")
Back to Archived Journals » Journal of Receptor, Ligand and Channel Research » Volume 8
Role of calcitonin gene-related peptide and brain natriuretic peptide to modulate the excitability state of trigeminal neurons: relevance to migraine pathology and treatment
Authors Vilotti S, Fabbretti E, Nistri A
Received 27 October 2014
Accepted for publication 28 November 2014
Published 19 January 2015 Volume 2015:8 Pages 31—41
DOI https://doi.org/10.2147/JRLCR.S50993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Trevor W. Stone
Sandra Vilotti,1 Elsa Fabbretti,2 Andrea Nistri1
1Neuroscience Department, International School for Advanced Studies (SISSA), Trieste, Italy; 2Center for Biomedical Sciences and Engineering, University of Nova Gorica, Nova Gorica, Slovenia
Abstract: Hyperactivity of trigeminal sensory neurons is a major process to generate recurrent headache, typical of migraine attacks. How physiological nociception is converted into strong pathological pain remains, however, poorly understood. In recent years, certain neuropeptides and their receptors have been shown to modulate sensory neuron nociception and to contribute to the persistent hyperalgesia due to the sensory stimulus sensitization that defines the clinical experience of chronic pain syndromes, including migraine. Using calcitonin gene-related peptide (CGRP) and brain natriuretic peptide (BNP) as examples, this review addresses the mechanisms through which neuropeptides might modulate nociceptor activity. One attractive notion is that pain signaling by trigeminal sensory neurons is potently regulated by the ambient levels of these peptides: CGRP is thought to facilitate neuronal firing responsible for trigeminal sensitization necessary to trigger headache, whereas BNP is proposed to act as a negative regulator of trigeminal neuron activity. For either peptide, the key target appears to be the ATP-gated P2X3 receptor that, widely expressed by trigeminal sensory neurons, generates fast, large excitation to release glutamate onto second-order brain neurons. The fine balance between the activities of these peptides is suggested to ultimately determine whether nociception is perceived at higher center as a physiological or pathological response. Hence, the clinical goal of CGRP antagonism using either pharmacological receptor blockers or monoclonal antibodies (to sequester this peptide or to directly inhibit its receptor) is currently considered a novel approach for migraine prophylaxis and to treat acute headache attacks.
Keywords: trigeminal ganglion, headache, sensory neurons, P2X3, TRPV1
Introduction – migraine pain can arise via chronic dysfunction of nociceptive neurons
While the molecular mechanisms of nociceptive neuron signaling have been widely investigated,1 the pathophysiological basis of chronic pain is less understood. A prototypical case is the recurrent chronic headache of migraine, which is a highly prevalent and disabling neurovascular disorder2 affecting a significant proportion of the adult population worldwide, and representing an enormous socioeconomic and health care burden for both the individual and society.3–5
Migraine is characterized by attacks of headache associated with autonomic nervous system dysfunction, and in about one-third of the cases, with transient neurological symptoms termed “aura”, whereby aberrant visual perceptions are the commonest complaint.6 Despite the fact that the migraine attack etiology remains largely unknown, evidence points to the notion that migraine headache results from strong activation of primary afferent neurons (trigeminal ganglion [TG] nociceptors) that innervate craniofacial tissues (including meninges and their blood vessels) and project to the trigeminal nucleus of the brainstem and spinal cord with further signal processing at higher brain centers.7–9 In physiological conditions, membrane depolarization (arising from mechanical, thermal, or chemical stimuli) of the peripheral terminals of TG nociceptors widely distributed over the craniofacial territory will, if sufficiently large to reach a certain threshold, evoke action potentials that propagate to the central nervous system. To decode such stimuli and convert them into sensory information, TG neurons express a range of receptors/ion channels that confer stimulus-related excitation. The depolarization induced by sensory stimuli is then modulated by membrane ion channels (e.g. K+ channels) that constrain the excitation propagating via voltage-gated Na+ channels responsible for the generation of action potentials. Conveying painful signals to the brain, therefore, demands a delicate balance between excitatory and inhibitory processes at the level of TG neurons. When such a fine equilibrium is pathologically disrupted, sensitization develops, whereby TG neurons become hyperresponsive even to mild stimuli: in the case of migraine, this is an important phenomenon to trigger headache.10 Furthermore, allodynia, namely pain generation by non-noxious stimuli, can occur in response to peripheral and central inputs.10–12 Symptoms of sensitization during migraine include throbbing headache and its aggravation during routine physical activities that increase intracranial pressure such as coughing and bending over.13,14
The question then arises as to whether sensitization can originate from functional changes in the receptor proteins transducing the sensory stimulus and/or in the voltage-dependent ion channels controlling neuron excitability. Recent studies have demonstrated that trigeminal sensory neurons express a large number of integral membrane proteins that sense strong external stimuli like pungent odors, chemicals, or temperature changes. The interest in their role related to migraine resides in the possibility that such proteins can be important triggers of migraine pain attacks either via direct activation of trigeminal neuron firing or via release of local mediators, in turn, exciting trigeminal neurons. This complex scenario presumably requires a delicate equilibrium between mechanisms dampening or enhancing neuronal excitability, and its derangement may lead to acute and/or chronic headache.15 Due to space constraints, the present short review focuses on the mechanisms of trigeminal pain transduction and how two endogenous neuropeptides, namely calcitonin gene-related peptide (CGRP) and brain natriuretic peptide (BNP), may up- or downregulate it. These (and other) neuropeptides might also be the intermediaries of the action of certain external stimuli on trigeminal sensory neurons as outlined earlier. Updated reviews are currently available for general involvement of CGRP in pain processing,16,17 while the role of BNP in maladaptive sensory signaling is a more recent finding.18,19
Molecular mechanisms of nociceptors involved in migraine pain signaling: shifting targets can facilitate pain onset
A large body of evidence has identified membrane proteins expressed on dural afferent nerve endings and on the soma of TG neurons and suggested to play an important role in migraine pathophysiology.20 Immunolabeling and retrograde-labeling experiments in rodents have revealed that most small- and medium-sized TG neurons innervating the dura express voltage-insensitive acid-sensing cationic channels (ASICs) as well as transient receptor potential (TRP) channels and purinergic P2X receptors.21–23 Although pharmacological block of cortical-spreading depression (the underlying process mediating aura) and TG activation was experimentally elicited by the potassium-sparing diuretic amiloride via an action proposed to suppress ASIC activity,23 there is little evidence that prior to (or during) a migraine attack, the local extracellular pH values can fall to a level sufficient for strong ASIC activation.
TRP receptor/channels comprise a wide family of membrane proteins with multifarious functions of relevance to pain processing.15,24 For instance, the potential role of TRPA1 receptors in migraine has been considered because TRPA1 agonists may trigger migraine headache in susceptible individuals.25–28 Nevertheless, TRPA1 receptors, which are expressed only by a small minority of sensory neurons,29 are typically stimulated by reactive oxygen species, and it is currently unclear whether such agents are generated in sufficient amount to trigger migraine or are by-products of subsequent neurovascular dysfunction associated with headache and contributing to sustain it. Recent results indicate that chemical irritants found as environmental pollutants may also stimulate TRPA1 channels and evoke headache.30
Among the family of TRP receptors that can contribute to triggering migraine attacks, transient receptor potential vanilloid (TRPV)-4 is expressed by meningeal fibers: its activation by cold temperature31 or airborne irritants32 produces afferent nociceptive signaling from the head that may contribute to migraine headache. TRPM8 channels that are activated by cold are also proposed to be involved in triggering trigeminal neuron activity33 in accordance with the notion that a TRPM8 gene variant confers susceptibility to migraine.34 While several studies have demonstrated capsaicin (and heat)-sensitive TRPV1 channels to be important for the development of inflammatory, thermal pain conditions,35–37 their ultimate role in migraine remains unclear, and there are no current antimigraine drugs blocking TRPV1 receptors in clinical use.
Cannabinoids have been commonly associated with analgesic effects. In the case of headache, trigeminal neurons express the G-protein-coupled CB1 receptors that are the principal transducers of the neuronal action of endogenous endocannabinoids like anandamine and 2-acylglycerol.15 The hypothesis of the role of endocannabinoids in migraine was proposed as part of a syndrome (clinical endocannabinoid deficiency) with facilitated onset of pain.38 This concept was supported by the clinical observation of low endocannabinoid levels in the cerebrospinal fluid of migraine patients.39 The role of endocannabinoids in trigeminal pain processing is complex because anandamide can produce contrasting effects on the trigeminovascular unit activity, in which neurons, glia, and local vascular elements interact at meningeal level.40,41 Furthermore, anandamide exerts several cellular effects distinct from CB1 receptor activation, including activation of TRPV1 receptors.42 These multifarious actions by endocannabinoids may ultimately converge onto the modulatory effects of neuropeptides.
A number of studies have proposed that an acute attack of migraine is associated with a large release of ATP into the extracellular compartment to stimulate nociceptors.43 While ATP receptors constitute a heterogeneous population, those with ionotropic properties and therefore, producing rapid ion current responses, belong to the P2X subclass. Among ATP-sensitive P2X receptors, the P2X3 receptors are selectively and almost exclusively expressed by sensory ganglion neurons,44,45 suggesting their key role in processing pain. The subject of P2X3 receptors has been reviewed in detail.46,47 In the mouse TG, P2X3 receptors are expressed by the vast majority of sensory neurons that may also express receptors for capsaicin (TRPV1)48 and have been implicated in craniofacial pain,49,50 including migraine.47 These data highlight the role of P2X3 receptors in nociception, and suggest that they may be an important target for novel analgesic drugs.51 For instance, dihydroergotamine (DHE), a nonselective ergot alkaloid derivative extensively used for the acute treatment of migraine, depresses ATP-mediated sensitization of TG neurons via downregulation of P2X3 receptors:52 these data suggest that the effectiveness of DHE in treating migraine might be partly due to its inhibitory effect on P2X3 receptors.
Ca2+-activated potassium channels may also contribute to controlling the hyperactivity of the trigeminocervical complex. Activation of neuronal large-conductance calcium-activated potassium channels (BKCa or MaxiK) induces hyperpolarization of neurons,53 while intravenous administration of the MaxiK opener NS1619 dose-dependently inhibits neurogenic dural vasodilation in a model of trigeminovascular nociception.54 Among the group of voltage-independent K+ channels, the TRESK K2P channel modulates cellular excitability55 and is expressed in the TG. Although mutation in the KCNK18 gene that encodes a protein member of the TRESK subfamily of leak K+ channels was reported to be linked to familial migraine,56 it was later suggested that KCNK18 might act as a modifier instead of being sufficient to cause typical migraine.57
Certain genetic mutations associated with familial migraine and affecting channels directly involved in hyperexcitability have provided further information to understand the primary mechanisms underlying migraine pathophysiology. In particular, the genes known to cause familial hemiplegic migraine (FHM) are all involved in ion homeostasis across the neuronal cell membrane. Missense mutations in CACNA1A gene encoding the pore-forming subunits of the neuronal voltage-gated P/Q-type Ca2+ channel (Cav2.1) are reported to be the commonest cause of FHM type 1.58 Such mutation determines a major gain of function of Cav2.1 channels whose activation threshold is shifted to more negative membrane potential.59 Thus, not only this mutation favors neuronal excitability, but it also facilitates presynaptic release of transmitter, in particular glutamate.60
Migraine pain: sensitization process and its triggers
A number of studies and a large body of clinical as well as preclinical data have provided evidence for the involvement of sterile inflammation as one important mechanism for the activation of the migraine pain pathway.61 This process is purported to produce abnormal cross talk between neurons and nonneuronal cells in the TG and to reinforce activation and recruitment of immune cells releasing inflammatory agents to perpetuate sensitization.62–65 A recent study with in vitro TG cultures and a cell-line of macrophages has, in fact, shown how co-culturing these cells enhances phagocytic activity of macrophages and augments functional responses by P2X3 receptors.64 During neurogenic inflammation, several neuropeptides, neuromodulators, and other signaling molecules are released from neurons and glial and inflammatory cells of the trigeminovascular system. Pro-inflammatory cytokine levels have been found to be elevated in the plasma and in the cerebrospinal fluid of migraineurs.66–68 Bradykinin, histamine, serotonin (5-HT), and prostaglandin E2 cause mechanical sensitization and increase the excitability of somatic69 and meningeal nociceptors.10,11 Other inflammatory mediators like interleukins 1 (IL-1), 6 (IL-6), and 8 (IL-8) and tumor necrosis factor α exert their effects through the endogenous release of eicosanoids and sympathetic amines.70,71 Increased nitric oxide (NO) production within the meninges may also contribute to migraine headache in patients.72 These inflammatory mediators collectively modulate the activity of various ion channels.73,74
Neuropeptides as modulators of nociceptors in migraine
The seminal observation that a pharmacological antagonist of the endogenous neuropeptide CGRP was a potent antimigraine drug75 catalyzed a strong focus on the action of CGRP in migraine. CGRP is a 37-amino acid neuropeptide derived from the gene encoding calcitonin by alternative splicing of mRNA and proteolytic processing of its precursor.76,77 CGRP exists as two isoforms, α- and β-CGRP.78,79 α-CGRP is predominant in the central and peripheral nervous systems, while β-CGRP is mainly present in the enteric nervous system.80 Jugular plasma concentrations of CGRP are increased during attacks of migraine.81 In addition, release of CGRP is produced by electrical stimulation of TG fibers.82 Chemical stimuli that activate, for example, TRPA1 or TRPV1 receptors, or endogenous mediators liberated by neuroinflammatory cells increase the release of CGRP.82–84 These data have, therefore, led to consider CGRP as a major “migraine mediator” as recently reviewed.16,17,85,86
CGRP is known to mediate neurogenic inflammation in peripheral tissues by increasing blood flow, and by recruiting immune cells, that may, in turn, activate sensory neurons.87 In accordance with this notion, the peptide evokes strong stimulation of NO synthesis and release from trigeminal ganglia88 as well as liberation of inflammatory cytokines.89 Furthermore, CGRP can promote the release of endogenous algogenic mediators like bradykinin from TG satellite cells,63 thus contributing to the creation of a chemical milieu that facilitates sensory neuron activity. Its receptors are expressed in most peripheral components of the trigeminovascular system, including blood vessels, Schwann cells, dural mast cells, satellite glial cells, and a subpopulation of TG neurons.90 The importance of CGRP in the pathophysiology of migraine pain is supported by the fact that its intravenous administration causes delayed migraine-like headache in migraineurs, whereas CGRP receptor antagonists are proving effective in treating migraine,91 in line with the notion that migraine patients may somehow be hypersensitive to the CGRP-positive modulation of nociception.75,92 It has been suggested that CGRP may mediate cross talk between TG neurons and satellite glial cells63 resulting in the increased expression and/or membrane targeting of specific pain receptors, such as ATP-gated P2X3, as well as an increased production of other inflammatory mediators, like IL-1β, thus sensitizing nociceptors and reinforcing neuroinflammation.93,94
An interesting issue concerns the origin of endogenous CGRP release with particular relevance to the migraine attack. Recent studies have suggested that low levels of endocannabinoids may decrease their inhibitory control over the trigeminovascular system in migraine patients, a phenomenon that, in turn, may lead to increased CGRP.95 The action of endocannabinoids can also include a site in the brainstem to regulate trigeminal excitability.96 The mechanism of action of endocannabinoids in migraine remains, however, incompletely understood in view of the activation of TRPV1 receptors by anandamide and the implications of this result for the control of the local vasculature at meningeal level.
Gain of function of P2X3 receptors is an important mechanism of CGRP action
CGRP has been demonstrated to upregulate the membrane expression and function of P2X3 receptors86,94,97 through enhancing gene transcription and receptor membrane targeting.98 The mechanisms of P2X3 facilitation mediated by CGRP involve the activation of complex intracellular pathways and depend on protein kinase A (PKA) and protein kinase C (PKC) activity.94,98 It is noteworthy that only a minority of TG neurons can bind CGRP94 suggesting that the action of this peptide is discrete and can perhaps be amplified via further signaling processes within and between TG cells. In vivo studies confirm that CGRP injection into the temporomandibular joint capsule leads to early, PKA-dependent increase in P2X3 receptors of the spinal trigeminal nucleus neurons.97
The potentiating action by CGRP on P2X3 receptors is typically slow as it requires approximately 1 hour to develop fully, and it persists long after the peptide is eliminated.94 Importantly, this effect of CGRP is selective for P2X3 receptors because, in TG cultures, it does not affect TRPV1 receptors.94 Furthermore, it has been proposed that neuronal P2X3 receptor activation and ensuing depolarization can also release CGRP, thereby perpetuating a self-supporting process for TG neuron sensitization.20
A significant component of the action by CGRP occurs indirectly, through the release of the algogenic neurotrophin brain-derived neurotrophic factor (BDNF).98 In accordance with this view, P2X3 receptor expression is promoted by CGRP/BDNF-dependent activation of calcium/calmodulin-dependent kinase II and of the transcription factor CREB.98 Patients show significantly higher BDNF serum levels during migraine attacks.68,99 BDNF is a key mediator of plasticity in trigeminal nociceptive pathways,100 and it has been shown to facilitate P2X3 receptor function.98 Thus, CGRP and BDNF may be viewed as a combinatorial stimulus to facilitate TG sensitization.
The constitutive upregulation of P2X3 receptors in TG of the mouse model of FHM-1 is due to the higher level of CGRP release probably caused by increased Ca2+ inflow via gain of function of Cav2.1 channels.63,101,102 Furthermore, the perturbed intracellular Ca2+ homeostasis of such neurons enhances calcineurin activity with consequent change in the delicate intracellular balance between phosphorylation and dephosphorylation processes of P2X3 receptors with a positive impact on their function.101 Thus, preclinical and clinical data concur to show a pivotal role of CGRP in the migraine pain attack and suggest upregulated P2X3 receptors as a major contributor to TG sensitization.
Clinical pharmacology of CGRP antagonists to treat headache
The growing evidence linking migraine to neuropeptide signaling emphasizes the view that neuropeptide receptors represent an important area for pharmacological treatment. Despite recent advances in the structure of CGRP receptors obtained with crystallography, the design of effective CGRP antagonist remains a significant challenge in view of the heteromeric nature of the receptor complex.103 Table 1 summarizes a few recent data for CGRP inhibitors based on receptor antagonists (the “gepants”) or monoclonal antibodies used for current clinical investigations. Detailed reviews on this subject are also available.17,91,103
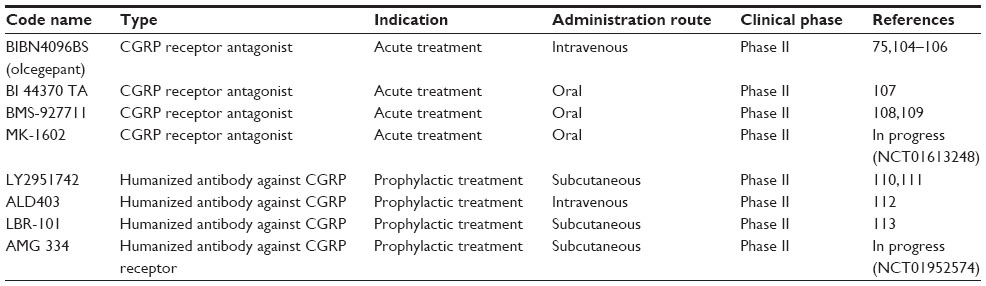 | Table 1 Current CGRP antagonists/antibodies in clinical trial for migraine therapy |
CGRP antagonists appear to be attractive new drugs because they lack vasoconstrictor effects that limit the use of other migraine therapies by patients with cerebral or coronary vascular disease.114–116 It is noteworthy that, since antagonists can possess distinct species specificity for the CGRP receptor,103 the present discussion is primarily centered on human studies. In a Phase II double-blind randomized clinical trial of 126 patients with migraine, the olcegepant response rate to block a migraine attack was comparable to that of triptans.75 Its tolerability was good, but this drug could only be administered by intravenous injection,75 thus representing a pitfall for treatment accessibility and repetition. More recently developed CGRP antagonists are available as oral formulation. Some of them (namely telcagepant/MK-0974 and MK-3207) were shown to be effective against migraine, but, due to liver toxicity concerns, their development was discontinued.117,118 Three new CGRP receptor antagonists (BI 44370 TA, BMS-927711, and MK-1602) have completed Phase II trials, although only two have been reported (BI 44370 TA and BMS-927711) (Table 1).
A recent positron emission tomography study in man has shown that telcagepant achieved only low receptor occupancy at an efficacious dose, suggesting that large-scale antagonism of brain CGRP receptors is unlikely to be required for migraine efficacy.119 It is, however, feasible that central CGRP receptor antagonism may provide additional therapeutic benefits.119
An alternative approach to pharmacological antagonists is the use of antibodies to target CGRP or its receptor. The sustained inhibition of CGRP signaling produced by monoclonal antibodies pharmacokinetically differentiates them from the CGRP antagonists, and renders the use of antibodies more suitable as prophylactic treatment rather than acute migraine medication.
One advantage with the antibody therapies is that they are structurally very different from the CGRP antagonists; thus, it is less likely that similar liver toxicity problems will be encountered. However, they are of high cost, and at least, some of them require an injectable route of administration. Moreover, the potential onset of immunological reactions should be borne in mind, and chronic depletion of CGRP may have adverse side effects actually induced by the lack of this peptide. While monoclonal antibodies are not expected to cross the blood–brain barrier in physiological conditions, the integrity of this barrier during an attack of migraine with associated release of circulating inflammatory factors remains a subject for future studies. Hence, it is not clear whether these antibodies exert their pharmacological effects simply through a peripheral site of action. There are, in fact, suggestions that CGRP antagonists may work partly through a central mode of action,120 even if this hypothesis is not without controversies121,122 and not fully consistent with recent functional imaging data.119 It should, however, be recalled that the TG is outside the blood–brain barrier even in physiological conditions, and it remains an accessible target for these antibodies (or, indeed, pharmacological blockers). Thus, it will be of great interest to examine if the CGRP antibodies display comparable long-term efficacy as the CGRP antagonists.
There are no clinical data based on multicenter trials, in which the efficacy of antibodies against the CGRP receptor (or the peptide itself) has been compared versus administration of CGRP pharmacological antagonists. Furthermore, it is not known if there is any advantage in terms of migraine control in using an antibody therapy directed against the CGRP receptor rather than the circulating peptides. These issues will also need extensive future studies in view of the possible long-term changes in CGRP receptor function, once the natural ligand activity has been inhibited in a persistent manner.
LY2951742 and ALD403 are fully humanized monoclonal antibodies that potently and selectively bind to CGRP.111,123 ALD403, in particular, binds to both α and β forms of human CGRP (affinity Kd <20 pM123), and LBR-101 binds to the peptide receptor-binding site of CGRP.113 After antibody-dependent sequestration of circulating CGRP, the peptide receptors might remain available to other endogenous ligands supporting ongoing receptor signal transduction in neurons and nonneuronal cells.
Advances in antibody design technology have paved the way for an improved generation of therapeutic antibodies.124 Thus, therapeutic antibodies can be modified to increase their specificity125 and decrease their risk of adverse reactions, for example, by modulating their immune effector functions, extending their half-life, and optimizing their antigen-binding domains. The antibody stability is an important issue since, in order to neutralize CGRP, whose plasma levels are fluctuating during the course of the illness, high antibody concentrations should be attained. This goal is, however, not without problems as mechanism-related and nonspecific adverse reactions have been reported, including immunogenicity.126 Guidelines for the therapeutic use of antibodies have recently been published (http://www.ema.europa.eu/ema/). It should be noted that certain pharmacological antagonists of CGRP can also induce adverse reactions as shown, for example, by interruption of the trials with the CGRP receptor antagonist telcagepant.127
BNP is a potential endogenous negative regulator of nociceptive signaling
While CGRP is the prototypical neuropeptide for the sensitization of TG neurons, it seems likely that there are also endogenous neuropeptides to downregulate the activity of P2X3 receptors and they would, thus, represent an intrinsic negative feedback system to control pain sensitivity. Recent evidence has been gathered in support of a potential involvement of the natriuretic peptide system in the modulation of peripheral sensory neuron nociceptive signaling.128–131 The natriuretic peptides are a family of structurally related hormones widely known for their important effects on the cardiovascular system and the regulation of water–electrolyte homeostasis.132 These include atrial natriuretic peptide (ANP), BNP, and the C-type natriuretic peptide (CNP).133 All three peptides showed distinct amino acid sequence that hints to their separate genetic origin.
Natriuretic peptides exert their physiologic effects through binding, with different affinities, to three membrane receptor subtypes: the natriuretic peptide receptors A (NPR-A), B (NPR-B), and C (NPR-C), which are also known as guanylate cyclase A (GC-A), B (GC-B), and the clearance receptor, respectively. NPR-A is activated mainly by ANP and BNP.133,134
Unlike CGRP, the functional consequences of BNP action on neurons are less understood. Nevertheless, while BNP and its receptor NPR-A are both expressed in the rat dorsal root ganglia (DRG), their signaling may attenuate inflammatory pain through a mechanism involving the opening of large-conductance Ca2+-activated K+ (BKCa) channels.129 Microarray gene profiling has indicated that chronic pain models enhance BNP and its NPR-A in rat DRG. NPR-C is also present in DRG, where it colocalizes with TRPV1 channels. Here, however, NPR-C activation by CNP displays diametrically opposite effects compared to NPR-A, as it enhances thermal hyperalgesia in a TRPV1- and PKC-dependent fashion.128 These results indicate the coexistence of functional NPRs in DRG that exhibit contrasting effects on pain transduction, which raises question on how the two functions are inter-related and whether the natriuretic peptide modulatory role can also be generalized to cranial ganglia and other pain conditions.
In relation to migraine mechanisms, ANP gene expression has been shown to increase following experimental cortical-spreading depression.135 Moreover, a recent clinical report documents that the BNP precursor (proBNP) levels are raised in the jugular vein blood during a migraine attack,136 indicating a potential involvement of BNP in migraine pathophysiology.
Many migraine mediators have, in common, the ability to alter both neuronal and vascular function (eg, CGRP, substance P, bradykinin). This property is also shared by natriuretic peptides that activate cGMP-dependent pathways via GC in vascular cells to induce vasodilation137,138 and to stimulate nitric oxide synthase and NO production probably via activation of the NPR-C receptor.138,139 Due to the vasoactive properties of natriuretic peptides, it has been recently postulated that these substances can induce headache.140 Future studies are, however, needed to determine the vasoactive properties of the natriuretic peptides on cerebral vessels and their potential role in migraine. Several lines of evidence show that vasodilation of cranial (meningeal) and extracranial arteries is not necessary to directly provoke migraine pain as reviewed.61
In rodent TG and DRG, the NPR-A receptor has been reported to be expressed only by sensory neurons. In mouse TG, NPR-A is present at membrane level in almost all (>95%) neurons, and application of exogenous BNP rapidly evokes a large increase in cGMP level, a canonical effector of BNP,133 and phosphorylation of AKT, a kinase downstream of NPR-A activation.141 Such a strong and widespread activity of NPR-A receptors outlines a major role of this system in controlling neuronal excitability. In fact, although application of exogenous BNP does not change P2X3 receptor activity, the same protocol depresses TRPV1-mediated currents in TG.142 Furthermore, anantin, a selective NPR-A antagonist, slowly and greatly enhances P2X3 receptor function, implying that ambient BNP produced a constitutive downregulation of P2X3 receptors, which could not be further intensified by exogenous BNP.142 These data indicate the role of this endogenous peptide as a negative regulator of trigeminal sensory neuron excitability to nociceptive stimuli and should prompt future clinical studies to explore the role of NPR-A receptors in migraine. Further studies are required to clarify how BNP and CGRP may interact in migraine pain: an idealized scheme of the potential regulation by CGRP and BNP of TG neuronal function is illustrated in Figure 1.
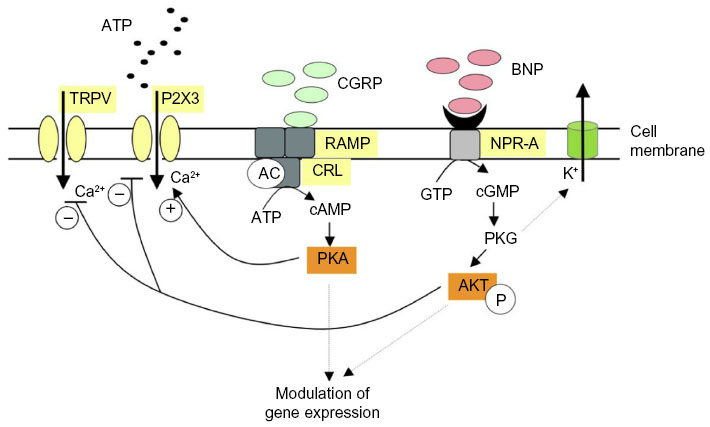 | Figure 1 Idealized scheme depicting modulation by CGRP or BNP of P2X3 and TRPV1 receptors of trigeminal sensory neurons. |
Conclusion
Somatosensory cortical maps show that the trigeminal territory is large and very sensitive to nociceptive stimuli. Thus, to prevent unwanted pain generation with associated allodynia and hyperalgesia as observed in chronic pain syndromes, potent intrinsic regulators should exist to counteract the sensitization elicited by neuropeptides like CGRP. While current interest in novel treatments for migraine is centered on the goal of blocking either CGRP or its receptors, future studies are necessary to find out whether boosting intrinsic negative regulators that may comprise K+ channels, endocannabinoids, or neuromodulatory peptides might be a viable approach to treat migraine.
Acknowledgments
This work was supported by the EU FP7 grant EuroHeadPain (#602633) and by the EU Crossborder Cooperation Programme Italy-Slovenia 2007–2013 (European Regional Development Fund and national funds; MINA).
Disclosure
The authors declare no conflict of interest.
References
Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288(5472):1765–1769. | |
Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2163–2196. | |
Stovner LJ, Hagen K. Prevalence, burden, and cost of headache disorders. Curr Opin Neurol. 2006;19(3):281–285. | |
Olesen J, Gustavsson A, Svensson M, Wittchen HU, Jönsson B, CDBE2010 study group; European Brain Council. The economic cost of brain disorders in Europe. Eur J Neurol. 2012;19(1):155–162. | |
Leonardi M, Steiner TJ, Scher AT, Lipton RB. The global burden of migraine: measuring disability in headache disorders with WHO’s classification of functioning, disability and health (ICF). J Headache Pain. 2005;6(6):429–440. | |
Lipton RB, Bigal ME, Rush SR, et al. Migraine practice patterns among neurologists. Neurology. 2004;62(11):1926–1931. | |
Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR. Neurobiology of migraine. Neuroscience. 2009;161(2):327–341. | |
Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: evidence for peripheral sensitisation. Lancet Neurol. 2009;8(7):679–690. | |
Levy D. Migraine pain and nociceptor activation – where do we stand? Headache. 2010;50(5):909–916. | |
Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384(6609):560–564. | |
Levy D, Strassman AM. Distinct sensitizing effects of the cAMP-PKA second messenger cascade on rat dural mechanonociceptors. J Physiol. 2002;538(pt 2):483–493. | |
Davis KD, Meyer RA, Campbell JN. Chemosensitivity and sensitization of nociceptive afferents that innervate the hairy skin of monkey. J Neurophysiol. 1993;69(4):1071–1081. | |
Blau JN, Dexter SL. The site of pain origin during migraine attacks. Cephalalgia. 1981;1(3):143–147. | |
Rasmussen BK, Jensen R, Olesen J. A population-based analysis of the diagnostic criteria of the international headache society. Cephalalgia. 1991;11(3):129–134. | |
Viana F. Chemosensory properties of the trigeminal system. ACS Chem Neurosci. 2011;2(1):38–50. | |
Bigal ME, Walter S, Rapoport AM. Calcitonin gene-related peptide (CGRP) and migraine current understanding and state of development. Headache. 2013;53(8):1230–1244. | |
Russell FA, King R, Smillie S-J, Kodji X, Brain SD. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev. 2014;94(4):1099–1142. | |
Akiyama T, Carstens E. Neural processing of itch. Neuroscience. 2013;250:697–714. | |
Mishra SK, Hoon MA. The cells and circuitry for itch responses in mice. Science. 2013;340(6135):968–971. | |
Yan J, Dussor G. Ion channels and migraine. Headache. 2014;54(4):619–639. | |
Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1):relationship to neuropeptides, the P2X3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11(3):946–958. | |
Simonetti M, Fabbro A, D’Arco M, et al. Comparison of P2X and TRPV1 receptors in ganglia or primary culture of trigeminal neurons and their modulation by NGF or serotonin. Mol Pain. 2006;2:11. | |
Holland PR, Akerman S, Andreou AP, Karsan N, Wemmie JA, Goadsby PJ. Acid-sensing ion channel 1: a novel therapeutic target for migraine with aura. Ann Neurol. 2012;72(4):559–563. | |
Benemei S, De Cesaris F, Fusi C, Rossi E, Lupi C, Geppetti P. TRPA1 and other TRP channels in migraine. J Headache Pain. 2013;14:71. | |
Kelman L. The triggers or precipitants of the acute migraine attack. Cephalalgia. 2007;27(5):394–402. | |
Nassini R, Materazzi S, Vriens J, et al. The “headache tree” via umbellulone and TRPA1 activates the trigeminovascular system. Brain. 2012;135(pt 2):376–390. | |
Wantke F, Focke M, Hemmer W, et al. Exposure to formaldehyde and phenol during an anatomy dissecting course: sensitizing potency of formaldehyde in medical students. Allergy. 2000;55(1):84–87. | |
Irlbacher K, Meyer B-U. Nasally triggered headache. Neurology. 2002; 58(2):294. | |
Huang D, Li S, Dhaka A, Story GM, Cao Y-Q. Expression of the transient receptor potential channels TRPV1, TRPA1 and TRPM8 in mouse trigeminal primary afferent neurons innervating the dura. Mol Pain. 2012;8:66. | |
Kunkler PE, Ballard CJ, Pellman JJ, Zhang L, Oxford GS, Hurley JH. Intraganglionic signaling as a novel nasal-meningeal pathway for TRPA1-dependent trigeminovascular activation by inhaled environmental irritants. PLoS One. 2014;9(7):e103086. | |
Wei X, Edelmayer RM, Yan J, Dussor G. Activation of TRPV4 on dural afferents produces headache-related behavior in a preclinical rat model. Cephalalgia. 2011;31(16):1595–1600. | |
Chen Y, Kanju P, Fang Q, et al. TRPV4 is necessary for trigeminal irritant pain and functions as a cellular formalin receptor. Pain. Epub 2014 Oct 2. | |
Mälkiä A, Morenilla-Palao C, Viana F. The emerging pharmacology of TRPM8 channels: hidden therapeutic potential underneath a cold surface. Curr Pharm Biotechnol. 2011;12(1):54–67. | |
Chasman DI, Schürks M, Anttila V, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;43(7):695–698. | |
Caterina MJ, Leffler A, Malmberg AB, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000; 288(5464):306–313. | |
Davis JB, Gray J, Gunthorpe MJ, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405(6783):183–187. | |
Gavva NR, Tamir R, Qu Y, et al. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313(1):474–484. | |
Russo EB. Clinical endocannabinoid deficiency (CECD):can this concept explain therapeutic benefits of cannabis in migraine, fibromyalgia, irritable bowel syndrome and other treatment-resistant conditions? Neuro Endocrinol Lett. 2008;29(2):192–200. | |
Sarchielli P, Pini LA, Coppola F, et al. Endocannabinoids in chronic migraine: CSF findings suggest a system failure. Neuropsychopharmacology. 2007;32(6):1384–1390. | |
Akerman S, Kaube H, Goadsby PJ. Anandamide acts as a vasodilator of dural blood vessels in vivo by activating TRPV1 receptors. Br J Pharmacol. 2004;142(8):1354–1360. | |
Akerman S, Kaube H, Goadsby PJ. Anandamide is able to inhibit trigeminal neurons using an in vivo model of trigeminovascular-mediated nociception. J Pharmacol Exp Ther. 2004;309(1):56–63. | |
Piomelli D. More surprises lying ahead. The endocannabinoids keep us guessing. Neuropharmacology. 2014;76(pt B):228–234. | |
Burnstock G. The role of adenosine triphosphate in migraine. Biomed Pharmacother. 1989;43(10):727–736. | |
Vulchanova L, Riedl MS, Shuster SJ, et al. Immunohistochemical study of the P2X2 and P2X3 receptor subunits in rat and monkey sensory neurons and their central terminals. Neuropharmacology. 1997;36(9):1229–1242. | |
Llewellyn-Smith IJ, Burnstock G. Ultrastructural localization of P2X3 receptors in rat sensory neurons. Neuroreport. 1998;9(11):2545–2550. | |
Burnstock G. Purinergic receptors and pain. Curr Pharm Des. 2009; 15(15):1717–1735. | |
Fabbretti E, Nistri A. Regulation of P2X3 receptor structure and function. CNS Neurol Disord Drug Targets. 2012;11(6):687–698. | |
Saloman JL, Chung M-K, Ro JY. P2X3 and TRPV1 functionally interact and mediate sensitization of trigeminal sensory neurons. Neuroscience. 2013;232:226–238. | |
Yang Z, Cao Y, Wang Y, et al. Behavioural responses and expression of P2X3 receptor in trigeminal ganglion after experimental tooth movement in rats. Arch Oral Biol. 2009;54(1):63–70. | |
Oliveira MCG, Parada CA, Veiga MCFA, Rodrigues LR, Barros SP, Tambeli CH. Evidence for the involvement of endogenous ATP and P2X receptors in TMJ pain. Eur J Pain. 2005;9(1):87–93. | |
Ford AP. In pursuit of P2X3 antagonists: novel therapeutics for chronic pain and afferent sensitization. Purinergic Signal. 2012;8(Suppl 1):3–26. | |
Masterson CG, Durham PL. DHE repression of ATP-mediated sensitization of trigeminal ganglion neurons. Headache. 2010;50(9):1424–1439. | |
Gribkoff VK, Starrett JE, Dworetzky SI. Maxi-K potassium channels: form, function, and modulation of a class of endogenous regulators of intracellular calcium. Neurosci Rev. 2001;7(2):166–177. | |
Akerman S, Holland PR, Lasalandra MP, Goadsby PJ. Inhibition of trigeminovascular dural nociceptive afferents by Ca2+-activated K+ (MaxiK/BK(Ca)) channel opening. Pain. 2010;151(1):128–136. | |
Enyedi P, Czirják G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. 2010;90(2):559–605. | |
Lafrenière RG, Cader MZ, Poulin JF, et al. A dominant-negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med. 2010;16(10):1157–1160. | |
Andres-Enguix I, Shang L, Stansfeld PJ, et al. Functional analysis of missense variants in the TRESK (KCNK18) K channel. Sci Rep. 2012;2:237. | |
Ophoff RA, Terwindt GM, Vergouwe MN, et al. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87(3):543–552. | |
van denMaagdenberg AM, Pietrobon D, Pizzorusso T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41(5):701–710. | |
Tottene A, Conti R, Fabbro A, et al. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron. 2009;61(5):762–773. | |
Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol. 2013;75:365–391. | |
Franceschini A, Vilotti S, Ferrari MD, van den Maagdenberg AMJM, Nistri A, Fabbretti E. TNFα levels and macrophages expression reflect an inflammatory potential of trigeminal ganglia in a mouse model of familial hemiplegic migraine. PLoS One. 2013;8(1):e52394. | |
Ceruti S, Villa G, Fumagalli M, et al. Calcitonin gene-related peptide-mediated enhancement of purinergic neuron/glia communication by the algogenic factor bradykinin in mouse trigeminal ganglia from wild-type and R192Q Cav2.1 Knock-in mice: implications for basic mechanisms of migraine pain. J Neurosci. 2011;31(10):3638–3649. | |
Franceschini A, Nair A, Bele T, van den Maagdenberg AM, Nistri A, Fabbretti E. Functional crosstalk in culture between macrophages and trigeminal sensory neurons of a mouse genetic model of migraine. BMC Neurosci. 2012;13:143. | |
Franceschini A, Hullugundi SK, van den Maagdenberg AMJM, Nistri A, Fabbretti E. Effects of LPS on P2X3 receptors of trigeminal sensory neurons and macrophages from mice expressing the R192Q Cacna1a gene mutation of familial hemiplegic migraine-1. Purinergic Signal. 2013;9(1):7–13. | |
Bø SH, Davidsen EM, Gulbrandsen P, et al. Cerebrospinal fluid cytokine levels in migraine, tension-type headache and cervicogenic headache. Cephalalgia. 2009;29(3):365–372. | |
Ishizaki K, Takeshima T, Fukuhara Y, et al. Increased plasma transforming growth factor-beta1 in migraine. Headache. 2005;45(9):1224–1228. | |
Tanure MTA, Gomez RS, Hurtado RCL, Teixeira AL, Domingues RB. Increased serum levels of brain-derived neurotropic factor during migraine attacks: a pilot study. J Headache Pain. 2010;11(5):427–430. | |
Steen KH, Reeh PW, Anton F, Handwerker HO. Protons selectively induce lasting excitation and sensitization to mechanical stimulation of nociceptors in rat skin, in vitro. J Neurosci. 1992;12(1):86–95. | |
Obreja O, Schmelz M, Poole S, Kress M. Interleukin-6 in combination with its soluble IL-6 receptor sensitises rat skin nociceptors to heat, in vivo. Pain. 2002;96(1–2):57–62. | |
Sachs D, Cunha FQ, Poole S, Ferreira SH. Tumour necrosis factor-alpha, interleukin-1beta and interleukin-8 induce persistent mechanical nociceptor hypersensitivity. Pain. 2002;96(1–2):89–97. | |
Olesen J, Thomsen LL, Iversen H. Nitric oxide is a key molecule in migraine and other vascular headaches. Trends Pharmacol Sci. 1994;15(5):149–153. | |
Belmonte C, Gallar J, Pozo MA, Rebollo I. Excitation by irritant chemical substances of sensory afferent units in the cat’s cornea. J Physiol. 1991;437:709–725. | |
Pozo MA, Gallego R, Gallar J, Belmonte C. Blockade by calcium antagonists of chemical excitation and sensitization of polymodal nociceptors in the cat’s cornea. J Physiol. 1992;450:179–189. | |
Olesen J, Diener HC, Husstedt IW, et al; BIBN 4096 BS Clinical Proof of Concept Study Group. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350(11):1104–1110. | |
Terenghi G, Polak JM, Ghatei MA, et al. Distribution and origin of calcitonin gene-related peptide (CGRP) immunoreactivity in the sensory innervation of the mammalian eye. J Comp Neurol. 1985;233(4):506–516. | |
Alevizaki M, Shiraishi A, Rassool FV, Ferrier GJ, MacIntyre I, Legon S. The calcitonin-like sequence of the beta CGRP gene. FEBS Lett. 1986;206(1):47–52. | |
Noguchi K, Senba E, Morita Y, Sato M, Tohyama M. α-CGRP and β-CGRP mRNAs are differentially regulated in the rat spinal cord and dorsal root ganglion. Brain Res. 1990;7(4):299–304. | |
Watkins HA, Rathbone DL, Barwell J, Hay DL, Poyner DR. Structure-activity relationships for α-calcitonin gene-related peptide. Br J Pharmacol. 2013;170(7):1308–1322. | |
Juaneda C, Dumont Y, Quirion R. The molecular pharmacology of CGRP and related peptide receptor subtypes. Trends Pharmacol Sci. 2000;21(11):432–438. | |
Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28(2):183–187. | |
Ebersberger A, Averbeck B, Messlinger K, Reeh PW. Release of substance P, calcitonin gene-related peptide and prostaglandin E2 from rat dura mater encephali following electrical and chemical stimulation in vitro. Neuroscience. 1999;89(3):901–907. | |
Bowen EJ, Schmidt TW, Firm CS, Russo AF, Durham PL. Tumor necrosis factor-α stimulation of calcitonin gene-related peptide expression and secretion from rat trigeminal ganglion neurons. J Neurochem. 2006;96(1):65–77. | |
Mason RT, Peterfreund RA, Sawchenko PE, Corrigan AZ, Rivier JE, Vale WW. Release of the predicted calcitonin gene-related peptide from cultured rat trigeminal ganglion cells. Nature. 1984;308(5960):653–655. | |
Messlinger K, Fischer MJM, Lennerz JK. Neuropeptide effects in the trigeminal system: pathophysiology and clinical relevance in migraine. Keio J Med. 2011;60(3):82–89. | |
Giniatullin R, Nistri A, Fabbretti E. Molecular mechanisms of sensitization of pain-transducing P2X3 receptors by the migraine mediators CGRP and NGF. Mol Neurobiol. 2008;37(1):83–90. | |
Raddant AC, Russo AF. Calcitonin gene-related peptide in migraine: intersection of peripheral inflammation and central modulation. Expert Rev Mol Med. 2011;13:e36. | |
Vause CV, Durham PL. CGRP stimulation of iNOS and NO release from trigeminal ganglion glial cells involves mitogen-activated protein kinase pathways. J Neurochem. 2009;110(3):811–821. | |
Thalakoti S, Patil VV, Damodaram S, et al. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. Headache. 2007;47(7):1008–1023; [discussion 24–25]. | |
Lennerz JK, Rühle V, Ceppa EP, et al. Calcitonin receptor-like receptor (CLR), receptor activity-modifying protein 1 (RAMP1), and calcitonin gene-related peptide (CGRP) immunoreactivity in the rat trigeminovascular system: differences between peripheral and central CGRP receptor distribution. J Comp Neurol. 2008;507(3):1277–1299. | |
Tso AR, Goadsby PJ. New targets for migraine therapy. Curr Treat Options Neurol. 2014;16(11):318. | |
Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia. 2002;22(1):54–61. | |
Capuano A, De Corato A, Lisi L, Tringali G, Navarra P, Dello Russo C. Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: relevance for migraine pathology. Mol Pain. 2009;5:43. | |
Fabbretti E, D’Arco M, Fabbro A, Simonetti M, Nistri A, Giniatullin R. Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene-related peptide. J Neurosci. 2006;26(23):6163–6171. | |
Greco R, Gasperi V, Maccarrone M, Tassorelli C. The endocannabinoid system and migraine. Exp Neurol. 2010;224(1):85–91. | |
Akerman S, Holland PR, Lasalandra MP, Goadsby PJ. Endocannabinoids in the brainstem modulate dural trigeminovascular nociceptive traffic via CB1 and “triptan” receptors: implications in migraine. J Neurosci. 2013;33(37):14869–14877. | |
Cady RJ, Glenn JR, Smith KM, Durham PL. Calcitonin gene-related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol Pain. 2011;7:94. | |
Simonetti M, Giniatullin R, Fabbretti E. Mechanisms mediating the enhanced gene transcription of P2X3 receptor by calcitonin gene-related peptide in trigeminal sensory neurons. J Biol Chem. 2008; 283(27):18743–18752. | |
Fischer M, Wille G, Klien S, et al. Brain-derived neurotrophic factor in primary headaches. J Headache Pain. 2012;13(6):469–475. | |
Buldyrev I, Tanner NM, Hsieh H, Dodd EG, Nguyen LT, Balkowiec A. Calcitonin gene-related peptide enhances release of native brain-derived neurotrophic factor from trigeminal ganglion neurons. J Neurochem. 2006;99(5):1338–1350. | |
Nair A, Simonetti M, Birsa N, et al. Familial hemiplegic migraine Ca(v)2.1 channel mutation R192Q enhances ATP-gated P2X3 receptor activity of mouse sensory ganglion neurons mediating trigeminal pain. Mol Pain. 2010;6:48. | |
Fioretti B, Catacuzzeno L, Sforna L, et al. Trigeminal ganglion neuron subtype-specific alterations of Ca(V)2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J Physiol. 2011;589(pt 23):5879–5895. | |
Moore EL, Salvatore CA. Targeting a family B GPCR/RAMP receptor complex: CGRP receptor antagonists and migraine. Br J Pharmacol. 2012;166(1):66–78. | |
Doods H, Hallermayer G, Wu D, et al. Pharmacological profile of BIBN4096BS, the first selective small molecule CGRP antagonist. Br J Pharmacol. 2000;129(3):420–423. | |
Li J, Wang DH. Development of angiotensin II-induced hypertension: role of CGRP and its receptor. J Hypertens. 2005;23(1):113–118. | |
Salvatore CA, Moore EL, Calamari A, et al. Pharmacological properties of MK-3207, a potent and orally active calcitonin gene-related peptide receptor antagonist. J Pharmacol Exp Ther. 2010;333(1):152–160. | |
Diener H-C, Barbanti P, Dahlöf C, Reuter U, Habeck J, Podhorna J. BI 44370 TA, an oral CGRP antagonist for the treatment of acute migraine attacks: results from a phase II study. Cephalalgia. 2011; 31(5):573–584. | |
Luo G, Chen L, Conway CM, et al. Discovery of (5S,6S,9R)-5-amino-6-(2,3-difluorophenyl)-6,7,8,9-tetrahydro-5H-cyclohepta[b]pyridin-9-yl 4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin-1-yl)piperidine-1-carboxylate (BMS-927711):an oral calcitonin gene-related peptide (CGRP) antagonist in clinical trials for treating migraine. J Med Chem. 2012;55(23):10644–10651. | |
Marcus R, Goadsby PJ, Dodick D, Stock D, Manos G, Fischer TZ. BMS-927711 for the acute treatment of migraine: a double-blind, randomized, placebo controlled, dose-ranging trial. Cephalalgia. 2014;34(2):114–125. | |
Benschop RJ, Collins EC, Darling RJ, et al. Development of a novel antibody to calcitonin gene-related peptide for the treatment of osteoarthritis-related pain. Osteoarthritis Cartilage. 2014;22(4):578–585. | |
Dodick DW, Goadsby PJ, Spierings ELH, Scherer JC, Sweeney SP, Grayzel DS. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2014;13(9):885–892. | |
Amgen. A phase 2 study to evaluate the efficacy and safety of AMG 334 in migraine prevention; 2013. Available from: http://clinicaltrials.gov/show/NCT01952574 NLM identifier: NCT01952574. Accessed August 30, 2013. | |
Bigal ME, Escandon R, Bronson M, et al. Safety and tolerability of LBR-101, a humanized monoclonal antibody that blocks the binding of CGRP to its receptor: results of the phase 1 program. Cephalalgia. 2013;34(7):483–492. | |
Petersen KA, Birk S, Lassen LH, et al. The CGRP-antagonist, BIBN4096BS does not affect cerebral or systemic haemodynamics in healthy volunteers. Cephalalgia. 2005;25(2):139–147. | |
Petersen KA, Lassen LH, Birk S, Lesko L, Olesen J. BIBN4096BS antagonizes human alpha-calcitonin gene related peptide-induced headache and extracerebral artery dilatation. Clin Pharmacol Ther. 2005;77(3):202–213. | |
Arulmani U, Schuijt MP, Heiligers JPC, Willems EW, Villalón CM, Saxena PR. Effects of the calcitonin gene-related peptide (CGRP) receptor antagonist BIBN4096BS on alpha-CGRP-induced regional haemodynamic changes in anaesthetised rats. Basic Clin Pharmacol Toxicol. 2004;94(6):291–297. | |
Negro A, Lionetto L, Simmaco M, Martelletti P. CGRP receptor antagonists: an expanding drug class for acute migraine? Expert Opin Investig Drugs. 2012;21(6):807–818. | |
Hewitt DJ, Aurora SK, Dodick DW, et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia. 2011;31(6):712–722. | |
Hostetler ED, Joshi AD, Sanabria-Bohórquez S, et al. In vivo quantification of calcitonin gene-related peptide receptor occupancy by telcagepant in rhesus monkey and human brain using the positron emission tomography tracer [11C]MK-4232. J Pharmacol Exp Ther. 2013;347(2):478–486. | |
Sixt M-L, Messlinger K, Fischer MJM. Calcitonin gene-related peptide receptor antagonist olcegepant acts in the spinal trigeminal nucleus. Brain. 2009;132(pt 11):3134–3141. | |
Asghar MS, Hansen AE, Kapijimpanga T, et al. Dilation by CGRP of middle meningeal artery and reversal by sumatriptan in normal volunteers. Neurology. 2010;75(17):1520–1526. | |
Asghar MS, Hansen AE, Larsson HBW, Olesen J, Ashina M. Effect of CGRP and sumatriptan on the BOLD response in visual cortex. J Headache Pain. 2012;13(2):159–166. | |
Dodick DW, Goadsby PJ, Silberstein SD, et al; ALD403 study investigators. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol. 2014;13(11):1100–1107. | |
Leavy O. Therapeutic antibodies: past, present and future. Nat Rev Immunol. 2010;10(5):297. | |
Sela-Culang I, Kunik V, Ofran Y. The structural basis of antibody-antigen recognition. Front Immunol. 2013;4:302. | |
Niebecker R, Kloft C. Safety of therapeutic monoclonal antibodies. Curr Drug Saf. 2010;5(4):275–286. | |
Ho TW, Connor KM, Zhang Y, et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. Neurology. 2014;83(11):958–966. | |
Loo L, Shepherd AJ, Mickle AD, et al. The C-type natriuretic peptide induces thermal hyperalgesia through a noncanonical Gβγ-dependent modulation of TRPV1 channel. J Neurosci. 2012;32(35):11942–11955. | |
Zhang FX, Liu XJ, Gong LQ, et al. Inhibition of inflammatory pain by activating B-type natriuretic peptide signal pathway in nociceptive sensory neurons. J Neurosci Off J Soc Neurosci. 2010;30(32):10927–10938. | |
Heine S, Michalakis S, Kallenborn-Gerhardt W, et al. CNGA3: a target of spinal nitric oxide/cGMP signaling and modulator of inflammatory pain hypersensitivity. J Neurosci. 2011;31(31):11184–11192. | |
Schmidtko A, Gao W, König P, et al. cGMP produced by NO-sensitive guanylyl cyclase essentially contributes to inflammatory and neuropathic pain by using targets different from cGMP-dependent protein kinase I. J Neurosci. 2008;28(34):8568–8576. | |
Woodard GE, Rosado JA. Natriuretic peptides in vascular physiology and pathology. Int Rev Cell Mol Biol. 2008;268:59–93. | |
Potter LR, Yoder AR, Flora DR, Antos LK, Dickey DM. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol. 2009;191:341–366. | |
Potter LR. Domain analysis of human transmembrane guanylyl cyclase receptors: implications for regulation. Front Biosci. 2005;10:1205–1220. | |
Choudhuri R, Cui L, Yong C, et al. Cortical spreading depression and gene regulation: relevance to migraine. Ann Neurol. 2002;51(4):499–506. | |
Uzar E, Evliyaoglu O, Yucel Y, et al. Serum cytokine and pro-brain natriuretic peptide (BNP) levels in patients with migraine. Eur Rev Med Pharmacol Sci. 2011;15(10):1111–1116. | |
Padayatti PS, Pattanaik P, Ma X, van den Akker F. Structural insights into the regulation and the activation mechanism of mammalian guanylyl cyclases. Pharmacol Ther. 2004;104(2):83–99. | |
Andrade FA, Restini CBA, Grando MD, Ramalho LNZ, Bendhack LM. Vascular relaxation induced by C-type natriuretic peptide involves the ca2+/NO-synthase/NO pathway. PLoS One. 2014;9(5):e95446. | |
D’Souza SP, Davis M, Baxter GF. Autocrine and paracrine actions of natriuretic peptides in the heart. Pharmacol Ther. 2004;101(2):113–129. | |
Guo S, Barringer F, Zois NE, Goetze JP, Ashina M. Natriuretic peptides and cerebral hemodynamics. Regul Pept. 2014;192–193:15–23. | |
Abdelalim EM, Tooyama I. NPR-A regulates self-renewal and pluripotency of embryonic stem cells. Cell Death Dis. 2011;2:e127. | |
Vilotti S, Marchenkova A, Ntamati N, Nistri A. B-type natriuretic peptide-induced delayed modulation of TRPV1 and P2X3 receptors of mouse trigeminal sensory neurons. PLoS One. 2013;8(11):e81138. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.