")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 6
Road map for fibrolamellar carcinoma: progress and goals of a diversified approach
Authors Kastenhuber ER, Craig J, Ramsey J, Sullivan KM, Sage J, de Oliveira S, Riehle KJ, Scott JD, Gordan JD, Bardeesy N, Abou-Alfa GK
Received 15 November 2018
Accepted for publication 18 December 2018
Published 31 January 2019 Volume 2019:6 Pages 41—48
DOI https://doi.org/10.2147/JHC.S194764
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Edward R Kastenhuber,1,2 John Craig,3 Jon Ramsey,4 Kevin M Sullivan,5 Julien Sage,6,7 Sofia de Oliveira,8 Kimberly J Riehle,5 John D Scott,5 John D Gordan,9 Nabeel Bardeesy,10,11 Ghassan K Abou-Alfa1,12
1Cancer Biology and Genetics Program, Memorial Sloan Kettering Cancer Center, New York, NY, USA; 2Louis V. Gerstner Jr. Graduate School of Biomedical Sciences, Memorial Sloan Kettering Cancer Center, New York, NY, USA; 3Fibrolamellar Cancer Foundation, Greenwich, CT, USA; 4Department of Biochemistry, University of Vermont Cancer Center, Burlington, VT, USA; 5Northwest Liver Research Program, University of Washington, Seattle, WA, USA; 6Department of Pediatrics, Stanford University, Stanford, CA, USA; 7Department of Genetics, Stanford University, Stanford, CA, USA; 8Department of Medical Microbiology and Immunology, University of Wisconsin, Madison, WI, USA; 9Department of Medicine, University of California, San Francisco, San Francisco, CA, USA; 10Center for Cancer Research, Massachusetts General Hospital, Boston, MA, USA; 11Department of Medicine, Harvard Medical School, Boston, MA, USA; 12Department of Medicine, Weill Cornell School of Medicine, New York, NY, USA
Abstract: Fibrolamellar carcinoma is a rare liver cancer, which primarily afflicts adolescents and young adults worldwide and is frequently lethal. Given the rarity of this disease, patient recruitment for clinical trials remains a challenge. In November 2017, the Second Fibrolamellar Cancer Foundation Scientific Summit (Stamford, CT, USA) provided an opportunity for investigators to discuss recent advances in the characterization of the disease and its surrounding liver and immune context. The Fibrolamellar Cancer Foundation has thus set out a road map to identify and test therapeutic targets in the most efficient possible manner.
Keywords: fibrolamellar carcinoma, liver cancer, adolescent, young adult, fusion oncogene, cyclic AMP-dependent protein kinase catalytic subunit, heat shock protein 40
Introduction
The Second Fibrolamellar Cancer Foundation Scientific Summit was held in Stamford, CT, USA, in 2017. The goals of this meeting were to gather a diverse set of investigators, stimulate new collaborations, and devise new ways to inspire additional interest in the field. Here, we outline the exciting work supported by the Foundation and presented at the meeting. Significant progress in understanding fibrolamellar carcinoma (FLC) has recently been made by attendees and others as summarized in Table 1.
 | Table 1 Key recent developments Abbreviations: FLC, fibrolamellar carcinoma; PKA, protein kinase A; TCGA, The Cancer Genome Atlas. |
The objective of the Foundation and all investigators remains to rapidly and substantially improve patient outcomes of a rare cancer in an environment of limited resources. On the basis of clinical experience and the detailed molecular characterization of patient tumors as well as a fundamental understanding of protein kinase A (PKA) pathway biochemistry, a collection of physiologically relevant disease models has been recently generated. These complementary approaches will converge on candidate drug target identification and evaluation. Medicinal chemistry and pharmacology will be required to exploit potential dependencies, which, if successful, will culminate in clinical trials. In this iterative process, the cross-fertilization of the clinic and the laboratory can inform further experiments to enhance the understanding of basic biology and to optimize therapeutic and diagnostic strategies (Figure 1).
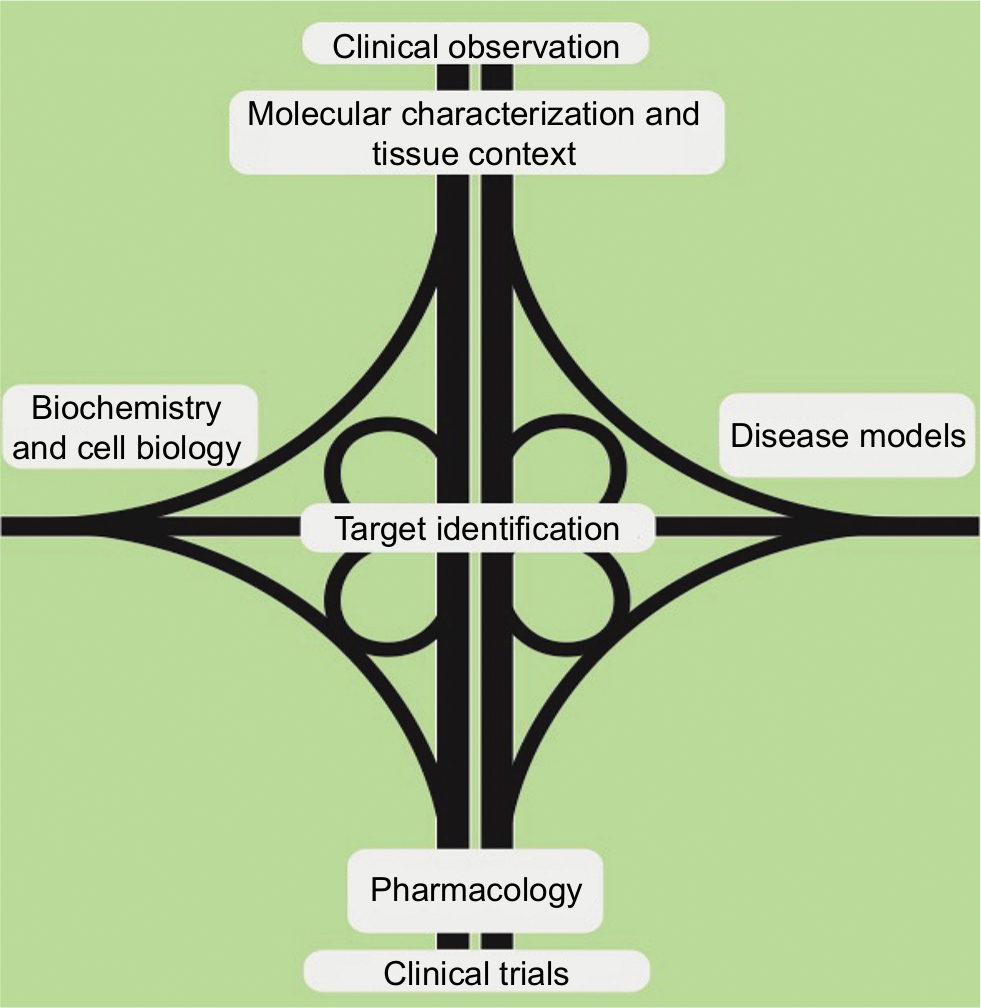 | Figure 1 The Second Fibrolamellar Cancer Foundation Scientific Summit developed a “road map” for a collaborative, multidisciplinary approach to improve outcomes for patients with fibrolamellar carcinoma. |
Clinical presentation and treatment
FLC afflicts adolescent and young adult patients without known liver cancer-associated risk factors such as chronic hepatitis infection, alcoholism, or exposure to chemicals or parasites, as originally described by John Craig (Fibrolamellar Cancer Foundation, Greenwich, CT, USA).8 This disease is recognizable based on its unique histological phenotype, with strands of lamellar fibrosis, and abundant and abnormal mitochondria on electron microscopy.
Manal Hassan (MD Anderson, Houston, TX, USA) discussed the epidemiology of the FLC population. Although the prognosis of FLC patients has been reported to be better than classic hepatocellular carcinoma (HCC), this no longer holds true when controlling for background liver cirrhosis.9 The current mainstay of FLC treatment is surgical resection, and no evidence-based systemic therapy exists. Differentiating between FLC and classic HCC is currently based on histology and will likely be aided by the recent development of a fluorescent in situ hybridization assay for sensitive and specific detection of the DNAJB1-PRKACA fusion,10 which is specific for FLC. The clinicopathologic characteristics and survival outcomes from this data set have already been reviewed with the support of the Foundation and are consistent with those reported in the literature.11 Surgical resection and disease extent were confirmed as important predictors of survival. The possibility of a negative association between female gender and prognosis could represent a clue for future therapeutic strategies.
Ghassan K Abou-Alfa (Memorial Sloan Kettering Cancer Center [MSKCC], New York, NY, USA) led the efforts of the meeting and discussed his clinical experience treating FLC and the challenges associated with launching successful clinical trials. Most patients undergo surgical resection, and some patients undergo liver transplantation. Many patients receive perioperative therapy, although no standard of care has been established.11 Relatively, few clinical trials have been conducted specifically for FLC, given the challenge of accruing a large number of patients with this rare disease. Retrospective epidemiological data do not support the conclusive superiority of any particular chemotherapeutic regimen.12 Dr Abou-Alfa led an FLC consortium including MSKCC, Harvard University, and University of California, San Francisco, which ran several clinical trials to evaluate different therapies for FLC. The first trial supported by the Foundation evaluated everolimus, leuprolide, and letrozole in patients with unresectable FLC.13 Currently, a clinical trial evaluating ENMD-2076, an aurora kinase and FLT3 inhibitor, is underway (NCT02234986). The fact that multicenter clinical trials can be conducted with sufficient enrollment is in itself an accomplishment, and the limited throughput to clinical adaptation requires ongoing careful prioritization of therapeutic strategies with the strongest rationale, aided by meaningful preclinical data.
Molecular characterization of the human disease
A major turning point in the study of this disease came with the discovery of DNAJB1-PRKACA gene fusion as a nearly universal characteristic of FLC, initially by the Simon laboratory,14 and subsequently confirmed by other groups.10,15,16 This gene fusion is the result of a somatic genetic deletion event, as it is not present in the adjacent normal liver,14 and usually does not occur in the setting of a heritable genetic disorder. These tumors harbor a low number of other mutations, and very few of which are seen in multiple patients, which is in line with observations in other malignancies that occur in young patients.17 Gene expression profiling and proteomics have confirmed that this tumor type is indeed biologically distinct from classic HCC and implicates a variety of biological processes that may underlie disease mechanisms.5 Praveen Sethupathy (Cornell University, Ithaca, NY, USA) offered that the comprehensive profiling of readouts such as the cancer cell epigenetic and metabolic state could generate new hypotheses regarding molecular mechanisms of disease and therapeutic vulnerabilities. Specifically, aberrantly expressed regulatory RNAs provide additional routes to dysregulating well-known cancer-associated pathways in FLC tumorigenesis.5 In collaboration with Jon Ramsey and Gary Stein (University of Vermont, Burlington, VT, USA), the possibility of targeting epigenetic degegulation13 was proposed by studying promoter bivalency in FLC. The bivalent marking of gene promoters by concomitant H3K4me3 and H3K27me3 is a prevailing mechanism used by pluripotent stem cells to “poise” lineage-specific genes for timely expression in the event of pro-differentiation signaling.18 It was hypothesized that phenotypic plasticity afforded to cells by promoter bivalency both is a critical feature of FLC and could nominate candidate genes that could be targeted to enforce differentiation and halt tumorigenesis.
A mechanistic explanation for remarkable specificity of DNAJB1-PRKACA to FLC remains elusive. Researchers speculated that tumorigenesis driven by this event may require high levels of activation of the DNAJB1 promoter in the cell of origin, a unique expression profile of regulators of the stability or localization of DNAJB1 or PRKACA, a unique expression profile of targets of phosphorylation by DNAJB1-PRKACA, or the requirement of a secondary extracellular signal present in particularly high levels in the liver. The fact that adolescent and young adult patients get FLC could be consistent with a yet-to-be defined reliance on a specific, puberty-associated, endocrine milieu for DNAJB1-PRKACA-mediated transformation. Anna Mae Diehl (Duke University, Durham, NC, USA) emphasized that the liver specificity of DNAJB1-PRKACA-mediated transformation supports the notion that aspects of the hepatic tissue environment may be a critical aspect of disease pathogenesis. Dr Diehl is engaged in the study of the stem cell niche in the liver microenvironment and how tumor–stroma cross talk supports both regeneration and tumorigenesis.19 Analogous to understanding the properties of tumor cells themselves, characterizing the surrounding tissue, including the types and activities of stromal cells and extracellular signaling molecules, will be of great value.
Tumor microenvironment and immunosurveillance
One major area of interest is the FLC microenvironment, including its potentially exploitable interaction with the immune system, as discussed by Amy Kim (Johns Hopkins, Baltimore, MD, USA), Kevin Sullivan (University of Washington, Seattle, WA, USA), and Kevin Barry (University of California San Francisco, San Francisco, CA, USA). There is considerable enthusiasm in exploring immunotherapy, specifically immune checkpoint blockade (PD-1/PD-L1 and others), based largely on the achievement of durable responses in lung cancer and melanoma.20,21 The finding of similar levels of lymphocytes in FLC as in background normal liver, and expression of markers of exhaustion, is provocative, but it remains unknown to what extent FLC is immunogenic. If infiltrating exhausted cytotoxic T cells are commonly found in FLC, immune checkpoint blockade could redirect a patient’s immune system to mount an effective immune response. Alternative strategies do not rely on endogenous antitumor immune recognition, but instead induce new immunity against a specific tumor antigen.22 It is unclear whether strategies aimed at activating phagocytosis of FLC cells by macrophages,23 or by modifying macrophage polarization,24 would be successful. Despite recent advances in the immunotherapy field, the challenge remains to define tumor-associated antigens in FLC patients which are not expressed in healthy tissues.25
Disease models
Studies based on observation and correlation are useful in hypothesis generation, yet controlled perturbation experiments are required to prove that a given tumor property is important for tumor development or a valuable therapeutic target. Thus, experimental model systems are needed to push the boundaries farther than what can be learned by the most careful examination of human tumor samples. Ideally, a panel of model systems with nonoverlapping strengths and caveats can be assembled to best predict which strategies will be most beneficial to patients.
The first FLC model system that was generated was a patient-derived xenograft (PDX) propagated in immunocompromised mice by Lola Reid (University of North Carolina, Chapel Hill, NC, USA).2 Consistent with the relatively indolent growth rate and resistance to cytotoxic drugs of primary FLCs, this xenograft has characteristics of biliary tree stem cells. PDX models provide a unique and powerful opportunity for perturbation experiments in vivo; a small number of additional PDX lines exist yet are not widely distributed or well characterized. For this reason, a central PDX repository for FLC has been proposed to accelerate the search for disease vulnerabilities. Harnessing such resources remains a challenge, given the substantial drift and mouse-specific evolution that has been observed over serial passage of PDXs.26
Independent studies by Francesco Niola (University of Copenhagen, Denmark) and Scott Lowe (MSKCC, New York, NY, USA) have functionally demonstrated that the DNAJB1-PRKACA gene fusion is sufficient to drive FLC tumorigenesis, using CRISPR-Cas9 to recreate the disease-associated deletion in mice.3,4 Spontaneous mouse models allow for the study of the entire process of tumor initiation and progression in the presence of an intact immune system, which may provide insights into the origin of the disease. Since methods of somatic genome editing are based on the use of wild-type inbred mouse strains, large-scale isogenic cohorts can be reproducibly generated. One major challenge of these mouse models of FLC, as described by Edward Kastenhuber (MSKCC, New York, NY, USA), is the long latency and low penetrance of tumor development driven by DNAJB1-PRKACA. He addressed this by additional genetic and chemical perturbations and found that the activation of Wnt signaling and the induction of fibrosis and liver regenerative signaling by the hepatotoxin 3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC) dramatically increased penetrance and decreased latency of lethal tumors, enabling future preclinical drug development.4
Julien Sage (Stanford University, Stanford, CA, USA) described a new, inducible mouse model of FLC. Exercising greater control over the cell type and timing in which the oncogenic driver is induced allows greater understanding of the origins and nature of the disease by lineage tracing.27 The Sage laboratory found that the expression of the PKA fusion protein during liver development leads to embryonic lethality and differentiation defects in the hepatocytic lineage. Metastasis is another significant challenge in the clinical management of FLC, which has not yet been adequately represented in existing in vivo models; novel or optimized models will be needed to study this aspect of the disease.
Along with the possibility of advancing liver organoid cultures,28 development of experimental systems in other simpler model organisms provides a unique set of opportunities. Sofia de Oliveira (University of Wisconsin, Madison, WI, USA) and Yi Guo (Mayo Clinic, Rochester, MN, USA) discussed that in zebrafish and fruit flies, respectively, one could maximize scalability and latency, while potentially maintaining some important aspects of the microenvironment.29,30 A high fecundity rate makes these models amenable to large-scale small molecule screening.31,32 Such platforms may help bridge the gap between high throughput in vitro assays and the most physiologically relevant in vivo experiments. Moreover, live imaging noninvasive techniques can be applied to intact zebrafish allowing the study of tumor microenvironment composition and cell dynamics in vivo at different stages of liver disease. Sofia de Oliveira is using a transgenic stable zebrafish FLC line that overexpresses the DNAJB1-PRKACA chimera specifically in the liver to enlighten the immune cell role in the development and progression of FLC. In her model, the overexpression of DNAJB1-PRKACA induces liver enlargement as well as increased recruitment of neutrophils and macrophages to liver area during early progression phase suggesting that innate immune cells might be involved in the progression of FLC. In the future, this transgenic model as well as zebrafish PDXs33 might be extremely useful tools to study not only FLC progression but also metastasis and help in the development of new targeted FLC therapies.
Biochemistry and cell biology
FLC-associated deletions result in the in-frame fusion of DNAJB1, which encodes a subunit of the heat shock protein chaperone HSP40, with PRKACA, which encodes for the PKAcα subunit of the cAMP-dependent protein kinase/PKA. Both the first exon of DNAJB1 and the kinase domain of PRKACA are required for tumorigenesis.4 The active site of the fusion protein retains similar structure and biochemical properties as the native kinase and results in the hyperactivation of kinase activity, at least in part by driving elevated expression of the PKAcα component.1,14,34 Thus, expanding our fundamental understanding of native PKA signaling reinforces the chances of productively interfering with pathological PKA signaling. Numerous context-specific substrates are modified by PKA phosphorylation, and it is not yet clear which of these PKA targets contribute to tumorigenesis.35
John Scott and Kimberly Riehle (University of Washington, Seattle, WA, USA) presented several fundamental aspects of the PKA pathway. Ligand-activated G-protein-coupled receptors transduce extracellular signals via G proteins such as Gs alpha (encoded by GNAS) and adenylyl cyclase, modulating levels of the second messenger cAMP and PKA activity.36 Although it was previously believed that cAMP bound the regulatory subunits of PKA, releasing and activating the catalytic subunits, Dr Scott presented his recent discovery that PKAcα can act in the context of a PKA holoenzyme and that, surprisingly, dissociation is not required for cAMP-mediated PKA activation.35 PKA is regulated by cellular localization through binding of the AKAP family of anchoring proteins, whose experimental manipulation could elucidate relevant downstream targets.7 Numerous ideas to perturb DNAJB1-PRKACA activity were heard (Drs Scott, Riehle, Ramsey, and Stein). Disrupting the regulation of AKAPs or PKA–AKAP interactions has been proposed as a pharmaceutical strategy.37 Endogenous PKI is an extremely potent and specific inhibitor of PKA and could guide rational drug design.36 The DnaJ segment of the fusion may lead to enhanced or novel phosphorylation targets of the fusion kinase, through its properties as a molecular chaperone or by altering subcellular localization or stability of the fusion protein. Thus, interfering with the DnaJ segment could provide specificity to the fusion over wild-type PKA.
John Gordon (UCSF, San Francisco, CA, USA) and Nabeeel Bardeesy (Harvard Medical School, Charlestown, MA, USA) posited that convergence in the biology of FLC and alternative paths to pathogenic PKA signaling could be enlightening. GNAS mutations, upstream of PKA, have been found in pancreatic cancer,38 and hot spot PRKACA mutations are found in adrenocortical tumors.39 Although the vast majority of FLCs are driven by the prototypical fusion, a case series identified FLCs alternatively caused by an inactivating germline mutation in PKA regulatory subunit PRKAR1A,40 supporting the notion that understanding the PKA pathway in other cancers could be applicable to FLC pathogenesis. Beyond these relatively uncommon instances of genetic alteration of the PKA pathway, nongenetic activation of PKA has been described to mediate drug resistance in breast cancer.41 Dr Gordon has commenced a comprehensive exploration of human tumor cell lines to discover alternative genetic mechanisms of activating the PKA pathway in other cancers. Dr Bardeesy elegantly demonstrated PKA dependency in a model of GNAS-mutant pancreas cancer, with further characterization of how this pathway impinges on the metabolic rewiring of cancer cells.42 Collectively, proteomics was used to nominate candidate therapeutic targets, which will next be evaluated in model systems.
Target identification and pharmacology
Small molecule inhibitors of PKA are in preclinical development by Blueprint Medicines and others (Stefanie Schalm, Blueprint Medicines, Cambridge, MA, USA). The fact that some PRKACA knockout mice are viable,43 and multiple in vivo studies of PKA inhibitors have been reported in the literature, suggests that PKA inhibition could have a therapeutic potential in the treatment of FLC, pending the availability of a suitable drug.44 Targeting fusion oncogenes has been effective in treating other cancers. For example, inhibiting another fusion kinase, BCR-ABL in chronic myeloid leukemia (CML), radically improved the long-term prognosis for CML patients.45 Likewise, targeting the gene fusion of PML (promyelocytic leukemia) and RARA (retinoic acid receptor-alpha) for degradation commonly cures acute promyelocytic leukemia,46 and kinase inhibition of ALK and ROS rearrangements has also induced clinical responses in heavily pretreated lung cancers.47 Drug development targeting alternative kinases unrelated to PKA, but that are aberrantly expressed in FLC, was discussed by Ken Ren (Casi Pharmaceuticals, Rockville, MD, USA) and Richard Cutler (Puma Biotechnologies, Los Angeles, CA, USA).
An iterative process to improve outcomes
As the field progresses, maintaining collaborative ties between clinicians and laboratory scientists will accelerate the iterative development of effective therapies and maximize the information gleaned from all clinical trials. John Craig called for the development of real-time monitoring of noninvasive biomarkers that could serve as early warning signs of disease progression, which would greatly benefit adaptive treatment decisions for individuals undergoing powerful cytotoxic or other experimental therapies.48 The utility of serum biomarkers including high serum vitamin B12 binding capacity,49,50 des-γ-carboxyprothrombin,51 and neurotensin52 has been reported. To best manage individual patient care, such markers could inform adjustments in dosing, treatment regimen, and the frequency of follow-up imaging studies. Strategies to improve imaging are also potentially valuable advances. By computed tomography (CT) imaging, FLC typically appears with a well-defined outline and heterogeneous contrast uptake with a large central scar and sometimes contains calcification.53 FLCs reportedly exhibit uptake of 18F-flourodeoxyglucose in positron emission tomography imaging studies.54
Although there is optimism that PKA inhibition could yield substantial clinical benefit, it is appropriate to anticipate the emergence of resistance. In response to single agents, innate resistance, where release of feedback inhibition reactivates a signaling pathway,55,56 and acquired resistance, where genetic mutations interfere allow for the outgrowth of drug-resistant subclones,57 have been recurrently encountered in many cancer therapies. This issue can be anticipated in vitro and ameliorated by rationally combining multiple drugs with nonoverlapping mechanisms of resistance.46,58
Outlook
We are optimistic that a multidisciplinary and collaborative approach to improving outcomes in FLC will be fueled by new technologies, comprehensive characterization of patient samples, advances in basic science and the opportunities for clinical adaptation and partnership with industry. Wide distribution of new insights and resources such as PDXs and cell lines will help accelerate the discovery process. Many challenges still lie ahead, but new technologies, strong patient engagement, and a community of interested researchers are actively investigating for a path forward.
Acknowledgments
We would like to thank the patients, their families, the Fibrolamellar Cancer Foundation, and their donors for their support. We are particularly grateful to Marna and Charles Davis for their dedication and founding of the Foundation, and John Hopper for his contribution and initiative for supporting the meeting and this report. We thank Praveen Senthupathy, Lola Reid, Kaloyan Tsanov, Pilar Mendoza, and Francisco Barriga for valuable critical feedback. ERK would like to thank Scott Lowe (MSKCC) for his mentorship. SDO is supported by the Cancer Research Institute/Fibrolamellar Cancer Foundation. ERK is supported by an F31 NRSA predoctoral fellowship from the NCI/National Institutes of Health under award number F31CA192835.
Disclosure
GKAA reports institutional research support from Acta Biologica, Agios, Array, Astra Zeneca, Bayer, Beigene, BMS, Casi, Celgene, Exelixis, Genentech, Halozyme, Incyte, Lilly, Mabvax, Novartis, OncoQuest, Polaris Puma, QED, and Roche, and consulting support from 3D Medcare, Agios, Alignmed, Amgen, Antengene, Aptus, Aslan, Astellas, Astra Zeneca, Bayer, Beigene, Bioline, BMS, Boston Scientific, Bridgebio, Carsgen, Celgene, Casi, Cipla, CytomX, Daiichi, Debio, Delcath, Eisai, Exelixis, Genoscience, Gilead, Halozyme, Hengrui, Incyte, Inovio, Ipsen, Jazz, Jansen, Kyowa Kirin, LAM, Lilly, Loxo, Merck, Mina, Newlink Genetics, Novella, Onxeo, PCI Biotech, Pfizer, Pharmacyte, Pharmacyclics, Pieris, QED, Redhill, Sanofi, Servier, Silenseed, Sillajen, Sobi, Targovax, Tekmira,Twoxar, Vicus, Yakult, and Yiviva. The authors report no other conflicts of interest in this work.
References
Cheung J, Ginter C, Cassidy M, et al. Structural insights into mis-regulation of protein kinase A in human tumors. Proc Natl Acad Sci U S A. 2015;112(5):1374–1379. | ||
Oikawa T, Wauthier E, Dinh TA, et al. Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat Commun. 2015;6(1):8070. | ||
Engelholm LH, Riaz A, Serra D, et al. CRISPR/Cas9 engineering of adult mouse liver demonstrates that the dnajb1-prkaca gene fusion is sufficient to induce tumors resembling fibrolamellar hepatocellular carcinoma. Gastroenterology. 2017;153(6):1662–1673. | ||
Kastenhuber ER, Lalazar G, Houlihan SL, et al. DNAJB1-PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2017;114(50):13076–13084. | ||
Dinh TA, Vitucci EC, Wauthier E, et al. Comprehensive analysis of the Cancer genome atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci Rep. 2017;7(1):44653. | ||
Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703–713. | ||
Smith FD, Esseltine JL, Nygren PJ, et al. Local protein kinase a action proceeds through intact holoenzymes. Science. 2017;356(6344):1288–1293. | ||
Craig JR, Peters RL, Edmondson HA, Omata M. Fibrolamellar carcinoma of the liver: a tumor of adolescents and young adults with distinctive Clinico-pathologic features. Cancer. 1980;46(2):372–379. | ||
Kaseb AO, Shama M, Sahin IH, et al. Prognostic indicators and treatment outcome in 94 cases of fibrolamellar hepatocellular carcinoma. Oncology. 2013;85(4):197–203. | ||
Graham RP, Jin L, Knutson DL, et al. DNAJB1-PRKACA is specific for fibrolamellar carcinoma. Mod Pathol. 2015;28(6):822–829. | ||
Ang CS, Kelley RK, Choti MA, et al. Clinicopathologic characteristics and survival outcomes of patients with fibrolamellar carcinoma: data from the fibrolamellar carcinoma Consortium. Gastrointest Cancer Res. 2013;6(1):3–9. | ||
Lim II, Farber BA, Laquaglia MP. Advances in fibrolamellar hepatocellular carcinoma: a review. Eur J Pediatr Surg. 2014;24(6):461–466. | ||
Ghassan K, Abou-Alfa RJM, Cosgrove D, et al. Randomized phase II study of everolimus (E), leuprolide + letrozole (ll), and E + ll (ELL) in patients (PTS) with unresectable fibrolamellar carcinoma (FLC). J Clin Oncol. 2015;33(Suppl 15):e15149. | ||
Honeyman JN, Simon EP, Robine N, et al. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science. 2014;343(6174):1010–1014. | ||
Xu L, Hazard FK, Zmoos AF, et al. Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum Mol Genet. 2015;24(1):50–63. | ||
Cornella H, Alsinet C, Sayols S, et al. Unique genomic profile of fibrolamellar hepatocellular carcinoma. Gastroenterology. 2015;148(4):806–818. | ||
Gröbner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555(7696):321–327. | ||
Messier TL, Boyd JR, Gordon JA, Stein JL, Lian JB, Stein GS. Oncofetal epigenetic bivalency in breast cancer cells: H3K4 and H3K27 tri-methylation as a biomarker for phenotypic plasticity. J Cell Physiol. 2016;231(11):2474–2481. | ||
Oh SH, Swiderska-Syn M, Jewell ML, Premont RT, Diehl AM. Liver regeneration requires Yap1-TGFβ-dependent epithelial-mesenchymal transition in hepatocytes. J Hepatol. 2018;69(2):359–367. | ||
Larkin J, Chiarion-Sileni V, Gonzalez R, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373(1):23–34. | ||
Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. | ||
Okada H, Kohanbash G, Zhu X, et al. Immunotherapeutic approaches for glioma. Crit Rev Immunol. 2009;29(1):1–42. | ||
Tseng D, Volkmer JP, Willingham SB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110(27):11103–11108. | ||
Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. | ||
Gee MH, Han A, Lofgren SM, et al. Antigen identification for orphan T cell receptors expressed on tumor-infiltrating lymphocytes. Cell. 2018;172(3):549–563. | ||
Ben-David U, Ha G, Tseng YY, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49(11):1567–1575. | ||
He L, Li Y, Li Y, et al. Enhancing the precision of genetic lineage tracing using dual recombinases. Nat Med. 2017;23(12):1488–1498. | ||
Broutier L, Andersson-Rolf A, Hindley CJ, et al. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat Protoc. 2016;11(9):1724–1743. | ||
Goessling W, Sadler KC. Zebrafish: an important tool for liver disease research. Gastroenterology. 2015;149(6):1361–1377. | ||
Asaoka Y, Terai S, Sakaida I, Nishina H. The expanding role of fish models in understanding non-alcoholic fatty liver disease. Dis Model Mech. 2013;6(4):905–914. | ||
Dar AC, Das TK, Shokat KM, Cagan RL. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature. 2012;486(7401):80–84. | ||
Evason KJ, Francisco MT, Juric V, et al. Identification of chemical inhibitors of β-Catenin-Driven liver tumorigenesis in zebrafish. PLoS Genet. 2015;11(7):e1005305. | ||
Fior R, Póvoa V, Mendes RV, et al. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc Natl Acad Sci U S A. 2017;114(39):E8234–E8243. | ||
Riggle KM, Riehle KJ, Kenerson HL, et al. Enhanced cAMP-stimulated protein kinase a activity in human fibrolamellar hepatocellular carcinoma. Pediatr Res. 2016;80(1):110–118. | ||
Kirschner LS, Yin Z, Jones GN, Mahoney E. Mouse models of altered protein kinase a signaling. Endocr Relat Cancer. 2009;16(3):773–793. | ||
Turnham RE, Scott JD. Protein kinase a catalytic subunit isoform PRKACA; history, function and physiology. Gene. 2016;577(2):101–108. | ||
Nygren PJ, Scott JD. Therapeutic strategies for anchored kinases and phosphatases: exploiting short linear motifs and intrinsic disorder. Front Pharmacol. 2015;6(e1001492):158. | ||
The Cancer Genome Atlas Research Network. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell. 2017;32(2):185–203.e13. | ||
Beuschlein F, Fassnacht M, Assié G, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N Engl J Med. 2014;370(11):1019–1028. | ||
Graham RP, Lackner C, Terracciano L, et al. Fibrolamellar carcinoma in the Carney complex: PRKAR1A loss instead of the classic DNAJB1-PRKACA fusion. Hepatology. 2018;68(4):1441–1447. | ||
Moody SE, Schinzel AC, Singh S, et al. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene. 2015;34(16):2061–2071. | ||
Patra KC, Kato Y, Mizukami Y, et al. Mutant Gnas drives pancreatic tumourigenesis by inducing PKA-mediated SIK suppression and reprogramming lipid metabolism. Nat Cell Biol. 2018;20(7):811–822. | ||
Skålhegg BS, Huang Y, Su T, Idzerda RL, Mcknight GS, Burton KA. Mutation of the Calpha subunit of PKA leads to growth retardation and sperm dysfunction. Mol Endocrinol. 2002;16(3):630–639. | ||
Lochner A, Moolman JA. The many faces of H89: a review. Cardiovasc Drug Rev. 2006;24(3–4):261–274. | ||
Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. | ||
de Thé H, Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer. 2010;10(11):775–783. | ||
Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015;16(3):280–296. | ||
Ajmera V, Perito ER, Bass NM, et al. Novel plasma biomarkers associated with liver disease severity in adults with nonalcoholic fatty liver disease. Hepatology. 2017;65(1):65–77. | ||
Paradinas FJ, Melia WM, Wilkinson ML, et al. High serum vitamin B12 binding capacity as a marker of the fibrolamellar variant of hepatocellular carcinoma. Br Med J. 1982;285(6345):840–842. | ||
Kanai T, Takabayashi T, Kawano Y, Kuramochi S, Miyazawa N. A case of postoperative recurrence of fibrolamellar hepatocellular carcinoma with increased vitamin B12 binding capacity in a young Japanese female. Jpn J Clin Oncol. 2004;34(6):346–351. | ||
Nakao A, Virji A, Iwaki Y, Carr B, Iwatsuki S, Starzl E. Abnormal prothrombin (des-gamma-carboxy prothrombin) in hepatocellular carcinoma. Hepatogastroenterology. 1991;38(5):450–453. | ||
Collier NA, Bloom SR, Hodgson HJF, Weinbren K, Lee YC, Blumgart LH. Neurotensin secretion by fibrolamellar carcinoma of the liver. The Lancet. 1984;323(8376):538–540. | ||
Ganeshan D, Szklaruk J, Kundra V, et al. Imaging features of fibrolamellar hepatocellular carcinoma. AJR Am J Roentgenol. 2014;202(3):544–552. | ||
Liu S, Wah Chan K, Tong J, Wang Y, Wang B, Qiao L. PET-CT scan is a valuable modality in the diagnosis of fibrolamellar hepatocellular carcinoma: a case report and a summary of recent literature. QJM. 2011;104(6):477–483. | ||
Manchado E, Weissmueller S, Morris JP, et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature. 2016;534(7609):647–651. | ||
Chandarlapaty S, Sawai A, Scaltriti M, et al. Akt inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71. | ||
Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by Bcr-Abl gene mutation or amplification. Science. 2001;293(5531):876–880. | ||
Glickman MS, Sawyers CL. Converting cancer therapies into cures: lessons from infectious diseases. Cell. 2012;148(6):1089–1098. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.