initiation of apoptosis and reduction of the inflammatory process in affected cells.101,102 Conversely, it also reported that caspase-1 can directly initiate inflammatory-induced cell death through the activation of the gasdermin-D-induced pyroptosis pathway, which in turn results in cardiomyocyte death, excessive scar formation, and poor ventricular remodeling.103 Other experimental studies on mice also reported that activation of NLRP3 inflammasome and caspase-1 may cause excessive cell death in cardiac ischemia. Thus, they suggested that inhibition of caspase-1 activation may have a great role in the progression of cardiac ischemia due to excessive cell death.102,104
Other reports also revealed that inflammasome can be involved in the development and expansion of CVDs through several mechanisms. For example, activation of IL-1β through inflammasome assembly contributes to induction of systemic hypertension and its progression to CVDs.105 Active IL-1β induces expression of renin-angiotensin-aldosterone system and ROS generation in the paraventricular nucleus, which helps the progression of hypertension into complications, including CVDs.106 In support of this, one study has reported that inhibition of IL-1β suppresses the deterioration of hypertension, rather results in good response of the disease.107 It is also reported that in vivo experimental study, caspase-1 deficient mice are protected from atherosclerosis.108 Similarly, the activation of IL-1β with NLRP3 inflammasome has a great contribution to atherosclerosis plaque formation and progression to CVDs.109–111 Thus, active IL-1β along with chemoattractants stimulate and attract more inflammatory cytokines to overcome the problem, but over time the plaque formation will be aggravated and lead to narrowing of the blood vessels. Then, there will be new development of coronary heart disease and associated complications. During this process, the release of damaged molecules and ROS further stimulates NLRP3 inflammasome activation and aggravates atherosclerosis plaque formation in positive loop pathways.112 Consequently, the activation of NLRP3 inflammasome by high level of cholesterol crystals, ROS, and other PAMPs and DAMPs further leads to the activation of IL-1β and gasdermin-D, which in turn causes the initiation of cardiac cells inflammation, pyroptosis, remodeling, hypertrophy and fibrosis formation. This condition further results in the deterioration of atherosclerosis to myocardial infarction and other cardiac complications.113 In addition to this, one study also reported that active caspase-1 with NLRP3 inflammasome can directly bind and degrade contractile proteins in smooth muscle cells. Then, the contractile property of these cells will be lost along with biochemical dysfunctions, which in turn leads to the formation of aortic destruction and aortic aneurysms and dissections.114 Thus, inhibition of caspase-1 expression with inflammasome inhibitors like glyburide (antidiabetic drug which blocks potassium efflux)98 significantly reduces angiotensin II–induced development of aortic aneurysms and dissections.114 Although the exact molecular mechanism is not clearly revealed, individuals with aortic aneurysms and dissections are usually associated with the formation of atherosclerosis and coronary heart disease.115 Therefore, we can generalize that caspase-1 could have a key role in the progression of hypertension and atherosclerosis into CVD complications, and can be helpful as a new insight of therapeutic strategies (Figure 2).
Although the role of caspase-1 in CVD remains controversial, the type of cardiac cells by which NLRP3 inflammasome and caspase-1 can be activated determines the role of NLRP3 inflammasome and caspase-1 in these cells. In leukocytes, the activation of NLRP3 inflammasome targets the maturation of pro-inflammatory cytokines, which in turn leads to inflammatory response-associated complications. In cardiomyocytes, the activation of NLRP3 inflammasome mainly targets the activation of gasdermin-D and other apoptotic caspases to induce caspase-1–mediated cell death without altering the production of increased IL-1β and IL-18. Surprisingly, in fibroblast cells, caspase-1 involves the maturation of IL-1β–mediated myofibroblast differentiation and collagen synthesis.103,116–118 In animals, fibroblasts are the most common cells of connective tissue that produce the structural framework of the tissue through the synthesis of extracellular matrix and collagen. Fibroblasts can also be involved in the inflammatory responses for tissue injuries or infections through the induction of chemokine synthesis.119
Role of Caspase-1 in Cancers
Recently, cancers have become one of the most common public health problems worldwide. Globally, about 18.1 and 9.6 million newly diagnosed cancer cases and cancer-related deaths, respectively have been reported in 2018.120 The molecular pathogenesis of most cancers are becoming the focus of many researchers, but not clearly understood yet. However, it has been understood that tumor cells are resistant to programmed cell death (apoptosis), have high proliferation rate and stimulate the immune cells to start their protective activities.121 The presence of chronic inflammation along with high intake of processed food, bad behavioral practices like alcohol drinking and cigarette smoking and poor physical activity are believed to be the major contributing factors for the increment of the incidence and mortality rate of most cancers.122–124 It has reported that the incidence rate of inflammation-associated human cancers reached around 15% in accordance to the total incidence rate of cancers.125 Although the exact molecular mechanism of chronic inflammation-associated cancer development is not clearly understood, it is believed that the generation of inflammation-associated cytokines such as IL-1 families, IL-6 and TNF-α and the generation of ROS are the main contributing factors of it. The tumor microenvironment is mainly coordinated by these inflammatory cells, which in turn aggravates the progression of tumor cells to advanced stages.126 The inflammatory cells are further activated in response of carcinogens and inflammatory-induced cell injuries. Initially, these cells are aimed to switch cellular stress and damage, but over time inability to manage the problem leads to obstinate interleukin production, which in turn brings the exacerbation of tissue destruction, thus, further leading to the development and progression of tumorigenesis.127 So far, it has been shown that caspase-1 activates of the proinflammatory ILs which may have a role in the formation and progression of tumorigenesis. Furthermore, the role of these ILs fighting against infection and autoimmune disease development are well-characterized, but their role in the development and progression of tumor residues remains controversial.128,129 The inhibition of SIRT1 expression through the noncanonical activity of caspase-1 may also partly contribute to the controversial effect of caspase-1 in tumor development and progression. The role of SIRT1 in tumorigenesis remains debatable although the inhibition of SIRT1 supports the therapeutic approach of some tumor cells like hepatocellular carcinoma.130
Moreover, the expression of caspase-1 is not equally observed in different tumor cells. For example, the expression of caspase-1 is low in colon, ovarian, and prostate cancers.131–133 in these cancers, the deficiency of caspase-1 may be contributed for the progression of tumor cell growth. This might be due to the effect of caspase-1 overexpression which induces the initiation of inflammatory programmed cell death (pyroptosis) and in turn lowers the growth of tumor cells.134 Programmed cell death (PCD) is the way of regulatory cell death that controlled by specific signaling pathways.135 Of the three PCD (apoptosis, necrosis, and pyroptosis), pyroptosis is targeted by the activation of caspase-1.136 Pyroptosis is a type of inflammatory-induced PCD commonly induced by the activation of gasdermin-D which is a noncanonical target molecule of active caspase-1.47,48 Pyroptosis is characterized by a lytic or membrane ruptured, pore forming and cell swelling type of cell death, which in turn releases inflammatory cytokine like IL-1β and IL-18 and may further induce inflammatory response.137 In the process, chromatin DNA fragmentation and nuclear condensation can also occurr.138 Therefore, we can generalize that caspase-1 may contributed to the reduction of abnormal cell growth through this pathway, and may in turn lower tumor cell growth. Moreover, the protective role of caspase-1 in tumorigenesis might be due to the tissue remodeling effect of IL-18 in the tumor microenvironment.132
However, caspase-1 expression and activation of IL-1β are frequently observed in melanoma, mesothelioma, and gastric cancers, in turn promotes tumorigenesis.139–141 In support of this, one study on animal and human breast cancer models revealed that activation of IL-1β is highly associated with the progression and metastasis of the tumor cells. They also reported that the growth of tumor cells and their metastasis into lung is significantly reduced in inflammasome components deficient in mice compared to the wild-type. These authors further revealed that the growth of a tumor cell is progressed through inflammasome-induced recruitment of myeloid cells such as tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs).142 But the molecular mechanism of inflammasome-induced myeloid cell recruitment is not fully understood. Therefore, caspase-1 may have both tumorigenic or antitumorigenic effects on cancer development and progression, but it depends on the type of inflammasome, methodology, and cancer. For instance, in many azoxymethane–dextran sulfate sodium (AOM/DSS)-induced cancer models, caspase-1 has a protective role in cancer progression. In this model, activation of NLRP3, NLRP6 and NLR12 have been shown to suppress tumor development, but not in NLRC4.143–145 In addition, cellular site of caspase-1 activation can also determine its role in tumorigenesis; caspase-1 activation in inflammatory cells promotes tumorigenesis, while in antigen-presenting cells, it suppresses carcinogenesis.139
With the presence of these controversies, we can summarize that the development and progression of inflammasome (caspase-1) induced tumorigenesis with five subdivisions (Figure 2). (i) Inflammatory induced release of ROS and RNS; maturation of IL-1β and IL-18 through inflammasome pathway attracts the release of more cytokines and chemokines, which brings cellular damage due to inflammatory reaction, then the inflamed tissues release ROS and RNS. These molecules are very toxic and may directly damage cellular proteins, lipids, carbohydrates, nucleic acid, and metabolic intermediates through oxidation or other way of reactions with them. Then, there will be a suppression of tumor suppressor genes, activation of tumor promoter pathways like Akt and beta-catenin signaling, then finally leads to replication mistakes and promotion of carcinogenesis.146,147 (ii) Inflammation-induced genomic instability; chronic inflammation in particular associates with the instability of genes such as microsatellite instability and shortening of telomeres. This favors the development and progression of tumorigenesis.148–150 (iii) Inflammation-induced impairment of DNA repair system; the DNA repair systems, such as nucleotide excision repair (NER), base excision repair (BER), and DNA mismatch repair (MMR) might be impaired due to inflammatory-induced overproduction of ROS/RNS and genomic instabilities, thereby facilitating the process of carcinogenesis.151 (iv) Inflammation induced alteration of signaling pathways; as DNA repair mechanism fails, there will be an initiation of signaling pathway alterations that are critical to control cell survival, proliferation, damage repair and apoptosis. Of the many signaling pathways, NF-κB and STAT3 signaling pathway alterations are frequently observed in this condition and promotes the process of carcinogenesis.152 For example, the activation and release of inflammasome induced NF-κB into the nucleus stimulates the inflammatory activity of the cell and cell survival genes.153 (v) Host defense induced carcinogenesis; when the epithelial cells of the host is not intact due to the inflammatory reaction or different stimuli, the normal flora found in the host becomes pathological and may activate carcinogenic-promoting genes like NF-κB pathway, leading to the progression of tumorigenesis.154
Physiologic and Pharmacological Inhibitors and/or Activators of Caspase-1 and Its Associated Cytokines, and Their Use as Therapeutic Targets in Obesity, Diabetes Mellitus, Cardiovascular Diseases and Cancers
As discussed earlier, the activation of inflammasome is a physiological process initiated by immune cells to prevent the host cells against stimulatory materials. But overexpression of inflammasome components may directly or indirectly contribute to the pathogenesis of several diseases, including obesity, DM, CVDs, and cancers. Therefore, it is expected that the activation of inflammasomes is regulated by several mechanisms physiologically. Among these, type-1 interferon (IFN), activated T cells, post-translational modifications of inflammasome components and immunoglobulin G (IgG) immune complexes showed a regulatory role of inflammasome activation.155–158 An experimental study revealed that IFN (IFN-α and IFN-β) inhibits the activation of NLRP1 and NLRP3 inflammasomes through the stimulation of IL-10 with a successive activation of signal transducer and activator of transcription (STAT1 and STAT1) and consequently suppresses the activation of caspase-1 with unclear mechanism.156 Moreover, activated T cells can have the ability to inhibit the activation of NLRP1 and NLRP3 inflammasomes through cell-to-cell interaction that initiated by activated T cell-induced expression of TNF-family ligands like RANKL and CD40L.157 One study also reported that post-translational modification of NLRP3 inflammasome (thiolnitrosylation of NLRP3 via the production of nitric oxide) due to the activation of IFN-γ by Mycobacterium tuberculosis inhibits the activation of caspase-1.159 The activation of inflammasome such as NLRP3, NLRC4, or AIM2 can also be inhibited by the IgG immune complexes formation. Based on the situation of stimulants, IgG immune complexes can adjust immune responses in either a pro- or anti-inflammatory direction. The production of IL-1α and IL-1β are suppressed through the ligation of activating Fcγ receptors (FcγR) by IgG immune complexes during the priming stage (in response to either microbial or endogenous danger signals) of inflammasome complex formation.158
Inflammasomes, especially NLRP3 inflammasome, are activated by several stimuli and their overexpression is highly associated with the development of NCDs. Due to this reason, investigating the potential pharmacological inhibitors of inflammasomes is becoming a target site to control the progression of various NCDs.160 Experimental studies in animal models revealed that the pharmacological inhibitors of inflammasomes can be targeted by the NLRP proteins, upstream signals, components of the inflammasome, target substrates of caspase-1 and receptors of the IL-1β and IL-18.161–164 The pharmacological inhibitors of NLRP3 inflammasome are extensively studied and some are specific for it. The specific inhibitors can suppress the activation of caspase-1 with NLRP3 inflammasome assembly but not AIM2, NLRP1 and NLRC4 inflammasome pathways (Figure 1). As NLRP3 inflammasome is the most common cause of NCDs, several in vivo and vitro studies have reported that inhibition of NLRP3 inflammasome helps to prevent the progression of obesity, DM, CVDs, and cancers.98,110,111,165,166 Therefore, from here, we summarized the pharmacological inhibitors of NLRP3 inflammasome into three major categories; (i) direct inhibitors of NLRP3 proteins. These inhibitors are designed to direct inhibition of NLRP protein expression, which in turn results in the blockage of NLRP3-NLRP3, NLRP3-NEK7 and NLRP3-ASC interaction (blockade of its ATPase activity) and activation of the inflammasome. Some these inhibitors are 3,4-methylenedioxy-b-nitrostyrene (MNS),161 OLT1177,162 MCC950,167 CY-09,163 oridonin168 and tranilast.163 (ii) Indirect inhibitors of the NLRP3 inflammasome. These inhibitors are indirectly blocking the activation of inflammasomes by targeting the P2X7 receptor, K+ efflux or the conformational changes of the NLRP3. Among these inhibitors, the most studied are glyburide,98 JC124,169 16673–34-0164 and FC11A-2.170 (iii) Inhibitors targeting the NLRP3 inflammasome components. Although all the target components of inflammasome is not clearly addressed, in vivo and vitro studies have reported that some inhibitors can be blocked the activity of caspase-1 by covalent modification of active caspase-1. Then, the target substrates of caspase-1 will not be activated. Other inhibitors can also suppress the NF-kB pathway via the inhibition of kinase activity of IKKβ. Some of the inhibitors targeting the inflammasome components are VX-740 (Pralnacasan) and its analog VX-765,171 parthenolide,172 β-Hydroxybutyrate (BHB)172 and Bay 11-7082.172
Collectively, inflammasome-induced activation of proinflammatory cytokines, gasdermin-D, and noncanonical target substrates are directly or indirectly associated with the development and progression of several NCDs, including obesity, DM, CVDs, and cancers. However, the studies have not shown similar results; some of them are opposite and controversial. Besides, the therapeutic effect of inflammasome inhibition is not equally contributed for the prevention of NCDs. Moreover, exact molecular mechanisms of inflammasome inhibition for some inhibitors are not clearly addressed yet.
Conclusion and Future Perspective
Activation of caspase-1 and inflammasome initially aims to regulate the immune cells to control the external and internal stimuli. If there is a failure to control the process, it may lead to inflammatory-induced secretion of other cytokines, chemokines, and other signaling pathways, which may further disturb the immune response against the insults. Although the effect of caspase-1 in different inflammatory-induced NCDs is distinct, inflammasome gives a new stand to look at the ambiguities of inflammation-associated chronic metabolic disorders, such as obesity, DM, CVDs, and cancer development. Therefore, to the extent of the current findings, we can generalize that caspase-1 may have both negative and positive effects on the initiation and progression of several NCDs which depends on several conditions. Furthermore, caspase-1 can control the occurrence of these diseases through the maturation of immune cells such as IL-18 or the regulation of inflammatory programmed cell death (pyroptosis) by activating gasdermin-D. On the contrary, caspase-1 can directly manipulate the expression of some protein/genes, such as SIRT-1, NF-Kβ and in turn disturbs the physiological activity of the cells, which further leads to the ignition of metabolic disorders. In this regard, we believed that this huge study variation will be extensively explored in the near future and achieve similar conclusions.
Abbreviations
NCDs, noncommunicable diseases; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; TLRs, toll-like receptors; IL-1β, interleukin 1 beta; NLRP, nucleotide-binding oligomerization domain, leucine rich repeat and pyrin domain containing; NBD, nucleotide-binding oligomerization domain; LRR, leucine rich repeat; ASC, apoptosis-associated speck-like protein containing a CARD; CARD, caspase activation and recruitment domains; ATP, adenosine triphosphate; RNS, reactive nitrogen species; NF-κB, nuclear factor kappaB; MAPK, mitogen-activated protein kinase.
Acknowledgment
The authors would like to acknowledge Mr Anteneh Ayelign for his unreserved contribution in language editing.
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed on the journal to which the article will be submitted; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Johnson RJ, Nakagawa T, Sanchez-Lozada LG, et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307–3315. doi:10.2337/db12-1814
2. McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5:a008656. doi:10.1101/cshperspect.a008656
3. Black RA, Kronheim SR, Merriam JE, March CJ, Hopp TP. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J Biol Chem. 1989;264:5323–5326.
4. Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi:10.1038/356768a0
5. Martinon F, Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 2007;14:10. doi:10.1038/sj.cdd.4402038
6. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10:417–426. doi:10.1016/S1097-2765(02)00599-3
7. Kayagaki N, Warming S, Lamkanfi M, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi:10.1038/nature10558
8. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi:10.1146/annurev.immunol.021908.132612
9. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi:10.1172/JCI57132
10. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi:10.1038/nature04516
11. Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi:10.1038/nature04515
12. Vandanmagsar B, Youm Y-H, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179. doi:10.1038/nm.2279
13. Kanneganti T-D, Özören N, Body-Malapel M, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440:233–236. doi:10.1038/nature04517
14. Zhong YK, Saleh M. FunctionsofNOD-likereceptorsinhumandiseases. Front Immunol. 2013;4:333.
15. Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi:10.1038/nature05485
16. Suganami T, Tanaka M, Ogawa Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr J. 2012;EJ12–E0271.
17. Faraj M, Lu HL, Cianflone K. Diabetes, lipids, and adipocyte secretagogues. Biochem Cell Biol. 2004;82:170–190. doi:10.1139/o03-078
18. Stumvoll M, Goldstein BJ, Van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet. 2005;365:1333–1346. doi:10.1016/S0140-6736(05)61032-X
19. Fève B, Bastard J-P. The role of interleukins in insulin resistance and type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:305. doi:10.1038/nrendo.2009.62
20. Netea MG, Joosten LA, Lewis E, et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat Med. 2006;12:650–656. doi:10.1038/nm1415
21. Ekpenyong C, Udokang NE, Akpan E, Samson T. Double burden, non-communicable diseases and risk factors evaluation in sub-Saharan Africa: the Nigerian experience. Eur J Sustain Dev. 2012;1:249. doi:10.14207/ejsd.2012.v1n2p249
22. Naghavi M, Abajobir AA, Abbafati C, et al. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1151–1210. doi:10.1016/S0140-6736(17)32152-9
23. Alzeidan R, Rabiee F, Mandil A, Hersi A, Fayed A. Non-communicable disease risk factors among employees and their families of a Saudi university: an epidemiological study. PLoS One. 2016;11:e0165036. doi:10.1371/journal.pone.0165036
24. Koenen TB, Stienstra R, Van Tits LJ, et al. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1β transcription in human adipose tissue. Diabetes. 2011;60:517–524. doi:10.2337/db10-0266
25. Organization WH. Preventing Noncommunicable Diseases (Ncds) by Reducing Environmental Risk Factors. World Health Organization; 2017.
26. Fernández DJ, Lamkanfi M. Inflammatory caspases: key regulators of inflammation and cell death. Biol Chem. 2015;396:193–203. doi:10.1515/hsz-2014-0253
27. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi:10.1016/j.cell.2014.04.007
28. Lamkanfi M. Emerging inflammasome effector mechanisms. Nat Rev Immunol. 2011;11:213–220. doi:10.1038/nri2936
29. Saleh M. The machinery of Nod‐like receptors: refining the paths to immunity and cell death. Immunol Rev. 2011;243:235–246. doi:10.1111/j.1600-065X.2011.01045.x
30. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi:10.1038/nri3452
31. Schroder K, Tschopp J. The inflammasomes. cell. 2010;140:821–832. doi:10.1016/j.cell.2010.01.040
32. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. nature. 2012;481:278–286. doi:10.1038/nature10759
33. Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi:10.1016/j.immuni.2013.05.016
34. Brodsky IE, Monack D NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens.
35. Próchnicki T, Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. 2017;26:71–93. doi:10.1016/j.cmet.2017.06.018
36. Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7:243–249. doi:10.1038/cmi.2010.10
37. Tenthorey JL, Kofoed EM, Daugherty MD, Malik HS, Vance RE. Molecular basis for specific recognition of bacterial ligands by NAIP/NLRC4 inflammasomes. Mol Cell. 2014;54:17–29. doi:10.1016/j.molcel.2014.02.018
38. Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi:10.1038/nature07710
39. Jin T, Perry A, Jiang J, et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36:561–571. doi:10.1016/j.immuni.2012.02.014
40. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165:792–800. doi:10.1016/j.cell.2016.03.046
41. von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31:73–106. doi:10.1146/annurev-immunol-032712-095944
42. Sharma D, Kanneganti T-D. The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617–629. doi:10.1083/jcb.201602089
43. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247.
44. Henao-Mejia J, Elinav E, Jin -C-C, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179. doi:10.1038/nature10809
45. Ke Z, Liang D, Zeng Q, et al. hsBAFF promotes proliferation and survival in cultured B lymphocytes via calcium signaling activation of mTOR pathway. Cytokine. 2013;62:310–321. doi:10.1016/j.cyto.2013.03.011
46. McGettrick A, O’neill L. NLRP3 and IL‐1β in macrophages as critical regulators of metabolic diseases. Diabetes Obes Metab. 2013;15:19–25. doi:10.1111/dom.12169
47. Keller M, Rüegg A, Werner S, Beer H-D. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi:10.1016/j.cell.2007.12.040
48. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. doi:10.1038/nrmicro2070
49. Molla MD, Ayelign B, Dessie G, Geto Z, Admasu TD. Caspase-1 as a regulatory molecule of lipid metabolism. Lipids Health Dis. 2020;19:1–7. doi:10.1186/s12944-020-01220-y
50. Skeldon AM, Faraj M, Saleh M. Caspases and inflammasomes in metabolic inflammation. Immunol Cell Biol. 2014;92:304. doi:10.1038/icb.2014.5
51. Geto Z, Molla MD, Challa F, Belay Y, Getahun T. Mitochondrial dynamic dysfunction as a main triggering factor for inflammation associated chronic non-communicable diseases. J Inflamm Res. 2020;13:97. doi:10.2147/JIR.S232009
52. Razani B, Chakravarthy MV, Semenkovich CF. Insulin resistance and atherosclerosis. Endocrinol Metab Clin North Am. 2008;37:603–621. doi:10.1016/j.ecl.2008.05.001
53. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–184. doi:10.1172/JCI29881
54. Stienstra R, Joosten LA, Koenen T, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12:593–605. doi:10.1016/j.cmet.2010.11.011
55. Stienstra R, Van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci. 2011;108:15324–15329. doi:10.1073/pnas.1100255108
56. Wang H, Capell W, Yoon J, Faubel S, Eckel R. Obesity development in caspase-1-deficient mice. Int J Obes. 2014;38:152–155. doi:10.1038/ijo.2013.59
57. Murphy AJ, Kraakman MJ, Kammoun HL, et al. IL-18 production from the NLRP1 inflammasome prevents obesity and metabolic syndrome. Cell Metab. 2016;23:155–164. doi:10.1016/j.cmet.2015.09.024
58. Lindegaard B, Matthews VB, Brandt C, et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes. 2013;62:3064–3074. doi:10.2337/db12-1095
59. Dinarello CA. IL-18: A TH1-inducing, proinflammatory cytokine and new member of the IL-1 family. J Allergy Clin Immunol. 1999;103:11–24. doi:10.1016/S0091-6749(99)70518-X
60. Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15:635–645. doi:10.1016/j.cmet.2012.04.001
61. Wellen K, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi:10.1172/JCI25102
62. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi:10.1172/JCI200319246
63. Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi:10.1172/JCI200319451
64. Wen H, Gris D, Lei Y, et al. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12:408. doi:10.1038/ni.2022
65. Lee Y-H, Pratley RE. The evolving role of inflammation in obesity and the metabolic syndrome. Curr Diab Rep. 2005;5:70–75. doi:10.1007/s11892-005-0071-7
66. Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi:10.1038/nri1937
67. Chalkiadaki A, Guarente L. High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 2012;16:180–188.
68. Yin Y, Li X, Sha X, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol. 2015;115:305282.
69. Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260:273–279. doi:10.1006/bbrc.1999.0897
70. Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-γ. Nature. 2004;429:771. doi:10.1038/nature02583
71. Yoshizaki T, Milne JC, Imamura T, et al. SIRT1 exerts anti-inflammatory effects and improves insulin sensitivity in adipocytes. Mol Cell Biol. 2009;29:1363–1374. doi:10.1128/MCB.00705-08
72. Walker AK, Yang F, Jiang K, et al. Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev. 2010;24:1403–1417. doi:10.1101/gad.1901210
73. Ponugoti B, Kim D-H, Xiao Z, et al. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J Biol Chem. 2010;285:33959–33970. doi:10.1074/jbc.M110.122978
74. Shibabaw T, Dessie G, Molla MD, Zerihun MF, Ayelign B. Assessment of liver marker enzymes and its association with type 2 diabetes mellitus in Northwest Ethiopia. BMC Res Notes. 2019;12:707. doi:10.1186/s13104-019-4742-x
75. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14:88.
76. Saeedi P, Petersohn I, Salpea P, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas. Diabetes Res Clin Pract. 2019;157:107843. doi:10.1016/j.diabres.2019.107843
77. Ogurtsova K, da Rocha Fernandes J, Huang Y, et al. IDF Diabetes Atlas: global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res Clin Pract. 2017;128:40–50. doi:10.1016/j.diabres.2017.03.024
78. Alberti KGM, Zimmet P. Epidemiology: global burden of disease—where does diabetes mellitus fit in? Nat Rev Endocrinol. 2013;9:258. doi:10.1038/nrendo.2013.54
79. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi:10.1038/nature05482
80. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi:10.1146/annurev-physiol-021909-135846
81. Liu C, Feng X, Li Q, Wang Y, Li Q, Hua M. Adiponectin, TNF-α and inflammatory cytokines and risk of type 2 diabetes: a systematic review and meta-analysis. Cytokine. 2016;86:100–109. doi:10.1016/j.cyto.2016.06.028
82. King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol. 2008;79:1527–1534. doi:10.1902/jop.2008.080246
83. Rivero-González A, Martín-Izquierdo E, Marín-Delgado C, Rodríguez-Muñoz A, Navarro-González JF. Cytokines in diabetes and diabetic complications. In: Cytokine Effector Functions in Tissues. Elsevier; 2017:119–128.
84. Williams MD, Nadler JL. Inflammatory mechanisms of diabetic complications. Curr Diab Rep. 2007;7:242–248. doi:10.1007/s11892-007-0038-y
85. Rodrigues KF, Pietrani NT, Bosco AA, Campos FMF, Sandrim VC, Gomes KB. IL-6, TNF-α, and IL-10 levels/polymorphisms and their association with type 2 diabetes mellitus and obesity in Brazilian individuals. Arch Endocrinol Metab. 2017;61:438–446. doi:10.1590/2359-3997000000254
86. Gupta S, Maratha A, Siednienko J, et al. Analysis of inflammatory cytokine and TLR expression levels in type 2 diabetes with complications. Sci Rep. 2017;7:7633.
87. Donath MY, Böni-Schnetzler M, Ellingsgaard H, Halban PA, Ehses JA. Cytokine production by islets in health and diabetes: cellular origin, regulation and function. Trends Endocrinol Metab. 2010;21:261–267. doi:10.1016/j.tem.2009.12.010
88. Donath MY, Dalmas É, Sauter NS, Böni-Schnetzler M. Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell Metab. 2013;17:860–872. doi:10.1016/j.cmet.2013.05.001
89. Stratford S, Hoehn KL, Liu F, Summers SA. Regulation of insulin action by ceramide dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J Biol Chem. 2004;279:36608–36615. doi:10.1074/jbc.M406499200
90. Ussher JR, Koves TR, Cadete VJ, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole-body oxygen consumption. Diabetes. 2010;59:2453–2464. doi:10.2337/db09-1293
91. Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002. doi:10.1172/JCI34260
92. Jager J, Grémeaux T, Cormont M, Le Marchand-brustel Y, Tanti J-F. Interleukin-1β-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology. 2007;148:241–251. doi:10.1210/en.2006-0692
93. Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes. 2003;52:2784–2789. doi:10.2337/diabetes.52.11.2784
94. Gao Z, Hwang D, Bataille F, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor κB kinase complex. J Biol Chem. 2002;277:48115–48121. doi:10.1074/jbc.M209459200
95. Stephens J, Pekala P. Transcriptional repression of the C/EBP-alpha and GLUT4 genes in 3T3-L1 adipocytes by tumor necrosis factor-alpha. Regulations is coordinate and independent of protein synthesis. J Biol Chem. 1992;267:13580–13584.
96. Bendtzen K, Mandrup-Poulsen T, Nerup J, Nielsen JH, Dinarello CA, Svenson M. Cytotoxicity of human pI 7 interleukin-1 for pancreatic islets of Langerhans. Science. 1986;232:1545–1547. doi:10.1126/science.3086977
97. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi:10.1038/ni.1831
98. Lamkanfi M, Mueller JL, Vitari AC, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187:61–70. doi:10.1083/jcb.200903124
99. Larsen CM, Faulenbach M, Vaag A, et al. Interleukin-1–receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356:1517–1526. doi:10.1056/NEJMoa065213
100. Mendis S, Puska P, Norrving B; Organization WH. Global Atlas on Cardiovascular Disease Prevention and Control. World Health Organization; 2011.
101. Merkle S, Frantz S, Schön MP, et al. A role for caspase-1 in heart failure. Circ Res. 2007;100:645–653. doi:10.1161/01.RES.0000260203.55077.61
102. Syed FM, Hahn HS, Odley A, et al. Proapoptotic effects of caspase-1/interleukin-converting enzyme dominate in myocardial ischemia. Circ Res. 2005;96:1103–1109.
103. Rauf A, Shah M, Yellon DM, Davidson SM. Role of caspase 1 in ischemia/reperfusion injury of the myocardium. J Cardiovasc Pharmacol. 2019;74:194–200. doi:10.1097/FJC.0000000000000694
104. Frantz S, Ducharme A, Sawyer D, et al. Targeted deletion of caspase-1 reduces early mortality and left ventricular dilatation following myocardial infarction. J Mol Cell Cardiol. 2003;35:685–694. doi:10.1016/S0022-2828(03)00113-5
105. Krishnan SM, Sobey CG, Latz E, Mansell A, Drummond GR. IL‐1β and IL‐18: inflammatory markers or mediators of hypertension? Br J Pharmacol. 2014;171:5589–5602. doi:10.1111/bph.12876
106. Fujita T. Mechanisms of salt induced hypertension: focus on adrenal and sympathetic nervous system. J Am Soc Nephrol. 2014;25:1148–1155. doi:10.1681/ASN.2013121258
107. Qi J, Zhao X-F, Yu X-J, et al. Targeting interleukin-1 beta to suppress sympathoexcitation in hypothalamic paraventricular nucleus in dahl salt-sensitive hypertensive rats. Cardiovasc Toxicol. 2016;16:298–306. doi:10.1007/s12012-015-9338-7
108. Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014;2014:1–8. doi:10.1155/2014/507208
109. Usui F, Shirasuna K, Kimura H, et al. Critical role of caspase-1 in vascular inflammation and development of atherosclerosis in Western diet-fed apolipoprotein E-deficient mice. Biochem Biophys Res Commun. 2012;425:162–168. doi:10.1016/j.bbrc.2012.07.058
110. Peng K, Liu L, Wei D, et al. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int J Mol Med. 2015;35:1179–1188. doi:10.3892/ijmm.2015.2129
111. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi:10.1038/nature08938
112. Lu J, Mitra S, Wang X, Khaidakov M, Mehta JL. Oxidative stress and lectin-like ox-LDL-receptor LOX-1 in atherogenesis and tumorigenesis. Antioxid Redox Signal. 2011;15:2301–2333. doi:10.1089/ars.2010.3792
113. Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2018;113:5. doi:10.1007/s00395-017-0663-9
114. Wu D, Ren P, Zheng Y, et al. NLRP3 (nucleotide oligomerization domain–like receptor family, pyrin domain containing 3)–caspase-1 inflammasome degrades contractile proteins: implications for aortic biomechanical dysfunction and aneurysm and dissection formation. Arterioscler Thromb Vasc Biol. 2017;37:694–706. doi:10.1161/ATVBAHA.116.307648
115. Golledge J, Norman PE. Atherosclerosis and abdominal aortic aneurysm: cause, response, or common risk factors? Am Heart Assoc. 2010.
116. Saxena A, Chen W, Su Y, et al. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol. 2013;191:4838–4848. doi:10.4049/jimmunol.1300725
117. Toldo S, Mezzaroma E, Mauro AG, Salloum F, Van Tassell BW, Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2015;22:1146–1161. doi:10.1089/ars.2014.5989
118. Kawaguchi M, Takahashi M, Hata T, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594–604. doi:10.1161/CIRCULATIONAHA.110.982777
119. Smith RS, Smith TJ, Blieden TM, Phipps RP. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am J Pathol. 1997;151:317.
120. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
121. Hanahan D, Weinberg RA. The hallmarks of cancer. cell. 2000;100:57–70. doi:10.1016/S0092-8674(00)81683-9
122. Vaughan TL, Davis S, Kristal A, Thomas DB. Obesity, alcohol, and tobacco as risk factors for cancers of the esophagus and gastric cardia: adenocarcinoma versus squamous cell carcinoma. Cancer Epidemiol Biomarkers Prev. 1995;4:85–92.
123. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi:10.1016/j.cell.2010.01.025
124. Schäfer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–638. doi:10.1038/nrm2455
125. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi:10.1038/nature07205
126. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867.
127. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. 2004;4:11–22. doi:10.1038/nrc1252
128. Wen H, Ting JP-Y, O’Neill LAJ. A role for the NLRP3 inflammasome in metabolic diseases—did Warburg miss inflammation? Nat Immunol. 2012;13(4):352–357. doi:10.1038/ni.2228
129. Guo B, Li Z. Endoplasmic reticulum stress in hepatic steatosis and inflammatory bowel diseases. Front Genet. 2014;5:242. doi:10.3389/fgene.2014.00242
130. Molla MD, Dessie G, Akalu Y, Ayelign B. Hepatocellular expression of SIRT1 and its effect on hepatocellular carcinoma progression: a future therapeutic perspective. Int J Hepatol. 2020;2020.
131. Winter RN, Kramer A, Borkowski A, Kyprianou N. Loss of caspase-1 and caspase-3 protein expression in human prostate cancer. Cancer Res. 2001;61:1227–1232.
132. Allen IC, TeKippe EM, Woodford R-MT, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med. 2010;207:1045–1056. doi:10.1084/jem.20100050
133. Feng Q, Li P, Salamanca C, Huntsman D, Leung PC, Auersperg N. Caspase-1α is down-regulated in human ovarian cancer cells and the overexpression of caspase-1α induces apoptosis. Cancer Res. 2005;65:8591–8596. doi:10.1158/0008-5472.CAN-05-0239
134. Xia X, Wang X, Cheng Z, et al. The role of pyroptosis in cancer: pro-cancer or pro-“host”? Cell Death Dis. 2019;10:1–13. doi:10.1038/s41419-019-1883-8
135. Sun Y, Peng Z. Programmed cell death and cancer. Postgrad Med J. 2009;85:134–140. doi:10.1136/pgmj.2008.072629
136. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907–1916. doi:10.1128/IAI.73.4.1907-1916.2005
137. Fink SL, Cookson BT. Caspase‐1‐dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006;8:1812–1825. doi:10.1111/j.1462-5822.2006.00751.x
138. Bergsbaken T, Cookson BT. Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 2007;3:e161. doi:10.1371/journal.ppat.0030161
139. Zitvogel L, Kepp O, Galluzzi L, Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol. 2012;13:343. doi:10.1038/ni.2224
140. Bruchard M, Mignot G, Derangère V, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19:57–64. doi:10.1038/nm.2999
141. Tu S, Bhagat G, Cui G, et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008;14:408–419. doi:10.1016/j.ccr.2008.10.011
142. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep. 2016;6:1–12.
143. Chen GY, Liu M, Wang F, Bertin J, Núñez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol. 2011;186:7187–7194. doi:10.4049/jimmunol.1100412
144. Allen IC, Wilson JE, Schneider M, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity. 2012;36:742–754. doi:10.1016/j.immuni.2012.03.012
145. Hu B, Elinav E, Huber S, et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci. 2010;107:21635–21640. doi:10.1073/pnas.1016814108
146. Marnett LJ. Oxyradicals and DNA damage. carcinogenesis. 2000;21:361–370.
147. Covey TM, Edes K, Coombs GS, Virshup DM, Fitzpatrick FA. Alkylation of the tumor suppressor PTEN activates Akt and β-catenin signaling: a mechanism linking inflammation and oxidative stress with cancer. PLoS One. 2010;5:e13545. doi:10.1371/journal.pone.0013545
148. Isokawa O, Suda T, Aoyagi Y, et al. Reduction of telomeric repeats as a possible predictor for development of hepatocellular carcinoma: convenient evaluation by slot‐blot analysis. Hepatology. 1999;30:408–412. doi:10.1002/hep.510300211
149. Hou L, Savage SA, Blaser MJ, et al. Telomere length in peripheral leukocyte DNA and gastric cancer risk. Cancer Epidemiol Biomarkers Prev. 2009;18:3103–3109. doi:10.1158/1055-9965.EPI-09-0347
150. Yang Y, Fruehauf J, Xiang S, Li CJ. Genomic instability in precancerous lesions before inactivation of tumor suppressors p53 and APC in patients. Cell Cycle. 2006;5:1443–1447. doi:10.4161/cc.5.13.2897
151. Güngör N, Haegens A, Knaapen AM, et al. Lung inflammation is associated with reduced pulmonary nucleotide excision repair in vivo. Mutagenesis. 2010;25:77–82. doi:10.1093/mutage/gep049
152. Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21:11–19. doi:10.1016/j.cytogfr.2009.11.005
153. Ghosh S, Karin M. Missing pieces in the NFkB puzzle. Cell. 2002;109:S81–S96. doi:10.1016/S0092-8674(02)00703-1
154. Porta C, Riboldi E, Sica A. Mechanisms linking pathogens-associated inflammation and cancer. Cancer Lett. 2011;305:250–262. doi:10.1016/j.canlet.2010.10.012
155. Billiau A. Anti-inflammatory properties of Type I interferons. Antiviral Res. 2006;71:108–116. doi:10.1016/j.antiviral.2006.03.006
156. Guarda G, Braun M, Staehli F, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–223. doi:10.1016/j.immuni.2011.02.006
157. Guarda G, Dostert C, Staehli F, et al. T cells dampen innate immune responses through inhibition of NLRP1 and NLRP3 inflammasomes. Nature. 2009;460:269–273. doi:10.1038/nature08100
158. Janczy JR Mechanisms for activation and inhibition of inflammasomes. 2014.
159. Mishra BB, Rathinam VA, Martens GW, et al. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome–dependent processing of IL-1β. Nat Immunol. 2013;14:52–60.
160. Zahid A, Li B, Kombe JK, Jin T, Tao J. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol. 2019;10:2538. doi:10.3389/fimmu.2019.02538
161. He Y, Varadarajan S, Muñoz-Planillo R, Burberry A, Nakamura Y, Núñez G. 3, 4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem. 2014;289:1142–1150. doi:10.1074/jbc.M113.515080
162. Marchetti C, Swartzwelter B, Gamboni F, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci. 2018;115:E1530–E9. doi:10.1073/pnas.1716095115
163. Lamkanfi M, Dixit VM. A new lead to NLRP3 inhibition. J Exp Med. 2017;214:3147. doi:10.1084/jem.20171848
164. Marchetti C, Toldo S, Chojnacki J, et al. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and non-ischemic injury in the mouse. J Cardiovasc Pharmacol. 2015;66:1. doi:10.1097/FJC.0000000000000247
165. Altaf A, Qu P, Zhao Y, Wang H, Lou D, Niu N. NLRP3 inflammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coron Artery Dis. 2015;26:409–421. doi:10.1097/MCA.0000000000000255
166. Karki R, Man SM, Kanneganti T-D. Inflammasomes and cancer. Cancer Immunol Res. 2017;5:94–99. doi:10.1158/2326-6066.CIR-16-0269
167. Perregaux DG, McNiff P, Laliberte R, et al. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J Pharm Exp Ther. 2001;299:187–197.
168. He H, Jiang H, Chen Y, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018;9:1–12. doi:10.1038/s41467-018-04947-6
169. Fulp J, He L, Toldo S, et al. Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: design, synthesis, and biological characterization. J Med Chem. 2018;61:5412–5423. doi:10.1021/acs.jmedchem.8b00733
170. Liu W, Guo W, Wu J, et al. A novel benzo [d] imidazole derivate prevents the development of dextran sulfate sodium-induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem Pharmacol. 2013;85:1504–1512. doi:10.1016/j.bcp.2013.03.008
171. Boxer MB, Shen M, Auld DS, Wells JA, Thomas CJ A small molecule inhibitor of Caspase 1. Probe Reports from the NIH Molecular Libraries Program [Internet]. National Center for Biotechnology Information (US); 2011.
172. Juliana C, Fernandes-Alnemri T, Wu J, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285:9792–9802. doi:10.1074/jbc.M109.082305
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.