")
Back to Journals » Infection and Drug Resistance » Volume 14
Role of Bacteria in the Incidence of Common GIT Cancers: The Dialectical Role of Integrated Bacterial DNA in Human Carcinogenesis
Authors Elagan SK, Almalki SJ, Alharthi MR, Mohamed MS, EL-Badawy MF
Received 1 March 2021
Accepted for publication 5 May 2021
Published 1 June 2021 Volume 2021:14 Pages 2003—2014
DOI https://doi.org/10.2147/IDR.S309051
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Héctor Mora-Montes
Sayed K Elagan,1 Saad J Almalki,1 MR Alharthi,1 Mohamed S Mohamed,1 Mohamed F EL-Badawy2
1Department of Mathematics and Statistics, College of Science, Taif University, Taif, 21944, Saudi Arabia; 2Department of Microbiology and Immunology, Faculty of Pharmacy, University of Sadat City, Sadat City, 32958, Egypt
Correspondence: Mohamed F EL-Badawy
Department of Microbiology and Immunology, Faculty of Pharmacy, University of Sadat City, Menoufia Governorate, Sadat City, 32958, Egypt
Tel +20-103-205-9964
Email [email protected]
Abstract: Despite the wide medical knowledge about the direct role of many viruses in the pathogenesis of certain cancers, there is still ambiguity and hazy vision about the direct role of bacteria in cancer incidence. Understanding the role of bacteria in carcinogenesis is no longer a scientific luxury, but it has become an urgent and extremely important necessity to realize the pathogenesis of cancer caused by oncogenic bacteria as an attempt to overcome the oncogenic mechanisms exhibited by these oncogenic bacteria. This review shed the light on the indirect role of the host’s inflammatory and immunological responses in the pathogenesis of bacteria-induced cancer. Also, this review discussed the indirect role of the bacterial toxins and virulence factors in the induction of common gastrointestinal cancers, such as gallbladder cancer (GBC), colorectal cancer (CRC), and gastric cancer (GC). Finally, this review dealt with the debate about the possibility of bacterial DNA integration into the human genome and cancer incidence.
Keywords: cancer, oncogenic bacteria, DNA, genome, gall bladder cancer
Introduction
The authors constructed this review in response to the obvious escalating number of gastrointestinal tract (GIT) cancer cases in Egypt every day in an attempt to sound the alarm and remind of the urgent need to prevent infection by oncogenic bacteria. Also, this review aimed to resolve the debate about the role of the integration of bacterial DNA into the human genome and cancer incidence. Microbial pathogens are involved in a variety of chronic human diseases. Currently, one of the most important diseases of concern to the medical community is cancer which arises from microbial origin.1 Microbial infection was found to cause 20% of all human tumors in which viral infection by oncogenic viruses constitutes the most tumors that develop from microbial origin. On the other hand, the role of bacteria in oncogenesis has been largely neglected2 for this reason, the current review was established to discuss the role of bacteria in the cancer incidence with particular emphasis on the mechanisms of bacterial carcinogenesis. To the best of our knowledge from cited literature, most studies solely attribute bacterial carcinogenesis to host inflammatory response without discussing the role of bacterial factors in carcinogensis in which many bacteria can exploit their host in various phases during their infection cycle with subsequent cell defects that affect cellular integrity and normal cellular functions resulting in cancer.2
In the current review, well-reported oncogenic bacteria in addition to bacterial and host factors contributing to cancer incidence were covered.Within this topic, the role of bacteria in the incidence of common GIT cancers like colorectal cancer (CRC), gallbladder cancer (GBC), and gastric cancer (GC) were mentioned. Besides, this review referred to the medical dispute about the possibility and role of the bacterial DNA integration into the human genome in cancer incidence also, the author’s point of view about this issue was mentioned.
Ethical Consideration Statement
All illustrated figures presented in this review are original and not published before in which the figures were created by Biorender® software after obtaining the paid license for the permission for publication .
Transmissibility of Cancer
The idea of cancer transmissibility was adopted in the 16th century, but this idea was hazy until the late 20th century in which widespread scientific progress has confirmed the validity of the previous theory that was proposed in the 16th century.3 Determination of the specific role of bacteria in carcinogenesis is difficult and challenging due to the following reasons (i) human colon harbors at least 500 bacterial species,4 (ii) the variable gap period between the incidence of bacterial infection and cancer presentation makes it is difficult to identify the culprit bacterial agent that caused the cancer.3
It has become certain that infection with some bacterial genera leads to the occurrence of different types of cancers that will be listed through this review, for instance, it was proved that the infection with Helicobacter pylori (H. pylori) is associated with gastric cancer,5 while the infection with enterotoxigenic Bacteroides fragilis (ETBF) is associated with CRC.6
The proposed etiology for the possibility of cancer-associated bacterial infection is attributed to both bacterial factors and host immune response.7,8 According to the data obtained from cited literature, it has become obvious that the incidence of cancer in the human being may arise from the chronic infection with certain types of bacteria that create a persistent inflammatory response with subsequent release of proinflammatory cytokines and tissue injury,leading to cancer incidence.3
Bacterial Infection and Oncogenesis
For several decades, it was reported that there is a relationship between certain bacterial species and various cancers (Figure 1) in which the prevailing belief is that there is a direct or indirect association between bacterial infection and the incidence of malignancy9 for instance, H. pylori indirectly contribute to GC and mucosal-associated lymphoid tissue (MALT) lymphoma via chronic inflammation of the gastric mucosa.10,11
 |
Figure 1 Reported human cancers by oncogenic bacteria. Created in ©BioRender.com. |
Complementing the previously mentioned association between infection by certain bacterial species and the incidence of certain cancers, it was reported that Fusobacterium spp. are associated with CRC,12 Salmonella enterica serovar Typhimurium is associated with GBC,13 Borrelia burgdorferi (B. burgdorferi) is associated with cutaneous B cell lymphoma,14 Escherichia coli (E. coli) is associated with esophageal and prostate cancers, Mycoplasma spp. are associated with lung cancer,15 Clostridia spp. and Ruminococcaceae are associated with breast cancer,16 and Chlamydia trachomatis and Mycoplasma genitalium are associated with ovarian cancer.15
Bacterial Factors Contributing to Carcinogenesis
Under this title, we will shed the light on the bacterial factors (Figure 2) that are confirmed to induce human carcinogenesis. It was reported that estrogen-metabolizing enzymes such as β-glucuronidases produced by Clostridia spp. contribute to the incidence of breast cancer.16 It was also reported that the cytolethal distending toxin (CDT) produced by E. coli15 and the downregulation of cyclin-dependent kinase inhibitor 2A (CDKN2) gene by uropathogenic E. coli17 contribute to the incidence of CRC and prostate cancer as described in the previous literature.15
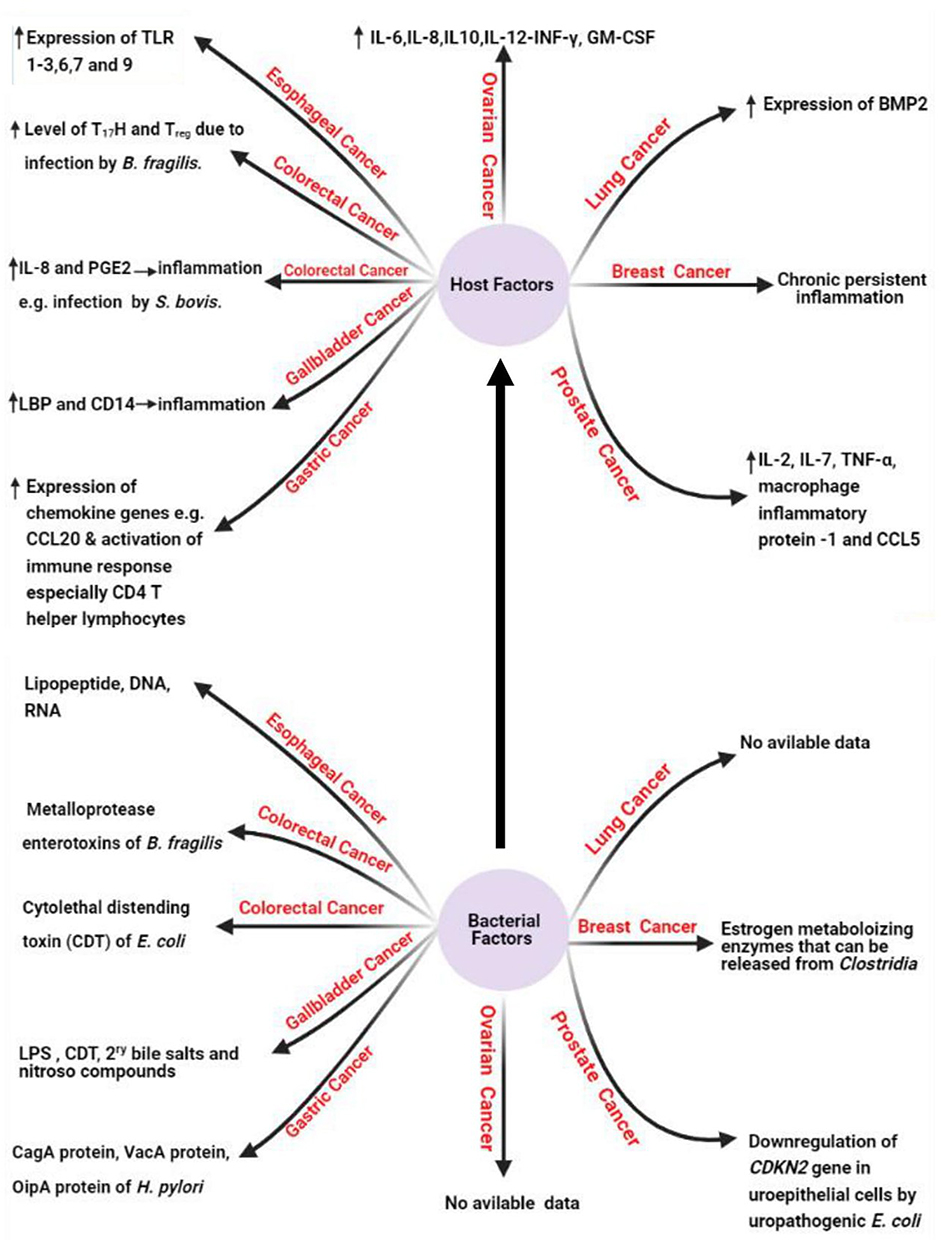 |
Figure 2 Host and bacterial factors contributing to cancer incidence. Created in ©BioRender.com. |
Also, it is reported that lipopolysaccharide (LPS) and CDT of S. typhi contribute to the incidence of GBC,15 while lipopeptides, DNA, and RNA of E. coli contribute to the incidence of esophageal cancer.18 The role of LPS in cancer incidence is mediated via the induction of the host immune response with subsequent release of specific cytokines including IL-1B, chemokine receptor 5 (CCR5), and IL-10 after the recognition of LPS by toll-like receptor (TLR). The released cytokines induce chronic inflammatory response that can result in somatic mutations with a subsequent great incidence of carcinogenesis.19 Among other bacterial factors that confirmed in the carcinogenesis is the production of bacterial oncogenic proteins such as cytotoxin-associated gene A (CagA) protein which encoded by cytotoxin-associated gene (cag) gene in H. pylori resulting in gastric adenocarcinoma via increased cellular proliferation and production of IL-8.5,20,21 In this context, no data are available about the bacterial factors that contribute to the incidence of lung and ovarian cancers.15
Also, it was found that microRNAs (miRNAs) are considered as the key inflammatory mediators that induce cancer through their effects on cell proliferation, cell death, DNA methylation, and DNA mutation resulting in carcinogensis via the host DNA damage.22–24 Altogether, the induced chronic inflammatory response by certain bacterial pathogens can accelerate DNA mutagenesis and induce cancer development.23
The Hypothesis of Chronic Irritation and Carcinogenesis
It is well known to all that, the history of the relationship between cancer and inflammation harkens back to 1800 years ago when Galenus postulated that the neoplastic tissue arose from tissue injury due to chronic inflammatory response.25,26 Seventeen centuries after, Virchow confirmed the hypothesis of Galenus in which chronic inflammation was found to cause lymphoreticular infiltration at the site of inflammation with subsequent malignant transformation,26 in which lymphoreticular infiltration was found to be related to pancreatic, esophageal and colon cancers in patients who suffered from chronic pancreatitis, chronic esophagitis, and chronic colitis, respectively.
Host Factors Contributing to Carcinogenesis
During the last decade, the potential role of bacteria in carcinogenesis was described by the British microbiologist Milton Wainwright27 in which it is proposed that about 20–25% of all cancers2 that affect humans arise from chronic inflammatory response due to persistent infection.28
Concerning host factors that induce carcinogenesis (Figure 2), it was found that continuous production of cytokines, reactive oxygen species, reactive nitrogen species, and growth factors from the recruited inflammatory cells at the inflammatory microenvironment can disturb normal biological and physiological processes leading to genomic instability with subsequent increased risk of cancer development.29
The induction of carcinogenesis by inflammatory mediators, immune cells, and pattern recognition receptors (PRRs) like TLR is reported worldwide in which increased expression of TLR-1,2, 3,6,7, and 9 is reported in esophageal cancer.15 Also, increased level of Th17 and Treg is reported in CRC caused by ETBF infection.30 Also, it was reported that increased expression of chemokine genes such as chemokine ligand 20 (CCL20) and increased activation of Th1 cells are associated with GC, while increased production of prostaglandin E2 (PGE2) and IL-8 are associated with CRC due to infection by Streptococcus bovis (S. bovis).31,32
Among the other host factors contributing to carcinogenesis is cytokine production by host immune cells like IL-6, IL-8, IL-10, interferon γ (INF-γ), and granulocyte/macrophage-colony stimulating factor (GM-CSF) that were reported to participate inthe incidence of ovarian cancer due to infection by M. genitalium.15
Common GIT Cancers Associated with Bacterial Infection
Infection by S. typhi and Incidence of GBC
One of the well-documented oncogenic bacteria is Salmonella enterica serovar Typhi (S. Typhi). The relationship between the infection by S. typhi and the incidence of GBC was investigated in Chile by Koshiol et al13 who reported a high antibody titer for Vi antigen among patients with GBC than the combined control group. Since S. Typhi was not isolated from all clinical specimens collected from GBC patients, it was proposed that chronic infection with S. Typhi could contribute to the incidence of GBC in which typhoid fever was endemic in Chile until the 1990s.3,13
It was reported that S. Typhi can cause genome instability and chromosomal defects33 through the production of cytolethal distending toxin (CDT) that gain the access to the nucleus of the host cell in murine gallbladder model15,34 as shown in (Figure 3).
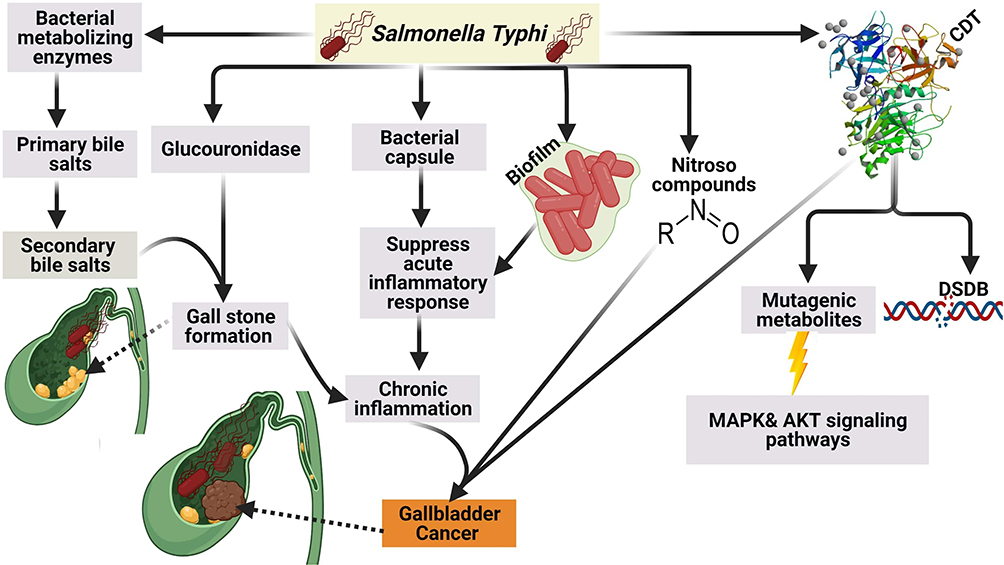 |
Figure 3 Mechanism of GBC by S. Typhi.Created in ©BioRender.com. |
In addition to CDT, it was found S. typhi produces certain mutagenic metabolites that stimulate the activation of mitogen-activated protein kinase (MAPK) and AKR thymoma (Akt) protein kinase pathways that induce and sustain malignant transformation.15 Among other carcinogens produced by S. typhi are nitroso compounds, glucuronidase, and secondary bile salts that are produced by the action of S. typhi on primary bile salts. It is thought that bacterial by-products and secondary bile salts play a pivotal role in the incidence of GBC.35,36
In addition to the aforementioned above, it is believed that the polysaccharide capsule of S. typhi contributes to the incidence of GBC by suppression of the acute mucosal inflammatory response and shifting to the sustained chronic inflammatory response.37 The authors strongly confirm that chronic typhoid carriers are more susceptible to GBC than healthy individuals based on our conviction that high endemicity with typhoid fever in Chile during the 1990s was correlated with a high incidence of GC.
Infection by B. fragilis and Incidence of CRC
B. fragilis is a strictly anaerobic Gram-negative bacterium that inhabits the human colon and is considered one of the normal human colon commensals. B. fragilis is classified according to toxin production into two types; the first type is non-toxigenic B. fragilis (NTBF), while the second type is enterotoxigenic B. fragilis (ETBF) which constitutively produce B. fragilis toxin (BFT) that is encoded by a chromosomal gene known as B. fragilis toxin (bft) gene.38
Fragilysin, or the so-called BFT, is a 20-kDa zinc-dependent metalloproteinase toxin which involved in colon carcinogensis through biofilm formation and intestinal inflammation that damages the tight junction of the intestinal epithelium, leading to increased intestinal permeability. It has been proven that chronic inflammation and tissue injury enhance and accelerate the process of carcinogenesis.38–41
Colon carcinogenesis by BFT is mediated by six main mechanisms involving the induction of the T-cell factor-dependent β-catenin pathway (TSF- β-catenin), cleavage of E-Cadherin, stimulation of IL-8 production leading to persistent proliferation of gut epithelial cells42 (Figure 4). Also, BFT induces the production of reactive oxygen species (ROS), DNA damage, and gut epithelial cell proliferation via the induction of spermine oxidase (SMO) and cellular inhibitor of apoptosis protein-2 (c-IAP2).43
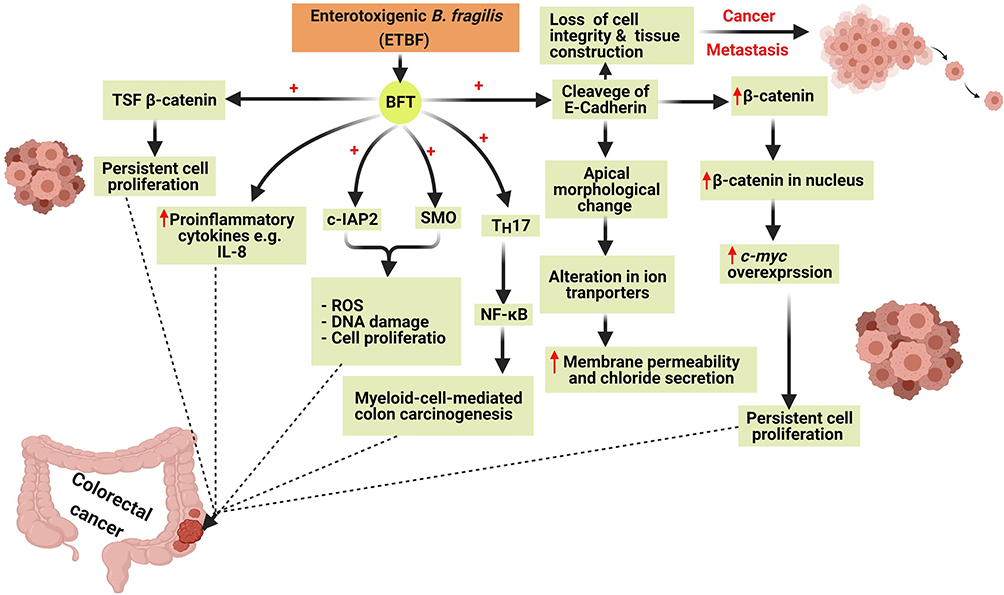 |
Figure 4 The six mechanisms involved in colon carcinogenesis by Bacteroides fragilis toxin (BFT). Colon carcinogenesis due to BFT is mediated by the induction of T-cell factor- dependent β-catenin pathway (TSF- β-catenin), cleavage of E-Cadherin, stimulation of IL-8 productionleading to the persistent proliferation of gut epithelial cells. BFT induces the production of reactive oxygen species (ROS), DNA damage, and gut epithelial cell proliferation via the induction of spermine oxidase (SMO) and cellular inhibitor of apoptosis protein-2 (c-IAP2). Created in ©BioRender.com. |
BFT also cleaves the epithelial cadherin (E-Cadherin) which is a calcium-dependent-adhesion protein, that is responsible for the adherence and the junction between epithelial cells, leading to loss of the cellular integrity and morphological changes of gut epithelial cells and therefore facilitating the spread of cancer cells to other sites.41
Cleavage of E-Cadherin also results in an elevated level of β-catenin inside the nucleus which in turn leads to overexpression of cellular-myelocytomatosis (c-myc) oncogene leading to persistent cell proliferation.42
In addition to the previously mentioned, the occurrence of CRC by BFT can be mediated via the induction of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) through activation of Th17 leading to myeloid-cell-mediated colon carcinogenesis.44
Another factor that contributes to the aetiopathogenesis of CRC is the increased expression of the proinflammatory cytokine IL-8 that was proved to play a pivotal role in colonic inflammation.45
Infection by H. pylori and Incidence of GC
H. pylori which formerly known as Campylobacter pylori (C. pylori) was categorized by the International Agency for Research on Cancer (IARC)46 as class I carcinogen, in which the previous studies revealed that approximately 75–78% of GC casses was attributed to H. pylori.5,47,48 Numerous studies have been established to explore the mechanisms by which H. pylori contributes to the incidence of GC.5,49 In this context, light will shed on on the molecular mechanisms of GC due to infection by H. pylori.
Induction of DNA Damage by H. pylori Infection
It is known that genome instability and accumulated somatic mutations are the characteristic features of cancer development.50 Genome instability due to H. pylori infection (Figure 5) can be mediated by direct DNA damage, failure of DNA repair mechanisms, or DNA methylation.49
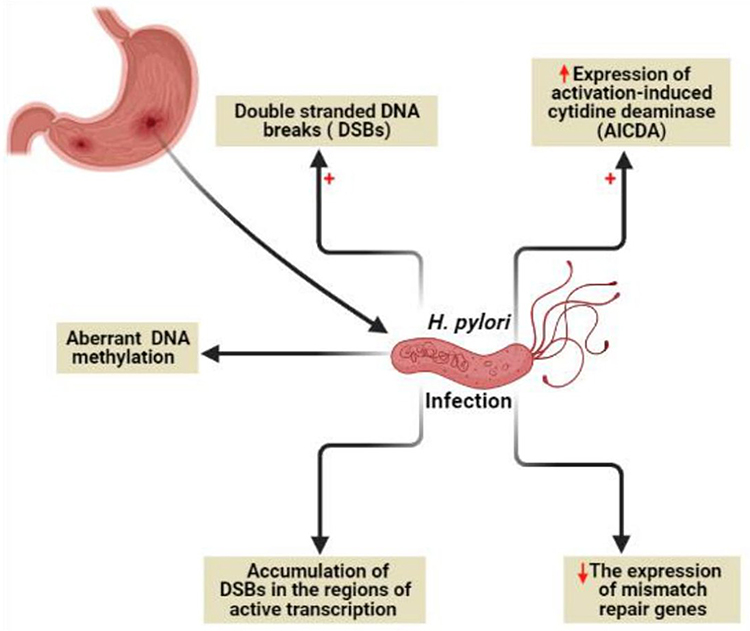 |
Figure 5 Genome instability due to H. pylori infection. Created in ©BioRender.com. |
H. pylori was found to target all the previously mentioned mechanisms via (i) induction of double-strand DNA breaks (DSBs),51 (ii) minimizing the expression of mismatch repair genes, (iii) aberrant DNA methylation,52 and (iv) maximizing the expression of activation-induced cytidine deaminase (AICDA) which also known as single-stranded DNA cytosine deaminase resulting in DNA mutation via deamination of cytosine nucleotides.53 Recently, a new characteristic mechanism of DNA damage by H. pylori through the accumulation of DSBs in the regions of active transcription near to telomeres was reported.54
Virulence Factors of H. pylori Contributing to Gastric Carcinogenesis
The most important virulence factors of H. pyloricontributing to the incidence of GC are (i) cytotoxin-associated gene A (CagA) protein,55 (ii) vacuolating cytotoxin A (VacA) protein,56 and (iii) outer inflammatory protein A (OipA) protein.57
Induction of GC by H. pylori CagA Protein
CagA protein is a 125–145 kDa protein encoded by a 40kb DNA fragment known as Cag pathogenicity island (Cag PI).58 The CagA protein is one of the most important oncoproteins produced by H. pylori which promotes gastric carcinogenesis via activation of the oncogenic signaling pathways and deactivation of tumor suppressor signaling pathways.48,55 The first evidence for the oncogenic role of CagA protein in the incidence of GC was reported by Ohnishi et al who first generated CagA transgenic mice that showed significant elevation for GC incidence.59
The Pathogenesis of Gastric Carcinogenesis by CagA Protein
The Cag PI encodes also for the functional components of the type IV secretion system (T4SS) which is a secretion protein complex that involved in translocation of the oncogenic CagA protein into the gastric epithelial cells through the binding with α5β1 integrin receptor on the surface of gastric epithelial cell.48
After the binding of T4SS with α5β1 integrin receptor, the CagA protein is injected inside the gastric epithelial cells initiating the production of IL-8 and dysregulation of the cell signaling via phosphorylation-dependent and phosphorylation-independent manners leading to induction of GC.48,60
The phosphorylated CagA which produced via the binding of CagA protein with src homology phosphatase 2 (SHP2) in addition to the growth factor receptor-bound protein 2 (Grb2)48 lead to the activation of many oncoproteins (Figure 6) like extracellular signal-regulated kinase (ERK), mitogen-activated protein kinase (MAPK), and CT10 (chicken tumor virus No. 10) regulator of the kinase (Crk) Crk/Crk-linker (Crk-L) signaling pathways21,61 that collectively lead to abnormal epithelial cell proliferation and hence, GC.48
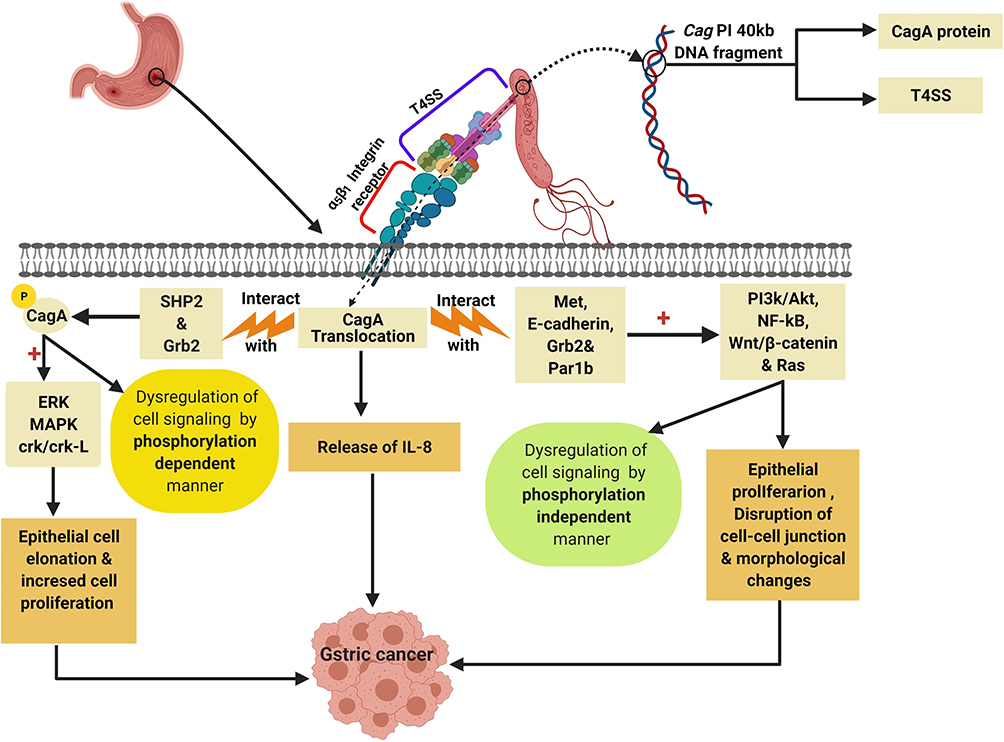 |
Figure 6 Role of CagA protein in induction of GC by dysregulation of cell signaling pathways. Created in ©BioRender.com. Abbreviations: ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; SHP2, src homology phosphatase 2; Grb2, Growth factor receptor-bound protein 2; RAS, rat sarcoma; Par1b, polarity-regulating kinase partitioning-defective 1b; Wnt/β-catenin, wingless integration-1/β-catenin; Crk/Crk-L, CT10 (chicken tumor virus No. 10) regulator of kinase/Crk-linker. |
On the other hand, the unphosphorylated portion of CagA protein interact with Met, E-cadherin, Grb2, and the polarity-regulating kinase partitioning-defective 1b (Par1b)48,62,63 signaling proteins resulting in activation of NF-κB, rat sarcoma (RAS), wingless integration-1 (Wnt)/β-catenin64 and phosphatidylinositol 3-kinase/Akt pathways leading to morphological changes and increased cell proliferation and therefore GC.48,65
The Dialectical Role of Bacterial DNA Integration in Human Carcinogenesis
Cancers are characterized by uncontrolled cell division. The main factor which enhances the transformation of normal cells into cancerous cells is the accumulation of somatic mutations.66 Integration of microbial DNA into the human chromosome is one of the causes of accumulated somatic mutations. The previous fact became obvious after the study of Schiffman et al67 which declared that approximately 90% of human cervical cancer genomes were found to harbor the DNA of human papillomavirus (HPV) so, it is well recognized that the viral DNA integration is a well-known mutagen.67
In contrast to the confirmed possibility of viral DNA integration to the human chromosome, most cancers that are proposed to be caused by certain bacterial species have been subjected to a major dispute in the medical community because it is difficult to determine if the established infection is a complication from the existing cancer (Figure 7) or the existing bacterial infection is the cause of the existing cancer.68
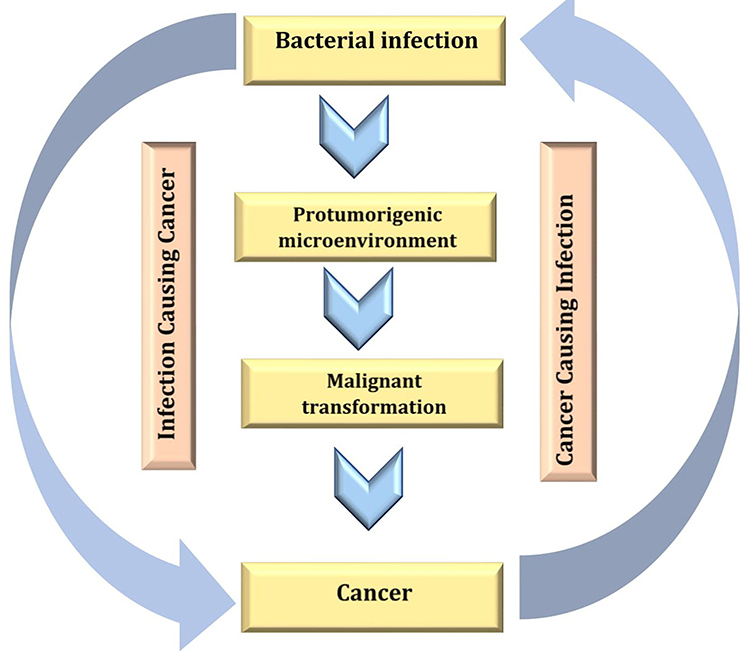 |
Figure 7 Ambiguous relationship between cancer and bacterial infection. Created in ©BioRender.com. |
In contrast to the fact that viral DNA integration into the human genome results in carcinogenesis, it has not been confirmed yet the possibility of bacterial DNA integration into the human genome and cancer incidence, so the idea of bacterial-induced carcinogenesis via DNA integration is still only a hypothesis that needs further more studies.69
It was proposed that bacterial carcinogenesis can occur via lateral gene transfer (LGT) between the human and bacteria.70 Only bacterial DNA and mRNA but not rRNA are recognized by the PRRs of the human immune system so, only bacterial mRNA and DNA molecules are attacked by different immune mechanisms.12
Based on the previous fact, only bacterial rRNA that is not recognized by the immune system can integrate into the human genome resulting in disruption and mutagenesis in proto-oncogenes and/or tumor suppressor genes leading to cancer.70–72
The hypothesis of bacterial DNA integration into the human genome has been taken into account after the report of The Cancer Genome Atlas (TCGA) which declared the detection of Acinetobacter-like DNA and Pseudomonas–like DNA in the myeloid leukemia (ML) and stomach adenocarcinoma clinical samples, respectively.70
It is hard to differentiate if the presence of the integrated bacterial DNA pieces on the human chromosome can result in driver mutations that directly drive cancer initiation and progression70,73 or the existing mutations are just merely as passenger mutations that occurred during cancer progression70 in which the cancerous cells are more susceptible to mutation during carcinogenesis than during the normal cellular state as cancer cells are more permissive to LGT.70
Resolving the Controversy About the Proper Aetiology of Bacterial Carcinogenesis
Through what was mentioned through this review, the authors strongly thought that the only confirmed real role of bacteria in the human carcinogenesis is mainly due to the induced chronic inflammatory processes with subsequent release of immune mediators that promote cell proliferation and inhibition of apoptosis.
Finally, the idea of carcinogenesis via the integration of bacterial DNA into the human genome is not accepted at least for the authors in this review due to the followig three reasons (i), the integration of the bacterial DNA into the human genome if it could happened it may be initiated after the cancer incidence by LGT as the DNA integration requires internalization of bacterial DNA to the nucleus, (ii),bacteria are mainly extracellular parasite in conrast viruses that are normally obligate intracellular parasites (iii), extracellular DNA is subjected to immune attack, (iv), all viruses are intracellular parasites, but not all of them are oncogenic.
Conclusion
Bacterial carcinogenesis is mainly mediated by host's inflammatory and immunological responses and bacterial oncogenic proteins. The hypothesis of bacterial carcinogenesis via DNA integration into the host chromosome remains unconfirmed and not acceptable as a predisposing factor for cancer incidence, so many cancer genome projects on different types of cancer should be encouraged to investigate such issues.
Acknowledgments
This study was funded by the Deanship of Scientific Research, Taif University, KSA [research project number 1-441-104].
Funding
The current work was funded by the Deanship of Scientific Research, Taif University, KSA, Program of Research Groups under Grant No. (1-441-104).
Disclosure
The authors declare no conflicts of interest.
References
1. Gargano LM, Hughes JM. Microbial origins of chronic diseases. Annual Review of Public Health. 2014;35(1):65–82. doi:10.1146/annurev-publhealth-032013-182426
2. Van Elsland D, Neefjes J. Bacterial infections and cancer. EMBO Rep. 2018;19(11):e46632. doi:10.15252/embr.201846632
3. Chang AH, Parsonnet J. Role of bacteria in oncogenesis. Clin Microbiol Rev. 2010;23(4):837–857.
4. Berg RD. The indigenous gastrointestinal microflora. Tren Microbiol. 1996;4(11):430–435.
5. Muzaheed. Helicobacter pylori oncogenicity: mechanism, prevention, and Risk Factors. Sci World J. 2020;3018326.
6. Haghi F, Goli E, Mirzaei B, Zeighami H. The association between fecal enterotoxigenic B. fragilis with colorectal cancer. BMC Cancer. 2019;19(1):1–4.
7. Basset C, Holton J, Bazeos A, Vaira D, Bloom S. Are Helicobacter species and enterotoxigenic Bacteroides fragilis involved in inflammatory bowel disease? Digest Dis An–d Sci. 2004;49(9):1425–1432.
8. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? lancet. 2001;357(9255):539–545.
9. Cummins J, Tangney M. Bacteria and tumours: causative agents or opportunistic inhabitants? Infect Agents Cancer. 2013;8(1):1–8.
10. Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136(6):1863–1873. doi:10.1053/j.gastro.2009.01.073
11. Polk DB, Peek RM. Helicobacter pylori: gastric cancer and beyond. Nature Reviews Cancer. 2010;10(6):403–414. doi:10.1038/nrc2857
12. Robinson KM, Sieber KB, Hotopp JCD. A review of bacteria-animal lateral gene transfer may inform our understanding of diseases like cancer. PLoS Genet. 2013;9(10):e1003877.
13. Koshiol J, Wozniak A, Cook P, et al. Salmonella enterica serovar Typhi and gallbladder cancer: a case–control study and meta-analysis. Cancer Med. 2016;5(11):33103235.
14. Schöllkopf C, Melbye M, Munksgaard L, et al. Borrelia infection and risk of non-Hodgkin lymphoma. Blood. 2008;111(12):5524–5529.
15. Eyvazi S, Vostakolaei MA, Dilmaghani A, et al. The oncogenic roles of bacterial infections in development of cancer. Microb Pathog. 2020;141:104019. doi:10.1016/j.micpath.2020.104019
16. Luu TH, Michel C, Bard J-M, Dravet F, Nazih H, Bobin-Dubigeon C. Intestinal proportion of Blautia sp. is associated with clinical stage and histoprognostic grade in patients with early-stage breast cancer. Nutr Cancer. 2017;69(2):267–275.
17. Tolg C, Sabha N, Cortese R, et al. Uropathogenic E. coli infection provokes epigenetic downregulation of CDKN2A (p16INK4A) in uroepithelial cells. Lab Invest. 2011;91(6):825–836.
18. Zaidi AH, Kelly LA, Kreft RE, et al. Associations of microbiota and toll- like receptor signaling pathway in esophageal adenocarcinoma. BMC Cancer. 2016;16(1):52. doi:10.1186/s12885-016-2093-8
19. Nemunaitis JM, Brown-Glabeman U, Soares H, et al. Gallbladder cancer: review of a rare orphan gastrointestinal cancer with a focus on populations of New Mexico. BMC Cancer. 2018;18(1):1–14.
20. Backert S, Tegtmeyer N, Selbach M. The versatility of Helicobacter pylori CagA effector protein functions: the master key hypothesis. Helicobacter. 2010;15(3):163–176.
21. Higashi H, Tsutsumi R, Muto S, et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science. 2002;295(5555):683–686.
22. Ohshima H, Bartsch H. Chronic infections and inflammatory processes as cancer risk factors: possible role of nitric oxide in carcinogenesis. Mutat Res. 1994;305(2):253–264.
23. Shacter E, Weitzman SA. Chronic inflammation and cancer. Oncol (Williston Park). 2002;16:
24. Schetter AJ, Heegaard NH, Harris CC. Inflammation and cancer: interweaving microRNA, free radical, cytokine and p53 pathways. Carcinogenesis. 2010;31(1):37–49.
25. Okada F. Inflammation-related carcinogenesis: current findings in epidemiological trends, causes and mechanisms. Yonago Acta Med. 2014;57(2):65–72.
26. Fishbein A, Hammock BD, Serhan CN, Panigrahy D. Carcinogenesis: failure of resolution of inflammation? Pharmacol Ther. 2020;107670. doi:10.1016/j.pharmthera.2020.107670
27. Agassandian M, Shurin GV. Bacterial Infections and Cancer Development. Infect Cancer. 2015;49–74.
28. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–867.
29. Pollard J. Bacteria, inflammation and cancer. Nat Rev Immunol. 2015;15(9):528.
30. Wu S, Rhee K-J, Albesiano E, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–1022.
31. Gold JS, Bayar S, Salem RR. Association of Streptococcus bovis bacteremia with colonic neoplasia and extracolonic malignancy. Arch Surg. 2004;139(7):760–765.
32. Biarc J, Nguyen IS, Pini A, et al. Carcinogenic properties of proteins with pro-inflammatory activity from Streptococcus infantarius (formerly S. bovis). Carcinogenesis. 2004;25(8):1477–1484.
33. Scanu T, Spaapen RM, Bakker JM, et al. Salmonella manipulation of host signaling pathways provokes cellular transformation associated with gallbladder carcinoma. Cell Host Microbe. 2015;17(6):763–774.
34. Shukla VK, Singh H, Pandey M, Upadhyay S, Nath G. Carcinoma of the gallbladder—is it a sequel of typhoid? Dig Dis Sci. 2000;45(5):900–903.
35. Goetze TO. Gallbladder carcinoma: prognostic factors and therapeutic options. World J Gastroenterol. 2015;21(43):12211–12217.
36. Di Domenico EG, Cavallo I, Pontone M, Toma L, Ensoli F. Biofilm producing Salmonella typhi: chronic colonization and development of gallbladder cancer. Int J Mol Sci. 2017;18(9)).
37. Espinoza JA, Bizama C, García P, et al. The inflammatory inception of gallbladder cancer. BBA Rev Cancer. 2016;1865(2):245–254.
38. Cheng WT, Kantilal HK, Davamani F. The Mechanism of Bacteroides fragilis Toxin Contributes to Colon Cancer Formation. Malays J Med Sci. 2020;27(4):
39. Snezhkina AV, Krasnov GS, Lipatova AV, et al. The dysregulation of polyamine metabolism in colorectal cancer is associated with overexpression of c-Myc and C/EBPβ rather than enterotoxigenic Bacteroides fragilis infection. Oxid Med Cell Longev. 2016;2016:2353560. doi:10.1155/2016/2353560
40. Boleij A, Hechenbleikner EM, Goodwin AC, et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis. 2015;60(2):208–215.
41. Sears CL, Geis AL, Housseau F. Bacteroides fragilis subverts mucosal biology: from symbiont to colon carcinogenesis. J Clin Invest. 2014;124(10):4166–4172.
42. Wu S, Morin PJ, Maouyo D, Sears CL. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124(2):392–400.
43. Yekani M, Baghi HB, Naghili B, Vahed SZ, Sóki J, Memar MY. To resist and persist: important factors in the pathogenesis of Bacteroides fragilis. Microb Pathog. 2020;149. doi:10.1016/j.micpath.2020.104506
44. Chung L, Orberg ET, Geis AL, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018;23(2):203–214.
45. Hwang S, Gwon S-Y, Kim MS, Lee S, Rhee K-J. Bacteroides fragilis toxin induces IL-8 secretion in HT29/C1 cells through disruption of E-cadherin junctions. Immu Netw. 2013;13(5):213–217.
46. Cancer IAfRo. Agents classified by the IARC Monographs, volumes 1-106. http://monographs.. iarc. fr/ENG/Classification/index. php. 2012.
47. Group I. Helicobacter pylori eradication as a strategy for preventing gastric cancer. IARC Working Group Reports. 2014.
48. Yong X, Tang B, Li B-S, et al. Helicobacter pylori virulence factor CagA promotes tumorigenesis of gastric cancer via multiple signaling pathways. Cell Commun Sign. 2015;13(1):1–13.
49. Servetas SL, Bridge DR, Merrell DS. Molecular mechanisms of gastric cancer initiation and progression by. Helicobacter Pylori. Curr Opin Infect Dis. 2016;29(3):304–310.
50. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674.
51. Hartung ML, Gruber DC, Koch KN, et al. H. pylori-induced DNA strand breaks are introduced by nucleotide excision repair endonucleases and promote NF-κB target gene expression. Cell Rep. 2015;13(1):70–79.
52. Maekita T, Nakazawa K, Mihara M, et al. High levels of aberrant DNA methylation in Helicobacter pylori–infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12(3):989–995.
53. Matsumoto Y, Marusawa H, Kinoshita K, et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nat Med. 2007;13(4):470–476.
54. Koeppel M, Garcia-Alcalde F, Glowinski F, Schlaermann P, Meyer TF. Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep. 2015;11(11):1703–1713.
55. Ansari S, Yamaoka Y. Helicobacter pylori virulence factor cytotoxin- associated Gene A (CagA)-mediated gastric pathogenicity. Int J Mol Sci. 2020;21(19):7430. doi:10.3390/ijms21197430
56. Ansari S, Yamaoka Y. Role of vacuolating cytotoxin A in Helicobacter pylori infection and its impact on gastric pathogenesis. Expert Rev Anti Infect Ther. 2020;18(10):987–996.
57. Zhao Q, Yin W, Zhao R, et al. Outer inflammatory protein of Helicobacter pylori impacts IL-8 expression, adherence, cell apoptosis and cell cycle of gastric cells independent of its copy number. Med Microbiol Immunol. 2020;209(5):621–630.
58. Hayashi T, Senda M, Morohashi H, et al. Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell Host Microbe. 2012;12(1):20–33.
59. Ohnishi N, Yuasa H, Tanaka S, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci. 2008;105(3):1003–1008.
60. Stein M, Rappuoli R, Covacci A. The cag pathogenicity island. Helicobacter Pylori: Physiol Gen. 2001;345–353.
61. Selbach M, Paul FE, Brandt S, et al. Host cell interactome of tyrosine- phosphorylated bacterial proteins. Cell Host Microbe. 2009;5(4):397–403.
62. Zeaiter Z, Cohen D, Müsch A, Bagnoli F, Covacci A, Stein M. Analysis of detergent-resistant membranes of Helicobacter pylori infected gastric adenocarcinoma cells reveals a role for MARK2/Par1b in CagA-mediated disruption of cellular polarity. Cell Microbiol. 2008;10(3):781–794.
63. Yamahashi Y, Hatakeyama M. PAR1b takes the stage in the morphogenetic and motogenetic activity of Helicobacter pylori CagA oncoprotein. Cell Adh Migr. 2013;7(1):11–17.
64. Murata-Kamiya N, Kurashima Y, Teishikata Y, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. 2007;26(32):4617–4626.
65. Suzuki M, Mimuro H, Kiga K, et al. Helicobacter pylori CagA phosphorylation-independent function in epithelial proliferation and inflammation. Cell Host Microbe. 2009;5(1):23–34.
66. Lei H, Tao K. Somatic mutations in colorectal cancer are associated with the epigenetic modifications. J Cell Mol Med. 2020;24(20):11828–11836.
67. Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. The Lancet. 2007;370(9590):890–907.
68. Arumugam M, Raes J, Pelletier E, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180.
69. Sieber KB Identification of bacterial DNA integration into the human cancer genome; 2016. https://archive.hshsl.umaryland.edu/handle/.
70. Riley DR, Sieber KB, Robinson KM, et al. Bacteria-human somatic cell lateral gene transfer is enriched in cancer samples. PLoS Comput Biol. 2013;9(6):e1003107. doi:10.1371/journal.pcbi.1003107
71. Häcker G, Redecke V, Häcker H. Activation of the immune system by bacterial CpG‐DNA. Immunology. 2002;105(3):245–251.
72. Sander LE, Davis MJ, Boekschoten MV, et al. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474(7351):385–389.
73. Wodarz D, Newell AC, Komarova NL. Passenger mutations can accelerate tumour suppressor gene inactivation in cancer evolution. J R Soc Interface. 2018;15(143):20170967. doi:10.1098/rsif.2017.0967
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.