")
Back to Journals » OncoTargets and Therapy » Volume 12
RNA-Seq Analysis Identified XLOC_009190 as Potential Therapeutic Target for Lung Adenocarcinoma
Authors Wang J, Wang N, Zhao W , Zhao L, Jing Y, Yang L, He J, Li J
Received 31 July 2019
Accepted for publication 31 October 2019
Published 18 December 2019 Volume 2019:12 Pages 11221—11229
DOI https://doi.org/10.2147/OTT.S225532
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjeev K. Srivastava
Jing Wang,1,* Ning Wang,1,* Wei-dong Zhao,2 Li-xian Zhao,2 Yong-guang Jing,3 Li-jie Yang,2 Jie He,2 Jun Li1
1Department of Surgery, School of Clinical Medicine, Dali University, Dali, Yunnan 671000, People’s Republic of China; 2The First Affiliated Hospital of Dali University, Dali, Yunnan 671000, People’s Republic of China; 3Department of Human Anatomy, School of Basic Medical Sciences, Dali University, Dali, Yunnan 671000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Li; Jing Wang
Department of Surgery, School of Clinical Medicine, Dali University, Xue-Ren Road, Xia Guang District, Dali, Yunnan 671000, People’s Republic of China
Email [email protected]; [email protected]
Background: The abnormal regulation on the expression of lncRNAs had been linked to multiple kinds of cancers, including lung adenocarcinoma.
Methods: In this study, we carried out RNA-Seq on the three tumors and their paired normal samples from Chinese patients with lung adenocarcinoma. All the transcripts were de novo assembled, among which all the possible lncRNAs were predicted by tools including PLEK, CNCI, CPC, Blastp, hmmscan, and so forth. Their expression levels, altogether with the annotated mRNAs, were quantified. The weighted correlation network analysis and analysis of differential expression were carried out to explain the biological function of these novel lncRNAs.
Results: The weighted correlation network analysis showed that the lncRNAs, which were highly correlated with protein-coding genes, participated in various pathways, including PI3K kinase pathways. These lncRNAs were important regulators in biological processes. Next, the differentially expressed lncRNAs were identified, including four known lncRNAs and one novel lncRNA (XLOC_009190). The cis-regulation of this novel lncRNA might act on MGST1, which protected cells by conjugation and glutathione peroxidase functions. The trans-regulation of this lncRNA was investigated by its correlated mRNAs. The results showed that it possibly played a role in transmembrane receptors like G protein-coupled receptors and potassium channels.
Conclusion: We proposed the potential biological function of XLOC_009190, but further experiments are needed to elucidate its roles and its potential to be the therapeutic target.
Keywords: lung adenocarcinoma, RNA-Seq, lncRNAs, de novo assembly
Introduction
Approximately 20,000 protein-coding genes accounted for less than two percent of the human genome.5 However, it was regarded that over seventy percent of the genome can be transcribed into RNA, most of which are non-coding RNAs.19 Long non-coding RNAs (lncRNAs), defined as non-coding RNAs with over 200 nucleotides, are the most critical non-coding RNAs. In the past, lncRNAs were considered as valueless noises during transcription, while they are proved to be essential regulators in the cell and also new targets in the disease research.23
The abnormal regulation on the expression of lncRNAs had been linked to multiple kinds of cancers. For example, MALAT1, which is expressed abundantly in the nucleus, was shown to participate in the metastasis and recurrence by regulating the biological activity of the E2F1 transcription factor.30 Tano et al26 reported that the depletion of MALAT1 would reduce tumorigenicity. Another lncRNA, lincRNA-p21, served as a repressor in p53-dependent transcriptional responses through the physical binding with hnRNP-K.8 H19, which is an estrogen-inducible lncRNA, was reported to be involved in cell survival and estrogen-induced cell proliferation.25
Actually, evidence indicating the functions of lncRNAs in the initiation and progression of lung adenocarcinoma are emerging. HOTAIR was highly expressed in lung cancer and it targeted chromatin repressor polycomb proteins, which resulted in increased cancer invasiveness and metastasis.18 Yan-Rong et al33 reported that the expression of lncRNA PVT1 was elevated in lung cancer cells and the increased expression was associated with the progression and metastasis. MEG3 was suppressed in lung cancer cells and the re-constitution of MEG3 led to a decrease in cell proliferation and increased apoptosis.11
It was estimated that lung adenocarcinoma led to 150,000 deaths in the US in 2018, which makes it one of the deadliest tumors in the world. When lung cancer is diagnosed, it is usually in locally advanced status or metastasis. As a result, the 5-year survival rates of patients with lung adenocarcinoma are lower, which was estimated to be approximately 18%, compared with other primary cancers. As a result, more studies were needed to understand the progression and prognosis of lung adenocarcinoma in order to help the targeted treatment. Moreover, lncRNAs were usually neglected in a lot of studies.
In this study, we collected three tumors and three paired normal samples from Chinese patients with lung adenocarcinoma, and RNA-Seq was carried out. We aimed to figure out the known, as well as novel, lncRNAs that participated in the initiation and progression of lung adenocarcinoma. Our results can provide some insights into the biological causes of lncRNAs on lung cancer and also new targets in lung cancer research.
Methods
Sample Collection, RNA Isolation, and NGS Sequencing
The tumor and paired normal samples were collected from three lung adenocarcinoma patients with written informed consent. Total RNA was prepared by the PureLink Total RNA purification kit (Invitrogen, USA). The quality and integrity of RNA were accessed by the Qubit3.0 Fluorometer (Life Technologies, USA) and Agilent 2100 RNA Nano 6000 Assay Kit (Agilent Technologies, USA). Three micrograms of RNAs per sample were used for the following steps. The rRNA in the library were removed by Ribo-Zero GoldKits (Illumina, USA). Twelve libraries for six samples with 2 replicates each were constructed using NEBNext Ultra DirectionalRNA Library Prep Kit for Illumina (NEB, USA) according to the product instruction. They were finally sequenced by Illumina HiSeq2500 for 150 bp pair-end sequencing.
Transcriptome Assembly and Quantification
Quality control was first executed for the RNA-Seq data from each sample by fastp.4 The RNA-Seq data, all passing the step of quality control, were then mapped to the human GRCh38 genome by using TopHat.28 The mapped reads then were de novo assembled using Cufflinks.29 The assembled transcripts were merged and quantified by Cuffmerge and Cuffnorm, respectively. On the other hand, the quantification of mRNAs was executed by RSEM with Gencode v29 as the annotation.15
Identification of Novel lncRNAs
First, the transcripts that owned only one exon were excluded. The sequences of assembled transcripts that were longer than 200bp were extracted from the merged GTF file by an internal python script. PLEK,14 which could predict lncRNAs or mRNAs based on a scheme of k-mer, and CNCI,16 which profiled adjoining nucleotide triplets, was utilized to distinguish lncRNAs from mRNAs. Only the transcripts that were predicted as non-coding transcripts by both tools were kept to the next step.
The protein-coding potential of the remaining transcripts was assessed by CPC.13 The possible open read frames (ORF) were extracted from the transcripts using TransDecoder (https://github.com/TransDecoder/TransDecoder). Any transcripts containing an ORF longer than 100 amino acids were excluded. The protein sequences of the longest ORFs for each left transcript were extracted. These proteins were searched against Pfam A by hmmscan6 and the protein database from NCBI by Blastp.17 All transcripts with E values lower than 1E-6 were removed because the lncRNAs should not be similar to any known proteins.
WGCNA Correlation Network Analysis
The expected count of the lncRNAs and mRNAs was combined together and was normalized by R package DESeq.1 Next, we extracted all the lncRNA–mRNA pairs with the absolute value of correlation over 0.9 in order to prevent a too large network. Weighted correlation network analysis (WGCNA) was carried out to find modules of highly correlated genes by R package WGCNA.12,34 The statistical overrepresentation tests and pathway annotation were carried out on the protein-coding genes in each module by PANTHER web server.20,21 Fisher’s exact test was carried out with FDR multiple test correction and 0.05 was set as the cutoff for significance.
Differential Expression Analysis of Tumor-Related lncRNAs
After normalization, the differentially expressed genes, including mRNAs and lncRNAs, were identified by DESeq.1 The p-value for lncRNAs was corrected using FDR, and the lncRNAs with FDR lower than 0.05 were regarded as tumor-related lncRNAs. We re-mapped these lncRNAs to the human genome with BLAT and decided whether these lncRNAs were already known or novel. For the novel lncRNA, its functional analysis contained two parts. First, considering the cis-regulation of lncRNA, the 100k bp upstream or downstream of the lncRNAs were scanned for its possible targets. Second, the mRNAs whose expression levels were highly correlated with this novel lncRNA were identified and Slimmer gene ontology enrichment27 was carried out by PANTHER web server.20
Results
Genome-Wide Identification of lncRNAs
Understanding the characteristics of lncRNAs in patients with lung adenocarcinoma would be valuable for exploring functional mechanisms and new therapeutic targets. To obtain a de novo assembled transcriptome of lncRNAs, we totally generated 12 RNA-Seq datasets from three patients. A step-wise protocol was applied to ensure a high-confident discovery of lncRNAs (Figure 1A).
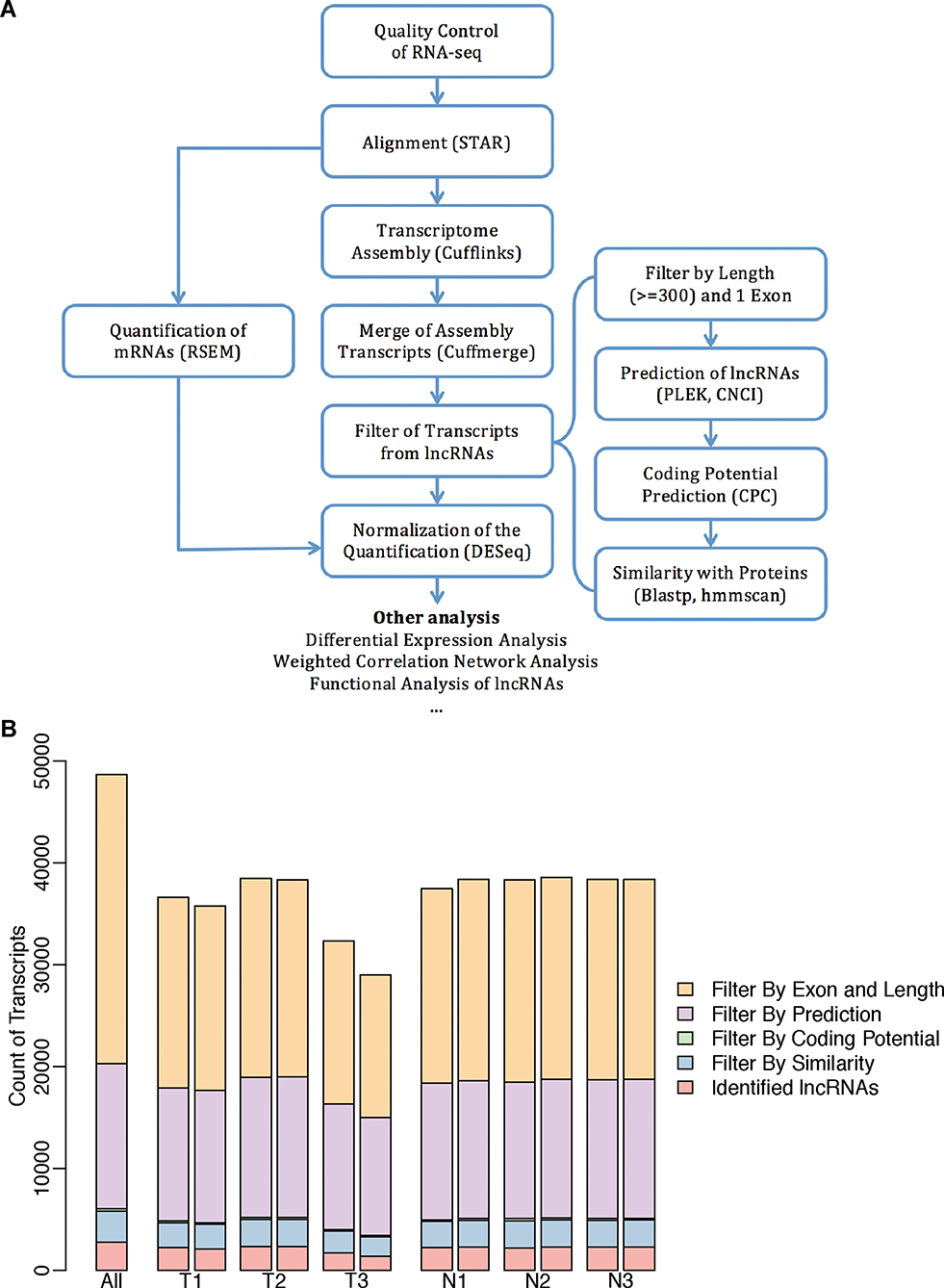 |
Figure 1 The genome-wide identification of lncRNAs. (A) The flow chart for the identification and following analysis of lncRNAs. (B) The bar plots displayed the count of transcripts from each of the six samples and the addition of all samples that were filtered out in the four steps from the flow chart “Filter of Transcripts from lncRNAs”. |
The libraries were first mapped to the reference genome and then the successfully mapped high-quality reads were used for transcriptome assembly. The de novo assembly produced 48,680 transcripts in total (Figure 1B). Totally, 27,161 transcripts with only one exon were removed considering the possible assembly error. Further analysis based on the length and the coding potential suggested that 1,891 of the transcripts were candidate lncRNAs. The proportions of transcripts filtered out in each step were similar in all the samples (Figure 1B).
Co-Expression Analysis Between lncRNAs and mRNAs
Preventing too large networks, the lncRNA-mRNA pairs were filtered by their correlation coefficients using a cutoff as 0.8. The lncRNA–mRNA pairs with a high correlation coefficient including 2372 mRNAs and 284 lncRNAs were retained for WGCNA. The WGCNA analysis identified 3 modules of highly correlated genes (Figure 2). The lncRNAs had related biological functions with the other mRNAs within the same modules. The genes belonging to the module colored as blue seemed to own a higher overlap.
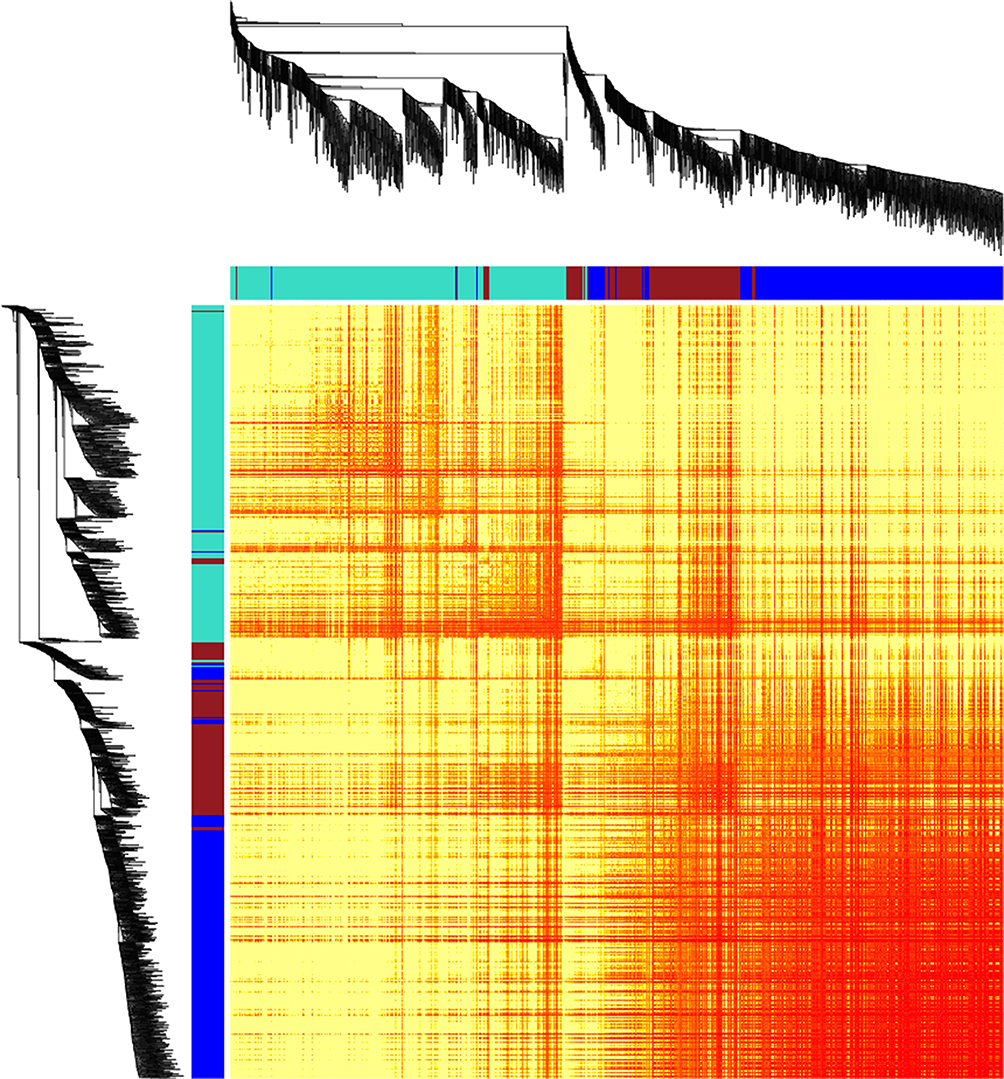 |
Figure 2 The gene network for lncRNA-associated mRNAs by WGCNA. The heatmap shows the topological overlap matrix among the lncRNA-associated mRNAs. The light yellow represents low overlap and the red color represents higher overlap. |
Statistical overrepresentation test hinted that the mRNAs in blue module were enriched in phosphoinositide 3-kinase (PI3 kinase) pathway (Table 1). On the other hand, the mRNAs in turquoise modules were associated with ubiquitin-proteasome pathway and platelet-derived growth factor (PDGF) signaling pathway. There was no pathway enriched for the mRNAs in the brown module. These results provided a basis for exploration of the lncRNA functions that some lncRNAs participated in the regulation of PI3K kinase, ubiquitin, and PDGF.
 |
Table 1 The Pathway Enrichment of Protein-Coding Genes in Identified Modules |
Identification of Stress-Related lncRNAs
We performed differential expression analysis on the contrast of tumor and normal tissues. In total, five lncRNAs were significantly up-regulated in tumor samples (Figure 3). Among the five lncRNAs, four of them were previously annotated (Table 2), while XLOC_009190 was never described in the past. The mapping of four known lncRNAs on the genomic coordinates showed their similarity to these known lncRNAs (Figure 4). XLOC_009190 was located on chromosome 12.
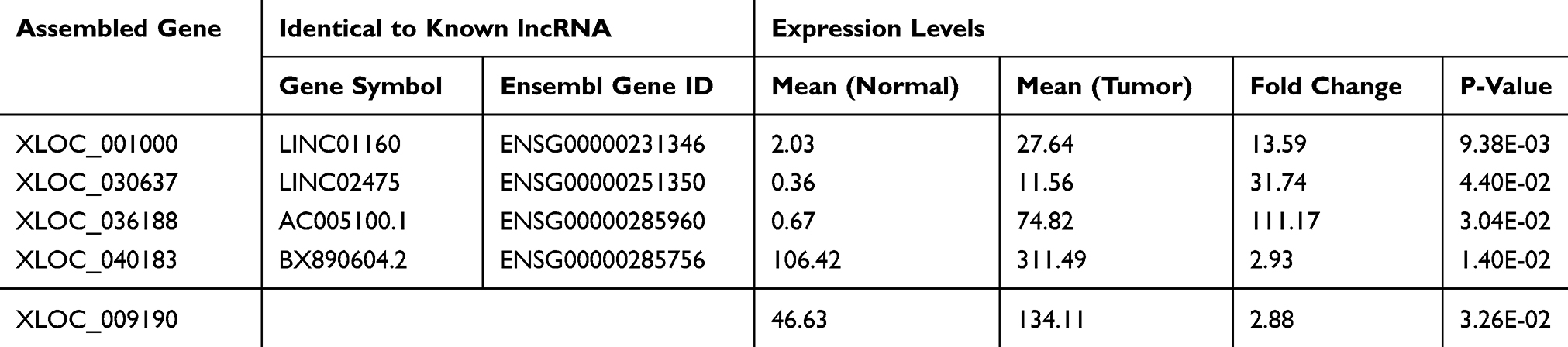 |
Table 2 The Details of the Differentially Expressed lncRNAs Between Normal and Tumor |
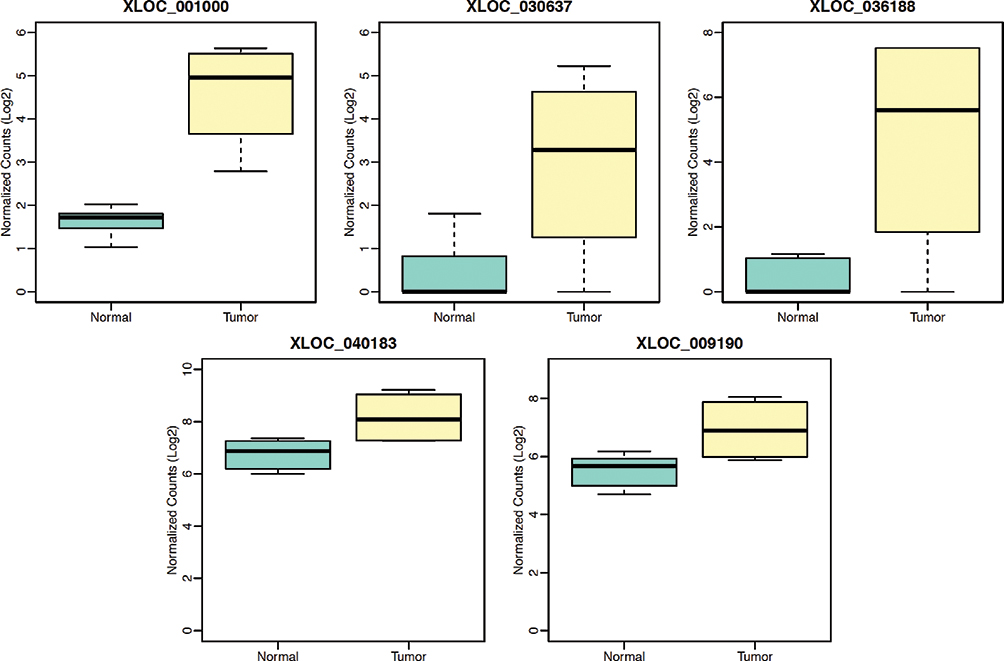 |
Figure 3 The differentially expressed lncRNAs between normal and tumor. In total, five lncRNAs were significantly up-regulated in tumor samples. |
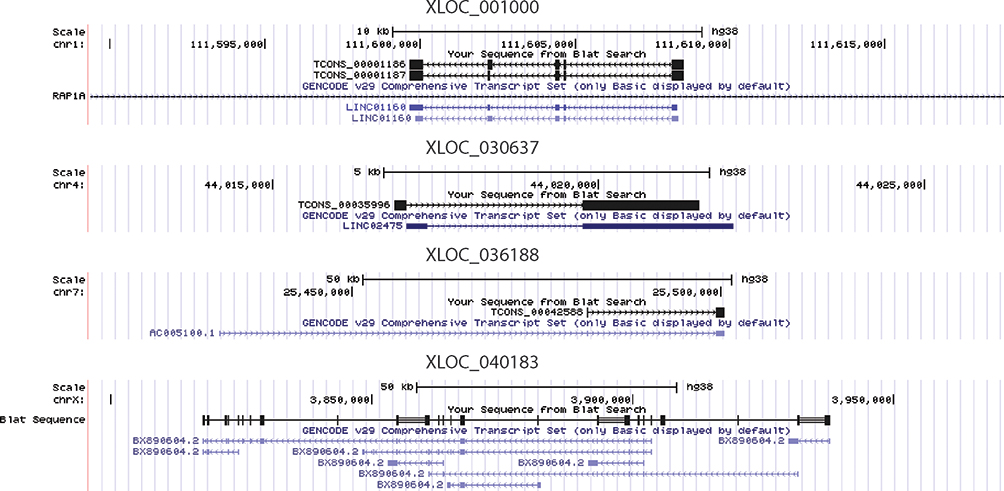 |
Figure 4 The mapping of four differentially expressed lncRNAs, whose sequences were similar to known lncRNAs, on the genomic coordinates. |
In order to figure out the potential biological function of XLOC_009190, the protein-coding genes located within ± 100k bp were identified (Figure 5A). Only MGST1 was located upstream, which might serve as the target of XLOC_009190 via cis-regulation. On the other hand, the potential mRNAs via trans-regulation were obtained by the correlation of their expression levels. The enriched gene ontology of these mRNAs hinted that XLOC_009190 might act in the detection of stimulus and transmembrane signaling receptor activity (Figure 5B).
 |
Figure 5 The biological function of novel lncRNA XLOC_009190. (A) The genomic coordinates showed that MGST1 was located between ±100k of XLOC_009190. (B) The GO term enrichment for the mRNAs whose expression levels were associated with XLOC_009190. |
Discussion
In this study, we assembled 1,891 lncRNAs for three patients with lung adenocarcinoma and identified four known and one novel lncRNAs that played a role in cancer.
Our results hinted that some lncRNAs were correlated closely with protein-coding genes, which could be categorized into three different modules. In the first module, the mRNAs and lncRNAs were significantly enriched in the pathway of PI3K kinase. Genes in the PI3K pathway are involved in cell survival, regulation of gene expression and cell metabolism, and cytoskeletal rearrangements.21 These genes are also known to be frequently altered in multiple cancers, including carcinomas of the lung. Because their high frequency of alterations include mutations and amplifications, which resulted in over-activation of certain upstream/downstream mediators, targeting genes in the PI3K signaling pathway can be a promising therapeutic approach in cancer treatment.32 The lncRNAs assignment into this module may be the potential targets as well.
Protein-coding genes in another module were associated with ubiquitin-proteasome pathway and PDGF signaling pathway. Ubiquitin-proteasome pathway involved in the degradation by the proteasome machinery. Numerous studies have shown that this pathway was occasionally the targets of tumor-related deregulation and can underlie the oncogenic transformation, tumor progression, metastasis, and drug resistance.24 PDGF, whose receptors belong to the family of receptor protein tyrosine kinases, plays a critical role in cellular proliferation and development.21 The expression of PDGF is commonly found in different human tumors, including lung adenocarcinoma. Pietras, Pahler, Bergers, and Hanahan2 confirmed the ability of tumor cell–derived PDGFs to make a contribution to the recruitment of both fibroblastic and vascular tumor stroma. Through its effects on fibroblastic and vascular tumor stroma, PDGF signaling may directly or indirectly promote the growth of tumor, the metastasis, and drug resistance.2
These results provided a basis for exploration of the lncRNA functions that they participated in the tumor progression, positively or negatively, by affecting different pathways.
Next, we identified five differentially expressed lncRNAs, which were up-regulated in the tumor samples. Four of them were known lncRNAs and one was novel.
We tried to identify the potential biological function of XLOC_009190. First, it might act by cis-regulating its upstream gene MGST1. MGST1 encodes a protein that catalyzes the conjugation of glutathione and the reduction of lipid hydroperoxides. According to the Human Protein Altas, MGST1 can serve as a prognostic marker in thyroid cancer and renal cancer.31 Some specific polymorphisms of MGST1 were also regarded to increase the risk of multiple cancers.9,35 Furthermore, Johansson, Jarvliden, Gogvadze, and Morgenstern10 reported that MGST1 protected cells, as well as mitochondria, by conjugation and glutathione peroxidase functions.
Second, the potential mRNAs regulated by XLOC_009190 were identified by their correlation with its expression levels. These correlated mRNAs were enriched in several Slimmer gene ontology terms. These terms included detection of stimulus, transmembrane signaling receptor activity, and other related molecular function.
For example, G protein–coupled receptors are one of the largest signal-conveying receptor families, which mediate various physiological processes. Moreover, they are also involved in the biological mechanisms of carcinoma. Many evidence have linked G protein-coupled receptors and other genes in the downstream with cancer growth and development. In fact, G protein–coupled receptors controlled various features of tumorigenesis, including tumor immunity, proliferation, invasion, and metastasis.3 Simultaneously, potassium channels were only enriched.
They are pore-forming transmembrane proteins controlling potassium flow across cell membranes. There was a study that hinted the antitumor efficacy of potassium channel–modulating agents.7 Potassium channels were able to regulate tumor proliferation and migration through both ion permeation–dependent or ion permeation–independent functions. Huang and Jan7 proposed that targeting potassium channel could be potential cancer therapeutics considering their cell surface localization and well-known pharmacology.
As a result, the novel lncRNA XLOC_009190 affected the patients with lung adenocarcinoma by two possible pathways. It might act on MGST1 that protected cells by conjugation and glutathione peroxidase functions. Otherwise, it played a role in transmembrane receptors like G protein–coupled receptors and potassium channels.
In summary, we de novo assembled the lncRNAs for the Chinese patients with lung adenocarcinoma, which were associated with PI3K kinase pathway and other tumor-related pathways. Moreover, we identified four known lncRNAs, as well as one novel lncRNAs (XLOC_009190), which were differentially expressed between tumor and normal. We proposed the potential biological function of these novel lncRNAs, but further experiments are needed to elucidate its roles.
Ethical Statement and Clinical Trial Number
This study was approved by the Medical Ethics Committee of Dali University with a reference number: 04/DALI02/19. Written informed consents have been provided by the patients.
Data Availability Statement
The datasets used and/or analyzed during this study are available from the corresponding author on reasonable request.
Acknowledgements
This research was supported by the Initial Scientific Research Fund of Doctor at Dali University (KYBS2018018).
Author Contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors have declared no conflicts of interest in this work.
References
1. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi:10.1186/gb-2010-11-10-r106
2. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22(10):1276–1312. doi:10.1101/gad.1653708
3. Bar-Shavit R, Maoz M, Kancharla A, et al. G protein-coupled receptors in cancer. Int J Mol Sci. 2016;17(8):1320. doi:10.3390/ijms17081320
4. Chen S, Zhou Y, Chen Y, Gu J. FASTP: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi:10.1093/bioinformatics/bty560
5. Ezkurdia I, Juan D, Rodriguez JM, et al. Multiple evidence strands suggest that there may be as few as 19,000 human protein-coding genes. Hum Mol Genet. 2014;23(22):5866–5878. doi:10.1093/hmg/ddu309
6. Finn RD, Jody C, William A, et al. HMMER web server: 2015 update. Nucleic Acids Res. 2015;43(1):30–38. doi:10.1093/nar/gkv397
7. Huang X, Jan LY. Targeting potassium channels in cancer. J Cell Biol. 2014;206(2):151–162. doi:10.1083/jcb.201404136
8. Huarte M, Guttman M, Feldser D, et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell. 2010;142(3):409–419. doi:10.1016/j.cell.2010.06.040
9. Iida A, Saito S, Sekine A, et al. Catalog of 46 single-nucleotide polymorphisms (SNPs) in the microsomal glutathione S-transferase 1 (MGST1) gene. J Hum Genet. 2001;46(10):0590–0594. doi:10.1007/s100380170026
10. Johansson K, Jarvliden J, Gogvadze V, Morgenstern R. Multiple roles of microsomal glutathione transferase 1 in cellular protection: a mechanistic study. Free Radic Biol Med. 2010;49(11):1638–1645. doi:10.1016/j.freeradbiomed.2010.08.013
11. Kruer TL, Dougherty SM, Reynolds L, et al. Expression of the lncRNA Maternally Expressed Gene 3 (MEG3) contributes to the control of lung cancer cell proliferation by the Rb pathway. PLoS One. 2016;11(11):e0166363. doi:10.1371/journal.pone.0166363
12. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008. doi:10.1186/1471-2105-9-559
13. Lei K, Yong Z, Zhi-Qiang Y, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(Web Server issue):W345. doi:10.1093/nar/gkm391
14. Li A, Zhang J, Zhou Z. PLEK: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k- mer scheme. BMC Bioinformatics. 2014;15(1):311. doi:10.1186/1471-2105-15-311
15. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323. doi:10.1186/1471-2105-12-323
16. Liang S, Haitao L, Dechao B, et al. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013;41(17):e166–e166. doi:10.1093/nar/gkt646
17. Lobo I. Basic Local Alignment Search Tool (BLAST). J Mol Biol. 2012;215(3):403–410.
18. Loewen G, Jayawickramarajah J, Zhuo Y, Shan B. Functions of lncRNA HOTAIR in lung cancer. J Hematol Oncol. 2014;7(1):90. doi:10.1186/s13045-014-0090-4
19. Mattick JS, Rinn JL. Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol. 2015;22(1):5–7. doi:10.1038/nsmb.2942
20. Mi H, Huang X, Muruganujan A, et al. PANTHER version 11: expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45(D1):D183–D189. doi:10.1093/nar/gkw1138
21. Mi H, Thomas P. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol Biol. 2009;563:123–140. doi:10.1007/978-1-60761-175-2_7
22. Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5(1):e19. doi:10.1371/journal.pmed.0050019
23. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi:10.1146/annurev-biochem-051410-092902
24. Spataro V, Norbury C, Harris AL. The ubiquitin-proteasome pathway in cancer. Br J Cancer. 1998;77(3):448–455. doi:10.1038/bjc.1998.71
25. Sun H, Wang GUO, Peng YAN, et al. H19 lncRNA mediates 17β-estradiol-induced cell proliferation in MCF-7 breast cancer cells. Oncol Rep. 2015;33(6):3045–3052. doi:10.3892/or.2015.3899
26. Tano K, Mizuno R, Okada T, et al. MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS Lett. 2010;584(22):4575–4580. doi:10.1016/j.febslet.2010.10.008
27. The Gene Ontology Consortium. Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res. 2017;45(D1):D331–D338. doi:10.1093/nar/gkw1108
28. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi:10.1093/bioinformatics/btp120
29. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi:10.1038/nbt.1621
30. Tripathi V, Shen Z, Chakraborty A, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9(3):e1003368. doi:10.1371/journal.pgen.1003368
31. Uhlen M, Zhang C, Lee S, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357(6352):eaan2507. doi:10.1126/science.aan2507
32. Wojtalla A, Arcaro A. Targeting phosphoinositide 3-kinase signalling in lung cancer. Crit Rev Oncol Hematol. 2011;80(2):278–290. doi:10.1016/j.critrevonc.2011.01.007
33. Yan-Rong Y, Shu-Zhi Z, Chun-Lei Z, Yun-Xia L, Sha-Sha Z, Xian-Jun F. Increased expression of the lncRNA PVT1 promotes tumorigenesis in non-small cell lung cancer. Int J Clin Exp Pathol. 2014;7(10):6929.
34. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi:10.2202/1544-6115.1128
35. Zhang H, Liao LH, Liu SM, et al. Microsomal glutathione S-transferase gene polymorphisms and colorectal cancer risk in a Han Chinese population. Int J Colorectal Dis. 2007;22(10):1185–1194. doi:10.1007/s00384-007-0308-9
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.