")
Back to Journals » International Journal of Nanomedicine » Volume 11
RNA interference-based nanosystems for inflammatory bowel disease therapy
Authors Guo J , Jiang XJ, Gui SY
Received 10 July 2016
Accepted for publication 5 September 2016
Published 12 October 2016 Volume 2016:11 Pages 5287—5310
DOI https://doi.org/10.2147/IJN.S116902
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lei Yang
Jian Guo,1 Xiaojing Jiang,1 Shuangying Gui1,2
1Department of Pharmaceutics, College of Pharmacy, Anhui University of Chinese Medicine, 2Institute of Pharmaceutics, Anhui Academy of Chinese Medicine, Hefei, Anhui, People’s Republic of China
Abstract: Inflammatory bowel disease (IBD), which includes ulcerative colitis and Crohn’s disease, is a chronic, recrudescent disease that invades the gastrointestinal tract, and it requires surgery or lifelong medicinal therapy. The conventional medicinal therapies for IBD, such as anti-inflammatories, glucocorticoids, and immunosuppressants, are limited because of their systemic adverse effects and toxicity during long-term treatment. RNA interference (RNAi) precisely regulates susceptibility genes to decrease the expression of proinflammatory cytokines related to IBD, which effectively alleviates IBD progression and promotes intestinal mucosa recovery. RNAi molecules generally include short interfering RNA (siRNA) and microRNA (miRNA). However, naked RNA tends to degrade in vivo as a consequence of endogenous ribonucleases and pH variations. Furthermore, RNAi treatment may cause unintended off-target effects and immunostimulation. Therefore, nanovectors of siRNA and miRNA were introduced to circumvent these obstacles. Herein, we introduce non-viral nanosystems of RNAi molecules and discuss these systems in detail. Additionally, the delivery barriers and challenges associated with RNAi molecules will be discussed from the perspectives of developing efficient delivery systems and potential clinical use.
Keywords: RNA interference, siRNA, miRNA, nanoparticles, inflammatory bowel disease, target therapy
Introduction
Inflammatory bowel disease (IBD) is a chronic intestinal disease with a high incidence of recurrence and low mortality, and there are two subtypes of IBD: ulcerative colitis (UC) and Crohn’s disease (CD).1 The Western world has a higher incidence and prevalence of IBD than the East.2 Nevertheless, epidemiological studies suggest that the incidence of IBD in Asia, South America, and the Middle East has increased annually.3 Over the past decade, the incidence rate of IBD in the People’s Republic of China has increased by approximately threefold.4 Currently, the etiology and pathogenesis of IBD are not clear. It is generally considered that the causative factors of IBD are attributed to complex factors of oxidative stress disorder, innate and adaptive immune disturbances, genetic variations, and microbial and dietary environmental factors.5 A permanent cure has not yet been developed, and thus patients require lifelong medicinal therapy, which creates a substantial economic burden.6 In the US, the mean cost associated with CD was ~$8,265 per adult patient per year, and 35% of this cost was spent on pharmaceutical therapy. The mean cost for patients with UC was $5,066, and the corresponding proportion spent on pharmaceutical therapy was ~27%.7
The primary goal of IBD treatment is to maintain clinical remission and mucosal concrescence.5 Conventional medicinal therapeutics for IBD can be divided into three categories: 1) non-steroidal anti-inflammatory drugs of classic agents (eg, sulfasalazine, mesalazine, and olsalazine), which inhibit the production of leukotriene, prostaglandin E, and free radicals; 2) glucocorticoids (eg, budesonide), which block the upstream metabolic pathways of arachidonic acid and have impressive anti-inflammatory and immunosuppressive effects; and 3) synthetic immunosuppressants (eg, azathioprine, cyclosporine A, and tacrolimus), which induce the apoptosis of T-lymphocytes to downregulate inflammation.8 Conventional medicinal therapies are limited due to their systemic adverse effects or toxicity in response to the long-term treatment process.9 Recently, biological therapy (natalizumab and tumor necrosis factor [TNF] antagonists of infliximab, adalimumab, and certolizumab pegol) has quickly advanced in IBD treatment. However, this approach has increased risks of opportunistic infections, autoimmunity, infusion reactions, and lymphoma, thus warranting sustained vigilance on the part of physicians and patients.10
The RNA interference (RNAi) technique modulates the expression of susceptibility genes and the secretion of proinflammatory cytokines related to IBD, thereby leading to the therapeutic effects of mucosal restoration and immune balance recovery in disease sites. Compared with conventional agents, RNAi treatment selectively exerts its therapeutic effects in intestinal tissues rather than causing immunosuppression in the entire body.9
In general, the RNAi molecules used in IBD treatment include short interfering RNA (siRNA) and microRNA (miRNA). MiRNA is a double-stranded endogenous regulator encoded in the genome. Mature miRNA consists of 21–25 nt segments with a 2 nt 3′ overhang.11 Mature miRNA incorporates with the multi-protein complex, RNA-induced silencing complex (RISC), and then the guide strand (antisense strand) of miRNA binds to the target mRNA within the 3′-untranslated region. Assisted by the Ago protein, the targeted mRNA is eventually translationally repressed or degraded by the mRNA–RISC complex.12 Inspired by the natural RNAi process, siRNA was developed as an exogenous double-stranded RNA for targeting mRNA degradation through the RNAi pathway. SiRNA is a specific chemosynthetic RNA duplex with a length of 19–23 nt and a 2 nt 3′ overhang. Alternatively, siRNA can also be endogenously derived from the dicer cleavage of double-stranded RNA in the cytoplasm.13,14 The incorporation of siRNA with RISC and the subsequent mRNA degradation mechanism are similar to those for miRNA.15 The siRNA sequence can be artificially designed to exquisitely regulate single gene expression because siRNA is completely complementary to the targeted mRNA sequence.
The physical and chemical characteristics of RNA have constrained the development of RNAi therapeutics for IBD. The hurdle associated with RNA is low transfection efficiency due to several anatomical and physical barriers, such as the depurination of nucleic acids in the stomach and endogenous ribonucleases in the gut lumen.16,17 The net negative charge of RNA impedes cytomembrane penetration because of the electrostatic repulsion from the anionic cell membrane.18 Therefore, chemical modification or protective vectors of RNA are necessary for RNAi molecules to reach the inflammatory site to exert curative efficacy. Chemical modifications enhance the resistance to rapid ribonuclease degradation, which is responsible for the short half-life of RNA in vivo.19,20 Such modifications primarily focus on the phosphodiester bond in the RNA backbone, 2′-ribose (2′-O-methyl,2′-O-fluoro,2′,4′-methylene bridge and so forth) and conjugation with peptide, cholesterol, polyethylene glycol (PEG), or aptamer.21,22 In addition, chemical modifications also represent a strategy for reducing the off-target effects of RNAi molecules.23
Most viral vectors possess high transfection efficiency.24 However, immunotoxicity and the risk of gene insertion in host chromosomes have motivated scientists to search for safer vectors in gene therapy protocols. The poor reproducibility of viral vectors also needs to be considered in large-scale commercial production.22 Therefore, non-viral nanoparticles have attracted considerable attention.
Non-viral nanoparticles protect RNA from degradation by endogenous ribonucleases and reduce the immune stimulation of cytokine production. The field of non-viral nanoparticle research is expanding, with numerous nanomaterial-related studies in the literature, which forms a solid foundation for biological and medical exploration. At present, with the exception of the commercially available transfection reagent of cationic liposomes in vitro, there are no approved siRNA or miRNA drugs for clinical use in IBD therapy.
In this review, RNAi-based nanoparticle delivery systems will be discussed with the aim of providing assistance in designing or synthesizing non-viral nanosystems for IBD treatment and exploring their potential use in clinical applications.
Nanoparticle delivery systems for RNAi molecules
To confer stability to RNA during delivery in vivo, an alternative strategy is to use special delivery systems. As a hot topic in recent decades, the knowledge accumulated regarding nanoparticle delivery systems forms a solid foundation for siRNA or miRNA delivery.
Nanoparticle delivery systems have several potential advantages in RNA delivery. Nanosized particles are retained in the intestinal tract with a large surface area, which increases the residence time in inflamed intestinal regions. Therefore, nanoparticle delivery systems are preferentially taken up by endocytosis in the enterocytes or M-cells, which have a highly increased number of RNAi molecules at inflamed intestinal regions. This characteristic also avoids the elimination of nanoparticle delivery systems in vivo, which is caused by diarrhea, a common symptom in patients with IBD.25 Additionally, nanoparticles tend to accumulate in the gaps between epithelial cells through the epithelial enhanced permeability and retention (eEPR) effect. This benefit causes nanoparticles with ligand modifications, such as antibody and glycosyl at the surface, to target macrophages in inflamed intestinal mucosa (Figure 1). A large amount of TNF-α is secreted from mononuclear macrophages in the intestinal tracts of patients with IBD, and high levels of TNF-α may enhance proinflammatory cytokines such as interleukin (IL)-1, IL-6, and IL-8, which aggravate inflammatory injury in disease sites. Furthermore, compared to conventional dosage systems, nanoparticle delivery systems decrease the required effective drug dose, thereby reducing the side effects of the drug.26 The outstanding properties of nanoparticles described earlier have provided a stable and reliable approach for delivering RNA to target sites in the intestinal tract.
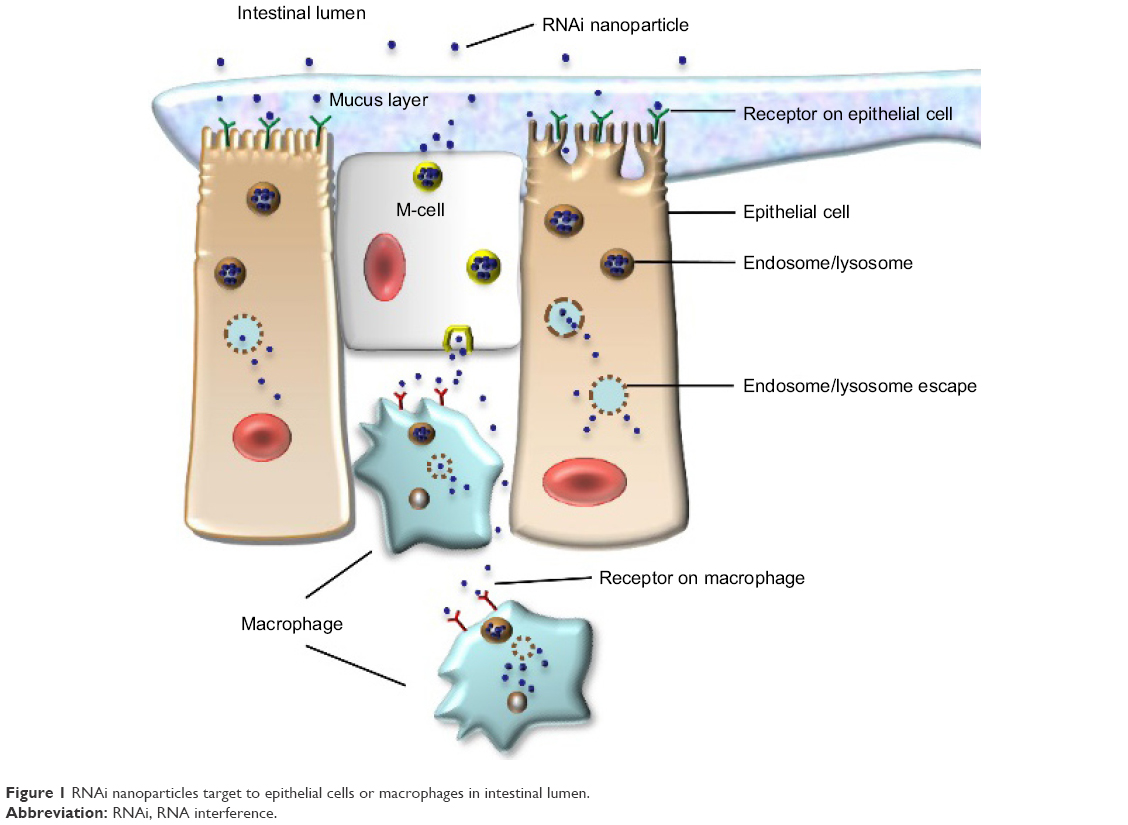 | Figure 1 RNAi nanoparticles target to epithelial cells or macrophages in intestinal lumen. |
A RNA nanoparticle delivery system should possess quite a few underlying principles; for example, it should have no stimulatory effects on immune systems, protect RNA from degradation induced by altered pH gradients and ribonucleases in the gastrointestinal tract, have a proper surface charge, possess a suitable binding strength with RNA, and escape endosomes or lysosomes before releasing siRNA or miRNA into the cytoplasm in target cells.
We review the current strategies for using siRNA and miRNA nanoparticles in IBD treatment, including liposomes, polysaccharide-based nanoparticles, polylactide (PLA)-based nanoparticles, calcium phosphate (CaP)/poly(D,L-lactide-co-glycolide acid) (PLGA)-based nanoparticles, nanoparticles-in-microsphere oral system (NiMOS), thioketal-based nanoparticles, and polyethylenimine (PEI)-based nanoparticles (Tables 1 and 2).
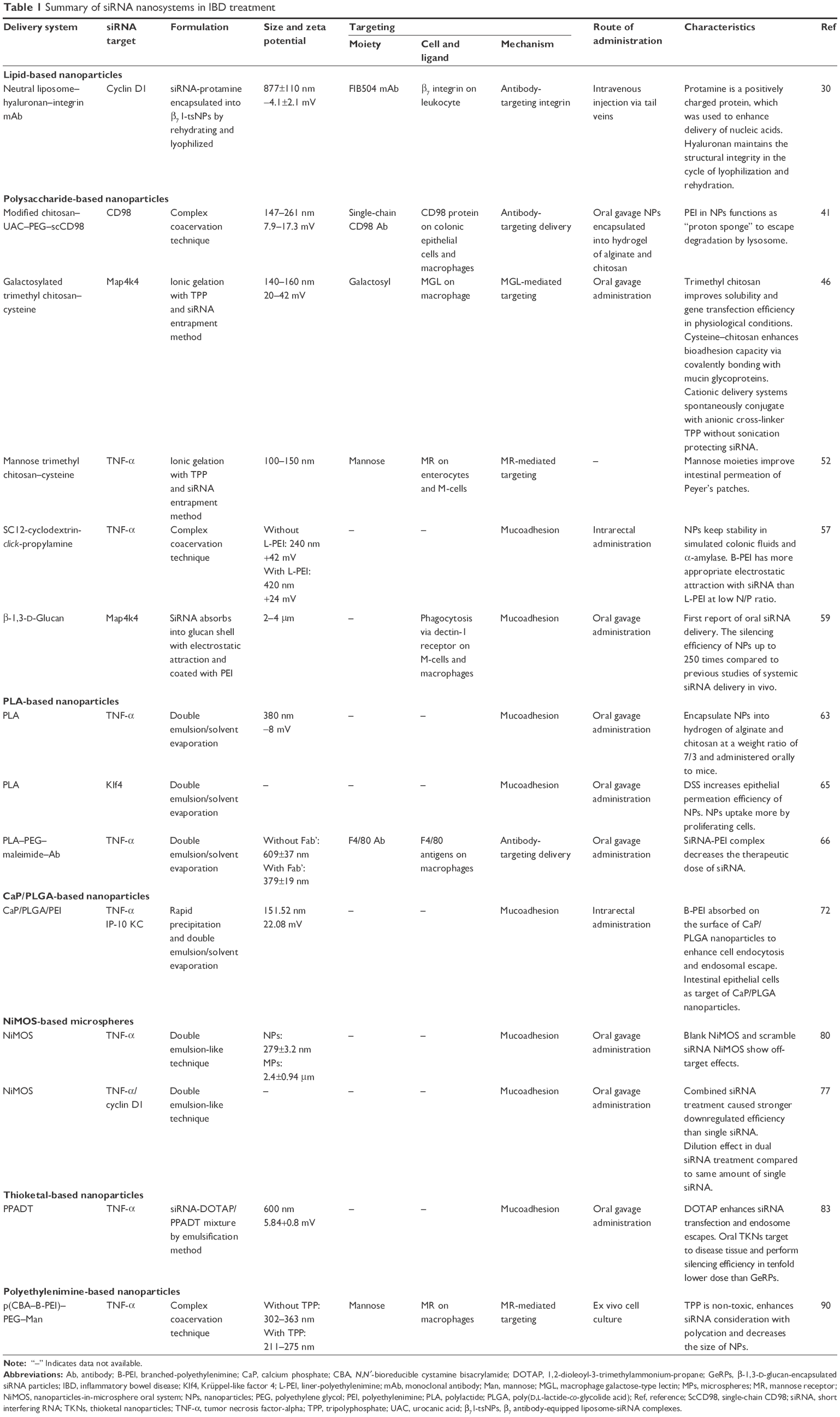 | Table 1 Summary of siRNA nanosystems in IBD treatment |
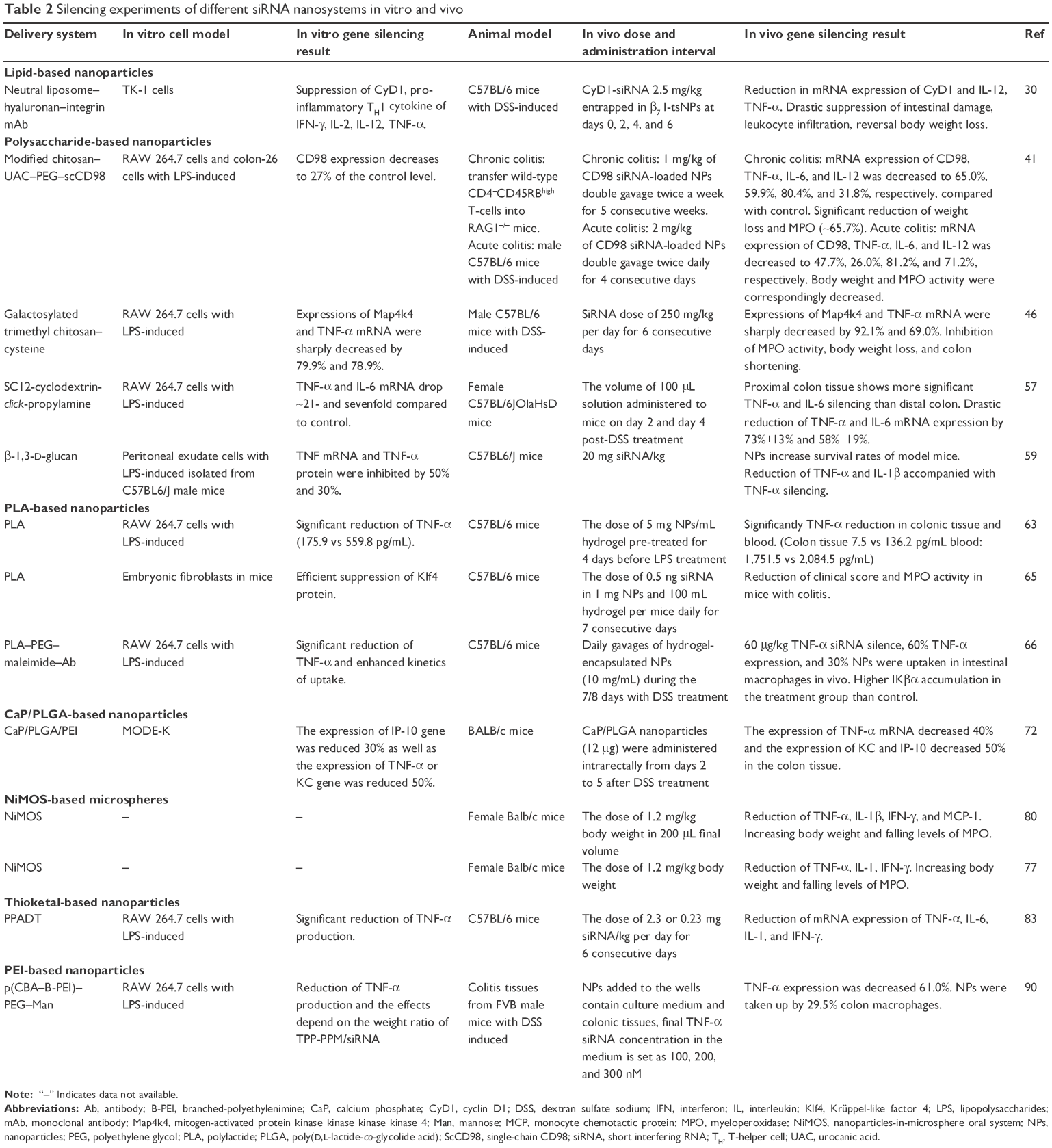 | Table 2 Silencing experiments of different siRNA nanosystems in vitro and vivo |
Nanoparticle delivery systems of siRNA
Liposomes
Liposomes are a type of vector that is biocompatible with cell membranes, and they are composed of a phospholipid bilayer and a hydrophilic core. The phospholipid bilayer assists liposomes in crossing the cell membrane into the cytoplasm, and the hydrophilic core allows liposomes to be loaded with RNAi molecules.27
In recent decades, cationic liposomes have been developed as standard transfection reagents for oligonucleotide or siRNA delivery. For example, antisense oligonucleotides of TNF-α mixed with lipofectin (Invitrogen, USA) transfect into the mouse macrophage cell line P388D1. The results showed an effective decrease in TNF-α mRNA in a dose- and sequence-dependent manner. In subsequent in vivo experiments, the administration of anti-TNF-α oligonucleotide via injection resulted in reduced disease activity index scores in both acute and chronic murine models of colitis.28 Furthermore, TNF-α siRNA liposomes (Lipofectamine 2000; Invitrogen) were rectally instilled in a murine model of IBD. The experimental results showed a relative mucosal resistance to experimental colitis.29 However, the practical silencing effects of transfection reagents in vivo are not as robust as those in vitro.
To obtain better gastrointestinal tract-targeting performance, liposomes were equipped with ligands, such as antibodies, on the liposome surface. These ligands allow liposomes to selectively bind to the receptors on the surface of enterocytes or immune cells. Peer et al30 developed integrin β7 antibody (FIB504)-equipped liposome–siRNA complexes (β7 I-tsNPs) and demonstrated that FIB504-equipped liposomes targeted leukocytes involved in colonic inflammation. The multi-lamellar liposomes were initially prepared using the lipid-film method with phosphatidylcholine, dipalmitoyl phosphatidylethanolamine (DPPE), and cholesterol in a molar ratio of 3:1:1. Subsequently, multi-lamellar liposomes were extruded to unilamellar nanoscale liposomes with the assistance of a Thermobarrel Lipex extruder. The liposomes were produced using a neutral phospholipid to avoid the toxicity of cationic lipids. High molecular weight (MW) hyaluronan (850 kDa) was then covalently linked to the surface of the liposomes in a ratio of 75 μg hyaluronan/μmol lipid with the assistance of 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride activation. The two considerations for introducing hyaluronan into nanoscale liposomes are as follows: 1) as a bridge linker, the FIB504 antibody subsequently attached to the hyaluronan and 2) hyaluronan was used to stabilize the particles during the cycle of lyophilization and rehydration. In another research report by Peer et al,31 the surface-binding ligand of hyaluronan on liposomes acted as a cryoprotectant during lyophilization, which may derive from providing a substitute structure to form stabilizing H bonds. It was found that with hyaluronan bound to antibodies, the change in the mean particle size during lyophilization and rehydration was 107±14 to 123±24 nm, whereas a typically growing aggregation of 110±21 to 1,330±750 nm was detected without hyaluronan. Liposome–siRNA delivery systems (CyD1-siRNA/β7 I-tsNPs) were synthesized using unloaded liposomes incorporated with cyclin D1 (CyD1) siRNA and protamine in a molar ratio of 5:1 through lyophilization and rehydration. Protamine provided positive charges to condensed siRNA with a positive shift from 24 to 18 mV in the nanoparticle formulation. Compared to naked siRNA and isotype control immunoglobulin G-attached stabilized nanoparticles (IgG-sNPs), β7 I-tsNPs targeted wild-type splenocytes but not β7 integrin knockout splenocytes. The therapeutic effects of CyD1 silencing were evaluated in murine models of dextran sulfate sodium (DSS)-induced colitis. Mice were intravenously injected with CyD1-siRNA/β7 I-tsNPs, and the CyD1 knockdown selectively decreased intestinal CyD1 mRNA 3–4-fold compared with mice administered with CyD1-siRNA/IgG-sNPs or the controls. Intestinal injury of leukocyte infiltration, body weight loss, and hematocrit decline were also reduced. In general, CyD1-siRNA/β7 I-tsNP has a higher siRNA entrapment capacity, in which each nanoparticle condensed ~4,000±1,200 siRNA molecules, and the lowest effective dose of siRNA was 2.5 mg/kg in systemic siRNA therapy in vivo. The stability of this liposome delivery system against ribonuclease was tested based on the silencing effect of Ku70 siRNA. Liposome–siRNA complexes entrapped Ku70 siRNA to retain its silencing effect up to 2 hours in serum compared with naked siRNA.30 Similarly, DEC205 mAb-decorated liposomes were manipulated to target dendritic cells for immunomodulation by silencing CD40 genes.32 The results indicated that neutral liposomes (1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphocholine) have insufficient encapsulation efficiency for siRNA. A cationic lipid (dimethyldioctadecylammonium bromide) was then added to the neutral liposome systems to obtain better encapsulation capacity. The positive charge of cationic liposomes is generally considered to increase electrostatic interactions with siRNA. However, cationic liposome vectors are typically toxic when delivered systemically in vivo due to abnormal aggregation with proteins in the circulation.33 During oral administration of liposomes, the lipid bilayer is degraded by bile salts and pancreatic lipases. Thus, liposomes could be coated with biodegradable or mucoadhesive polymers, such as chitosan and pectin, to resist the harsh environment in the gastrointestinal tract.34
Polysaccharide-based nanoparticles
Polysaccharides are composed of monosaccharide units joined by glycosidic bonds. Polysaccharides have favorable characteristics as natural biomaterials, such as low toxicity, biocompatibility, biodegradability, and low cost.35 The major advantage of polysaccharides as an RNA vector is their flexible characteristic of modifying with functional groups (hydroxyls, amines, and carboxylic acids) on the polysaccharide backbone. These modifications were intended to overcome the delivery obstacles of rapid mononuclear phagocyte system clearance, insufficient RNA binding, and endosome degradation.36 In addition, some polysaccharides (eg, chitosan, alginate, and hyaluronic acid) could achieve bioadhesion by forming non-covalent bonds among functional groups (hydroxyls, amines, and carboxylic acids) and epithelia on intestinal mucous.37 Polysaccharide-based delivery systems offer a special advantage over other systems. Polysaccharides retain their integrity during the course of transportation in the gastrointestinal tract. However, once polysaccharides arrive in the colon section, they are degraded by a wide range of enzymes (β-d-glucosidase, β-d-galactosidase, β-xylosidase, β-arabinosidase, azo reductase, and nitro reductase) produced by microflora.38 To date, chitosans, cyclodextrins, β-glucan, and cationic konjac glucomannan (cKGM) have been used to prepare siRNA nanosystems.
Chitosan
Chitosan is a linear cationic polysaccharide that consists of D-glucosamine and N-acetyl-D-glucosamine linked by β-(1,4)glycosidic bonds (Figure 2A). This polysaccharide is derived from the deacetylation of chitin and has many advantages in terms of biological properties, such as low toxicity, biocompatibility, and biodegradability. In particular, chitosan is frequently used as a mucosal delivery material because cationic chitosan easily interacts with anionic molecules or tissues, such as nucleic acids, mucosa, and other negatively charged polysaccharides.39
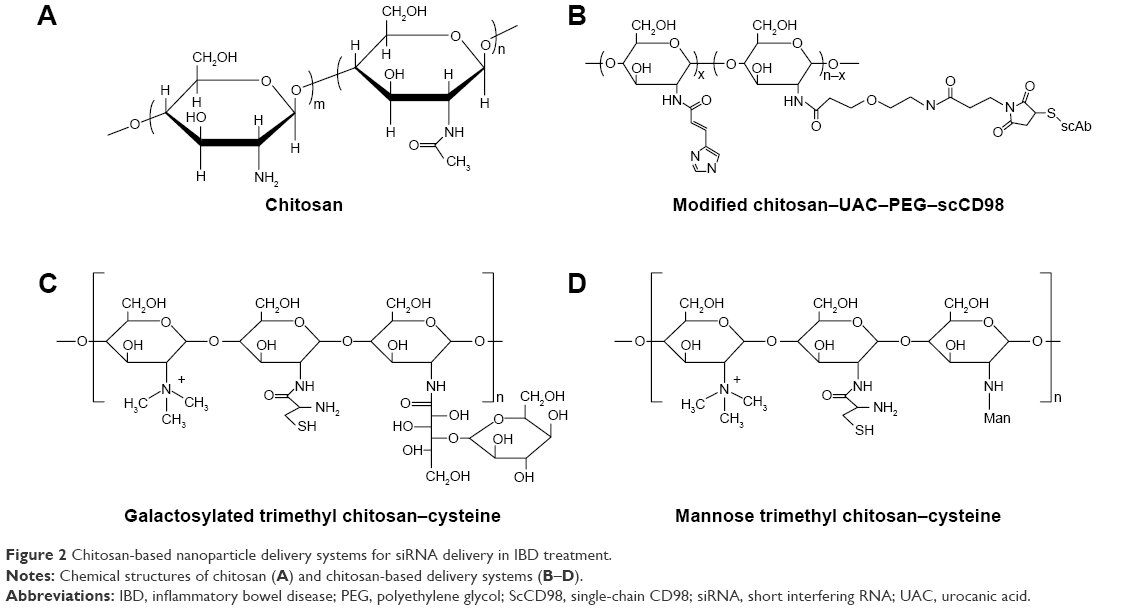 | Figure 2 Chitosan-based nanoparticle delivery systems for siRNA delivery in IBD treatment. |
There are some parameters that are highly related to the siRNA transfection efficiency of chitosan-based vectors, including MW, degree of deacetylation, and N/P charge ratio.40 The MW of chitosan influences the biochemical properties of nanoparticles, such as size, zeta potential, and chitosan/siRNA stability. The high MW chitosan prefers entanglement with siRNA compared to low MW chitosan. Thus, the high MW of chitosan increases the encapsulating capacity for negatively charged siRNA. The degree of deacetylation is related to the positive charge density when the nitrogen groups of chitosan are ionized under acidic conditions. Highly deacetylated chitosan has a higher positive charge and better siRNA binding capacity. Regarding the effect of the N/P ratio, which consists of the amino group charge of chitosan (N) and the phosphate group charge of siRNA (P), it determines the particle size, zeta potential, and gene silencing efficiency.
Intra- and intermolecular hydrogen bonding cause chitosan to form a rigid spatial structure. Therapeutic RNA molecules can be entrapped in chitosan-based matrices. Xiao et al41 formulated CD98 siRNA into chitosan polymeric nanoparticles and conjugated it with single-chain CD98 antibodies on its surface (Figure 2B). The chitosan was obtained from further deacetylation, and the final degree of deacetylation was 95.6%. The MW was 11.4 kDa. For the purpose of siRNA escape from endosomes/lysosomes, the hydroxyl of urocanic acid (UAC) then reacted with primary amine groups on the chitosan. To circumvent excessive viscoelasticity and adhesivity of nanoparticles trapped in mucus via the gastrointestinal tract and that could not reach the target site, UAC-Chitosan nanoparticles were coated with high-density bifunctional PEG derivatives (NHS-PEG-MAL). PEG modification decreased the mucoadhesion of the nanoparticles by enhancing the hydrophilicity and neutral charge on surfaces and facilitates “slipping” of nanoparticles through mucus.42 UAC–Chitosan–PEG nanoparticles were subsequently equipped with single-chain CD98 (scCD98) antibodies through covalently linking to maleimide on the nanoparticle surface at a molecular ratio of 1:2. The overexpression of CD98 on the surfaces of colonic epithelia cells and macrophages exerts a positive effect on the pathological process of IBD. The CD98 antibody-modified nanoparticles selectively targeted the cells in disease sites. ScCD98–PEG–Chitosan–UAC nanoparticles condensed CD98 siRNA with PEI by the complex coacervation technique. PEI is widely used to deliver nucleic acids not only because of their positively charged interaction with negatively charged siRNA but also as they are a proton buffering constituent. PEI induces the “proton sponge” effect to disrupt endosomal or lysosomal membranes and release siRNA into the cytoplasm. The results of fluorescence labeling indicated that scCD98–fluorescein isothiocyanate (FITC)–siRNA-loaded nanoparticles were taken up by Colon-26 and RAW 264.7 cells after 6 hours, and most nanoparticles were localized at the perinuclear region rather than the nucleus. The silencing efficiency of CD98 mRNA was tested in Colon-26 cells after treatment with lipopolysaccharide (LPS). The CD98 expression was decreased to 27% in the scCD98-functionalized siRNA nanoparticle group (ScCD98–PEG–UAC/PEI/siRNA weight ratio of 40:20:1). In particular, attenuated expression of proinflammatory cytokines mRNA such as TNF-α, IL-6, and IL-12 were also observed in response to CD98 silencing. The hydrogel containing scCD98-functionalized siRNA nanoparticles was orally administered in murine models of acute and chronic colitis. The nanoparticle therapeutic was obvious, which reduced the colitis index of weight loss, myeloperoxidase (MPO), and proinflammatory cytokines. The expression of CD98 mRNA was decreased 47.7% in the scCD98-functionalized siRNA group compared to the DSS control group. Pathological section of hematoxylin/eosin (H&E)-stained colonic sections showed that the treatment group had considerably less inflammation.41
Chitosan is not soluble in the physiological environment.43 This property is primarily because the D-glucosamine residue in chitosan has a pKa value of 6.5. Thus, chitosan–siRNA nanoparticle formulations are limited in acidic pH values to solubilize and ionize polymers. Furthermore, the chitosan–siRNA nanoparticles retain stability at physiological pH due to hydrophobic interactions.44 Nevertheless, the amino groups on chitosan are partially protonized at physiological pH, which provides an unstable combination between chitosan and siRNA. These properties limit the biomedical applications for chitosan. To overcome these problems, trimethyl chitosan (TC) was developed. Trimethylation modification refers to the quaternization of NH2 on the chitosan backbone, which increases the solubility of chitosan in a wider pH range and improves the transfection efficiency of the siRNA polyplex. Dehousse et al45 developed TC–siRNA nanoparticles using a simple complexation technique and compared their complexation efficiency and stability with those of chitosan nanoparticles, both assisted by sodium tripolyphosphate (TPP). It was found that TC nanoparticles have a highly spherical shape at pH 7.4 and no pH dependency on siRNA complexation. Furthermore, the inhibition rates of TC–siRNA nanoparticles were 1–2-fold higher than those of chitosan polyplexes.45
Mitogen-activated protein kinase kinase kinase kinase 4 (Map4k4) siRNA-loaded galactosylated-trimethyl chitosan–cysteine (GTC) nanoparticles were developed based on ionic gelation with TPP and hyaluronic acid (HA) (Figure 2C).46 Cationic GTC spontaneously conjugated with the anionic cross-linker TPP without sonication to protect the siRNA. Furthermore, cysteine-modified was used to enhance the bioadhesion capacity of nanoparticles through thiol-bearing compounds covalently bonding with mucin glycoproteins.47 The characterization of GTC/TPP (weight ratio of 10:1) revealed a particle size and zeta potential of 147.6±5.5 nm and 21.4±2.4 mV, respectively, whereas GTC/HA (weight ratio of 9:1) presented a particle size and zeta potential of 153.3±2.0 nm and 41.6±1.3 mV, respectively. Thus, GTC/HA shows a larger particle size and zeta potential than GTC/TPP. Furthermore, under high ionic strength conditions (0.2 M), the size of GTC/HA increases fivefold, and the zeta potential decreases by 26.1 mV. However, the particle size and zeta potential of GTC/TPP remained stable at 144.0±13.6 nm and 20.9±1.6 mV, respectively. Moreover, compared to GTC/HA, GTC/TPP also showed a more stable form in the pH range of 1.2–7.4, which simulates the gastrointestinal tract that the nanoparticles encountered.46 TPP is approved by the US Food and Drug Administration (FDA) as an excipient in food manufacturing due to its non-toxicity.48 In addition, TPP is widely used in forming ionic cross-linked chitosan nanoparticles because of its multi-valent anionic characteristic.49 The anionic TPP and cationic amino groups of chitosan interact with each other to form a gel through electrostatic interactions.50 The siRNA-loaded GTC nanoparticles tend to be taken up by macrophages due to the highly expressed macrophage–galactose-type lectin on the membrane of macrophages during inflammation.51 The microspectrofluorometry method was applied to investigate the cellular uptake of nanoparticles. The results suggested that the uptake amounts of carboxyfluorescein (FAM)–siRNA GTC/TPP were increased 5.8- and 1.6-fold compared to those of naked FAM–siRNA and TC/TPP, respectively. Similarly, in DSS-induced mouse colonic tissues, the inhibition ratio of Map4k4–siRNA GTC/TPP was 77.6%, which was also greater than the value of 61.1% for TC/TPP. Higher MPO activities were detected in UC tissues, and MPO is an inflammatory infiltration maker of UC pathogenesis because it is related to the activation of neutrophil granulocytes. In a pharmacodynamics assay, colitis in mice was induced by DSS, and the MPO activities were decreased 0.5–1.0-fold in the colon following the oral administration of Map4k4 siRNA-loaded GTC/TPP nanoparticles compared with the colitis control group. In the DSS-induced UC model, the mice exhibited sustained reduction of colon length and weight. However, in the Map4k4 siRNA GTC/TPP nanoparticle treatment group, colon reduction was inhibited, and the body weight curve exhibited less fluctuation compared with the colitis control group. UC histological remission of less epithelial cell damage and minimal mononuclear cell infiltration were also observed in H&E-stained colitis tissue from mice.46 The oral administration of siRNA nanoparticles is the most patient-compliant route and is cost efficient compared with other modes of administration (invasive injection or rectal administration) in IBD treatment. Nevertheless, low transfection limits its clinical application when siRNA transports in the gastrointestinal tract. It faces some challenges, such as siRNA degrades in intestinal physiological fluids and the epithelial barrier. To achieve successful oral delivery, mannose (Man)-modified trimethyl chitosan–cysteine TNF-α siRNA nanoparticles (MTC) were prepared by the siRNA entrapment method (Figure 2D).52 Man receptors express on the surfaces of macrophages, enterocytes, and M-cells, which highly combine with agents that contain terminal Man residues.53 In an ex vivo study, FAM–siRNA was demonstrated to show intestinal absorption by a fluorescence technique. FAM–siRNA can cross systems containing Peyer’s patches (FAE) and not containing Peyer’s patches (non-FAE). FAE are an important part of the intestinal mucosal immune system. The surface of FAE is covered by a layer of microfold cells, known as M-cells. M-cells can recognize many antigens in the gastrointestinal tract and present antigens to macrophages. The result shows that MTC enhanced the apparent permeability coefficients (Papp) of FAM–siRNA. Higher Papp was also observed in M-cell-enriched FAE compared to M-cell-free non-FAE, showing that M-cells are rapid and efficient absorption channels of MTC. The permeation of FAE was improved by Man moieties on the nanoparticles. Further gene silencing efficiency studies were conducted in rats with LPS-induced hepatic injury.52 To investigate the ideal Man ligand ratio, the MTC were prepared with different Man densities of 4%, 13%, and 21%. The results suggest that MTC with a Man density of 4% have maximum RNAi and therapeutic efficiency on the UC model.54
The aforementioned studies provide inspiration for designing target nanoparticles as IBD therapeutics. Modification of the chitosan backbone is a direction to optimize chitosan-based siRNA nanoparticles to obtain the powerful modified performance of chitosan with abundant hydroxyl and amino groups. Functional groups of antibody, galactose, Man, and trimethyl in the structure of nanoparticles enhance the intestinal absorption and cellular uptake, which improve the efficiency of gene silencing.
Other polysaccharides
Cyclodextrins are biocompatible cyclic polysaccharides composed of D-glucopyranose linked by α-(1,4) glycosidic bonds. Cyclodextrins have an amphiphilic structure, which is composed of a hydrophilic exterior and a hydrophobic cavity. Therefore, cyclodextrins have the ability to form inclusion complexes in which poorly water-soluble molecules (guests) are encapsulated in the interior of cyclodextrins (hosts). This characteristic of cyclodextrins increases the siRNA stability by resisting endonucleases.55 Furthermore, cyclodextrins reduce the immune stimulation of nucleic acid by avoiding non-specific interactions in physiological environments. It has been reported that cyclodextrins improve the permeability of oligonucleotides across the biological membranes.56
McCarthy et al57 developed amphiphilic cationic cyclodextrin vectors for TNF-α siRNA delivery and investigated the RNAi efficiency in vitro and in vivo. Amphiphilic cationic cyclodextrin was synthesized by modifying β-cyclodextrin. Bromo-cyclodextrin was reacted with tert-butyl 3-azidopropylcarbamate by cuprous-catalyzed azide–alkyne cycloaddition (“click” reaction). The propyl chain with a terminal Boc-protected primary amine was added to the secondary face. Subsequently, cyclodextrin was thioalkylated with dodecane thiol on the primary face at the 6-position. Finally, deprotection of the Boc-protected amine group afforded the resulting polymer (Figure 3A).58 The size of the modified cyclodextrin–siRNA complex (cyclodextrin/siRNA mass ratio of 10) was 240 nm, and the zeta potential was 42 mV.57 Modified cyclodextrin–siRNA complexes have ideal stability in the presence of simulated colonic fluids and α-amylases, which were secreted from the common organism Aspergillus oryzae in the intestinal tract of a patient with IBD. The expression of TNF-α mRNA decreased 21-fold after treatment with modified cyclodextrin–siRNA complexes (cyclodextrin/siRNA mass ratio of 20) in LPS-induced RAW 264.7 cells. Correspondingly, the concentration of TNF-α protein was decreased to 4.3±0.4 ng/mL compared with positive control of 18.3±0.9 pg/mL. In vivo, the modified cyclodextrin–siRNA complexes significantly decreased the proximal colon weight by 12.5 mg/cm compared with DSS controls. RT-PCR analysis was applied for investigating the silencing efficiency of modified cyclodextrin–siRNA complexes in a DSS-induced murine model. The results showed that the expressions of TNF-α and IL-6 were silenced by 73%±13% and 58%±19%, respectively, in proximal colon tissue. Proximal colon tissue showed a more significant silencing effect than the distal colon. In addition, the in vivo pharmacodynamics experiments performed using siRNA and linear-PEI (L-PEI) complexes of in vivo-jetPEI suggested that the charge of L-PEI was insufficient for forming nanoparticles with a high therapeutic effect compared with branched-PEI (B-PEI) at a low N/P ratio.57 The branched structure of primary, secondary, and tertiary amines in B-PEI provides a higher positive charge, which translates to a more appropriate electrostatic attraction with siRNA and increases transfection efficiency. Therefore, the charge of PEI is an important factor related to the nanoparticle performance in gene silencing.
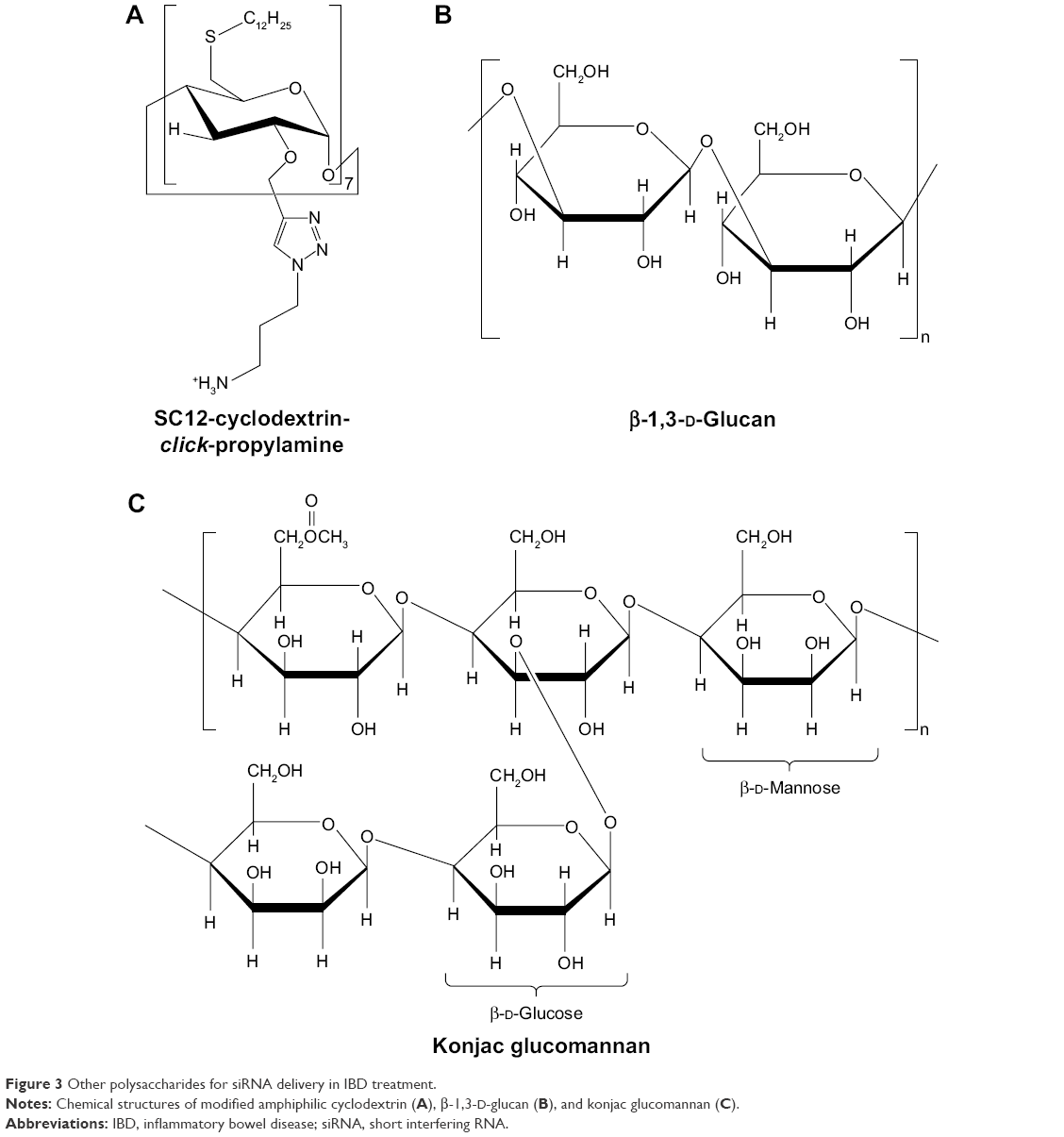 | Figure 3 Other polysaccharides for siRNA delivery in IBD treatment. |
β-Glucan is another type of polysaccharide composed of repeating D-glucose units linked by β-glycosidic bonds (Figure 3B). Aouadi et al59 developed the first oral siRNA delivery systems of micrometer-sized β-1,3-D-glucan-encapsulated Map4k4 siRNA particles (GeRPs) of a multi-layered formulation. β-1,3-D-Glucans were purified from baker’s yeast through solvent extraction. Negatively charged siRNA interacted with positively charged PEI and absorbed onto the shell of GeRPs. The GeRPs released the loaded Map4k4 siRNA, which was triggered by the acidic pH in phagosomes. The Map4k4 siRNA resulted in a 70%–80% knockdown of Map4k4 mRNA and 50% reductions of TNF-α in peritoneal exudate cells isolated from C57BL6/J mice. In vivo, oral administration of Map4k4 siRNA GeRPs increased the survival rates of mice and attenuated inflammatory injury by inhibiting TNF-α and IL-1β production in gut-associated lymphatic tissue.59
Another strategy of nucleic acid delivery through oral administration consists of utilizing phytagel and cKGM (Figure 3C).60 The swelling properties of cKGM are stronger than those of phytagel. The cKGM swells unrestrained by absorbing water until the microspheres burst and releases the contained oligonucleotide, whereas the phytagel offers sufficient strength to stabilize the microspheres during expansion. By adjusting the ratio of cKGM and phytagel, cKGM-based nucleic acid delivery microspheres selectively collapse in the colon section. The cKGM has abundant Man and β-glucan moieties, which specifically interact with Man and β-glucan receptors on the surface of macrophages. The cationic characteristic of cKGM conjugates to anionic antisense oligonucleotides. The cKGM-based TNF-α antisense oligonucleotide microspheres effectively decreased the concentration of TNF-α in the colon lumen and protected mice from injury in DSS-induced colitis.60
PLA-based nanoparticles
PLA is a type of aliphatic thermoplastic polyester that is widely used in medical applications approved by the US FDA, such as surgical sutures, implants, tissue culture, and controlled-release systems.61 This is primarily because PLA has characteristics of biodegradability, non-toxicity, and biocompatibility in the human body.62
In general, PLA-based siRNA nanoparticles are synthesized via a double emulsion/solvent evaporation technique. The group of Laroui et al has performed numerous studies in this area.63–66 The negatively charged siRNA combines with the positively charged PEI to form the internal aqueous phase. The internal aqueous phase is mixed with PLA or PLA derivatives in an organic phase, typically dichloromethane, to generate a water/oil (W/O) emulsion. The step of forming an emulsion is necessary. The first W/O emulsion is dropped in a second aqueous phase that contains a stabilizer agent, such as polyvinyl alcohol (PVA), to generate a water/oil/water emulsion (W/O/W). Finally, the nanoparticles are obtained by evaporating the organic solvent and freeze drying.
Laroui et al63 have used this technique to produce TNF-α siRNA/PEI-loaded PLA nanoparticles covered with PVA. Their studies indicate that the nanoparticle size and zeta potential were associated with the PVA surface characteristics (Figure 4). Pure PVA of 100% hydrolysis is non-soluble in water and physiological milieu. However, following the introduction of acetate groups into PVA at levels of 11%–14%, PVA is soluble and retains partial hydrophobicity, thus becoming an emulsifier in the nanoparticle formulation. In physiological milieu, less-soluble PVA effectively decreases the hydrodynamic radius of the nanoparticle shell, which spontaneously reduces the diameter of nanoparticles. The hydrodynamic radius of particles is related to pH in physiological milieu. It was found that the size of TNF-α siRNA-loaded PLA nanoparticle at neutral pH was smaller than that at acidic pH. In addition, the pH was also implicated in zeta potential of nanoparticle by PVA absorbed on the nanoparticle surfaces. PVAs with 86%–89% hydrolysis were applied in the nanoparticle formulations, which indicate that the contents of hydroxyl groups are tenfold higher than acetate groups of PVA. At pH values of 6–7.2, the deprotonation of hydroxyl groups creates a more negative zeta potential on the nanoparticles. The negative zeta potential of nanoparticles is beneficial to binding to cationic sites on the cell membrane and inducing endocytosis.67 Furthermore, in a LPS-stimulated macrophage model, TNF-α siRNA/PEI-loaded PLA nanoparticles exhibited 2–4-fold greater TNF-α silencing effects compared to lipofectamine vectors and the positive control.63 To investigate the silencing efficiency in colonic tissue, nanoparticles were encapsulated into a hydrogel of alginate/chitosan at a weight ratio of 7:3 and administered to LPS-treated mice by gavage.63,68 TNF-α siRNA-loaded PLA nanoparticles specifically release from the hydrogel in the colonic lumen rather than in the stomach and small intestine. It was found that TNF-α reduction was more obvious in the colon than in the liver and blood after pre-treatment with hydrogel nanoparticles, which indicates a desirable colon-specific delivery effect and therapy.63
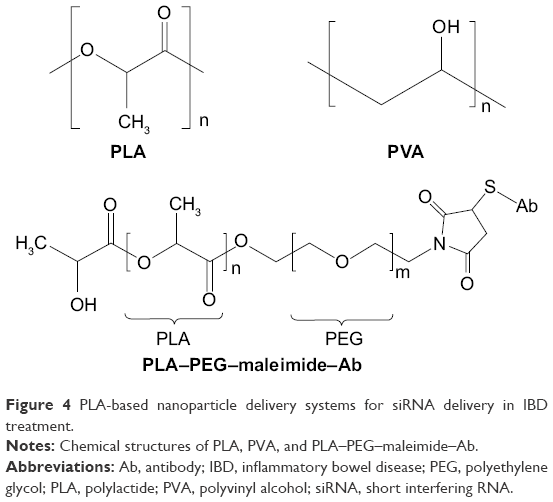 | Figure 4 PLA-based nanoparticle delivery systems for siRNA delivery in IBD treatment. |
A similar synthetic method for PLA nanoparticles was introduced by forming nanoparticles encapsulated in recombinant human prohibitin ameliorating acute murine colitis.64 Prohibitin is a type of positive therapeutic protein for attenuating UC and CD. Expression of prohibitin decreases the oxidative level and TNF-α-stimulated nuclear factor-κB (NF-κB) activation in intestinal epithelial tissue.69 Ghaleb et al65 also formed Krüppel-like factor 4 (Klf4)-siRNA-loaded PLA nanoparticles and encapsulated them in a hydrogel of alginate and chitosan. Klf4 is a zinc finger transcription factor expressed on macrophages and is considered to be proinflammatory mediator through the NF-κB signaling pathway. The expression of Klf4 was reduced more by DSS plus Klf4-siRNA-loaded PLA nanoparticles than by Klf4-siRNA-loaded PLA nanoparticles only in murine colonic epithelium, which indicated that DSS may enhance permeation efficiency in the process of nanoparticles crossing the colonic epithelium to some extent.65
To optimize the targeting performance to macrophages, F4/80 antibodies were grafted onto the surface of nanoparticles.66 The backbone of the nanoparticles was PLA. PEG-maleimide (Mal) was conferred to PLA (Figure 4). Regarding the nanoparticle structure, it was proposed that hydrophilic PEG–Mal was oriented to the second aqueous phase but that hydrophobic PLA was adjusted toward the organic phase; therefore, the nanoparticles were spontaneously decorated with PEG–Mal ligands on their surface. The nanoparticles were prepared using the double emulsion/solvent evaporation method, as described earlier. The inner aqueous phase contains cationic PEI and anionic siRNA, with an N/P ratio of 30 (N as the ammonium charge of PEI). Bovine serum albumin was then added to the internal aqueous phase and dichloromethane containing PLA–PEG–Mal to generate the first emulsion. Note that sodium cholate was chosen as the surfactant in the W/O/W emulsion preparation.66 Compared to PVA, sodium cholate was a poor stabilizer in PLA microspheres preparation, but it could be completely removed during the cleaning procedure.70 To obtain the Fab’ portion of F4/80 antibodies, they were initially digested by pepsin and treated with 2-mercaptoethanol to remove the Fc portion and cleave Fab disulfide bonds. The Fab’ portion of F4/80 antibodies subsequently interacted with the Mal moiety of nanoparticles (Fab’/Mal ratio of 1:4) via covalent thioether bonding. The size of the Fab’-bearing PLA–PEG nanoparticles was 376±19 nm as measured by dynamic light scattering. Furthermore, under scanning electron microscopy, Fab’-bearing PLA–PEG nanoparticles showed a more homogeneous distribution and less unwanted aggregation than PLA–PEG–Mal nanoparticles. This result may be attributed to the hydrophilicity of the Fab’-binding, which stabilizes the colloidal dispersion. In addition, surface plasmon resonance (SPR) experiments were conducted to confirm Fab’-binding and its integrity on the nanoparticles.66 Briefly, the Fab’-bearing PLA–PEG nanoparticles were “rolled” on a layer of carboxydextran in the SPR instrument. The amine groups of Fab’ interacted with the carboxyl group of carboxydextran, and a resonance angle shift was detected by the SPR instrument. However, the lack of amine groups had no response of a resonance angle shift. It was found that a resonance angle shift of 91 mDeg was generated by the Fab’-bearing PLA–PEG nanoparticles. Furthermore, to confirm the structural integrity of Fab’, the Fab’-bearing PLA–PEG nanoparticles were first attached on carboxydextran in the SPR instrument. Subsequently, RAW 264.7 (widely express F4/80 antigens) and Caco2 BBE cells (do not express F4/80 antigens) were “rolled” onto the nanoparticle surface. It was found that the Fab’-bearing PLA–PEG nanoparticles interacted more with the RAW 264.7 cells than with the Caco2 BBE cells, with shifts of 46.34 vs 1.23 mDeg, respectively. The results indicate that Fab’ retains its three-dimensional conformation and specifically binds to RAW 264.7 cells. In addition, the endocytosis of Fab’-bearing PLA–PEG nanoparticles was measured by fluorescent microscopy in RAW 264.7 cells. The FITC-siRNA was encapsulated in both Fab’-bearing nanoparticles and uncoated nanoparticles. The fluorescence intensity of the Fab’-bearing nanoparticles was 51,325 AU/μm2, and that of the uncoated nanoparticles was 10,279 AU/μm2. The results showed that the presence of Fab’ increased the phagocytosis of the nanoparticles. Furthermore, the Fab’-bearing PLA–PEG TNF-α siRNA nanoparticles attenuated colitis parameters such as weight loss, MPO, and Iκβα protein more efficiently than nanoparticles without Fab’ in vivo in a colitis mouse model. Similar results were observed in mice with colitis that were treated by alginate and chitosan hydrogel-encapsulated Fab’-bearing nanoparticles.66
CaP/PLGA-based nanoparticles
CaP is a component of biological hard tissues in bone, teeth, and tendons. Therefore, CaP has been employed as an ideal inorganic nanoparticle vector for nucleic acids or proteins based on its biodegradability and biocompatibility.71 CaP nanoparticles can permeate the cell membrane and release nucleic acid cargo into the cytosol by lysosomal escape. To provide reliable ribonuclease resistance and sustained release characteristics, nucleic acid-CaP has been further encapsulated into PLGA nanoparticles.
Frede et al72 developed siRNA-loaded CaP/PLGA nanoparticles against intestinal proinflammatory mediators for the local therapeutic treatment of IBD (Figure 5). A CaP dispersion was synthesized by mixing a solution of calcium-L-lactate and diammonium hydrogen phosphate. The siRNA was then mixed with the CaP dispersion, and siRNA–CaP nanoparticles were synthesized using a rapid precipitation method. The positive Ca+ was attracted to the negative phosphate groups of the nucleic acids by electrostatic attraction.73 In this step, siRNA was absorbed onto the surface of CaP and also acted as a stabilizing agent that inhibited crystal growth of CaP.74 Subsequently, siRNA–CaP was encapsulated into the PLGA matrix using a classic double emulsion solvent evaporation method. PLGA is a safe biodegradable material approved by the US FDA for use in various biomedical applications.75 The solution of siRNA–CaP was used as the internal aqueous phase, and the PLGA polymer was dissolved in the organic phase. PVA was chosen as a stabilizer added to the second aqueous phase in the formation of a W/O/W emulsion. It was reported that adding bovine serum albumin to the initial W/O emulsion inhibited aggregation of the emulsion and reduced nanoparticle diameter by acting as a surfactant in the PLGA nanoparticles formulation.74,76 In addition, to enhance the cell endocytosis of anionic siRNA–CaP–PLGA nanoparticles, the freeze-dried nanoparticles were finally resuspended in cationic B-PEI (25 kDa) solution at a weight ratio of 1:2 to perform a charge reversal to cationic nanoparticles. The final diameter of the siRNA-loaded CaP/PLGA nanoparticles was 151.52 nm, and the zeta potential was 22.08 mV. SiRNA-loaded CaP–PLGA nanoparticles do not activate endogenous immune to CD4+ T-cells or dendritic cells. Furthermore, siRNA-loaded CaP–PLGA nanoparticles showed a 30% reduced expression of IP-10 gene as well as 50% reduced expression of TNF-α or KC gene compared to controls in the murine epithelial cell line MODE-K. In addition, the siRNA-loaded CaP–PLGA nanoparticles still exhibited a silencing effect after incubation with murine colonic fluids or homogenate, which indicated that the CaP/PLGA nanoparticles protected the siRNA from degradation in the colon. The BALB/c mice were treated with DSS, and siRNA-loaded CaP–PLGA nanoparticles were administered intrarectally to examine their therapeutic effect. The results suggested that the expression of TNF-α mRNA decreased 40% and the expression of KC and IP-10 decreased 50% in the colon tissue compared to the controls after siRNA-loaded CaP–PLGA nanoparticle treatment.72
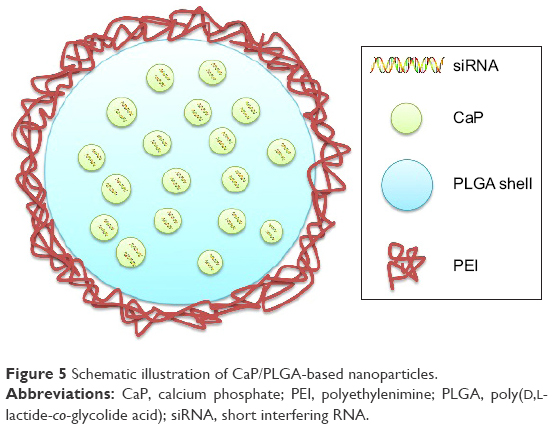 | Figure 5 Schematic illustration of CaP/PLGA-based nanoparticles. |
To locate the position of nanoparticles in mice, an in vivo imaging system was applied. BALB/c mice were intrarectally injected with AlexaFluor750-oligonucleotide CaP/PLGA nanoparticles (50 μg siRNA) after fasting overnight. The fluorescence was mainly concentrated in the distal part of the colon and mesenteric lymph node in mice. In addition, dendritic cells, macrophages, and T-cells were considered target cells of CaP/PLGA nanoparticles with promising cell uptake using a fluorescence-activated cell sorting technique. Similarly, enhanced nanoparticle uptake was observed in a colonic inflammatory model compared to healthy conditions.65,72 Ali et al77 administered identical doses of FITC-labeled budesonide–PLGA nanoparticles by gavage to mice under colonic inflamed and healthy conditions. The nanoparticle localization in colon tissue was determined using an in vivo imaging system. The fluorescence signals from nanoparticles were mainly concentrated in the inflamed area in the colonic inflamed model, whereas in the healthy model, nanoparticles were dispersedly distributed in the whole gut.77 This may be related to the high permeability of inflamed mucosa. These results suggest that nanoparticles tend to accumulate in the inflamed area in the colon, which favors an extended residence time and retards a rapid elimination of nanoparticles caused by diarrhea.
NiMOS
NiMOS is a type of solid-in-solid multi-compartmental system that is composed of a solid internal phase and further covered by a solid external phase (Figure 6). For example, siRNA was first encapsulated into type B gelatin nanoparticles and subsequently entrapped in poly-ε-caprolactone (PCL) microspheres utilizing the double emulsion solvent evaporation technique.78 Gelatin was used as a coating in pharmaceutical production. The type B gelatin was obtained from collagen via alkaline hydrolysis. It swelled in the aqueous phase and became a gel below 35°C. In addition, as synthetic polyester, PCL protects its payload from degradation during gastrointestinal transit. After the microspheres reached the colon section, PCL was degraded by lipase, and nanoparticles released into the colon lumen.79 For oral administration, the size of the microparticles should ideally be controlled to <5 μm.16 In general, there are three main factors related to the particle size of NiMOS: the amount of gelatin nanoparticles entrapped in NiMOS, the concentration of polymer in the organic phase, and the speed of homogenization during the double emulsion formation.80
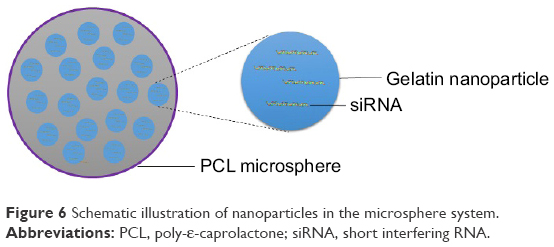 | Figure 6 Schematic illustration of nanoparticles in the microsphere system. |
Kriegel and Amiji81 fabricated TNF-α siRNA-containing NiMOS using type B gelatin nanoparticles with a size of 200 nm and subsequently entrapped them in PCL microspheres with a size of 2–4 μm. TNF-α siRNA was mixed with a gelatin solution, and the nanoparticles were obtained by ethanol precipitation and lyophilized. The nanoparticles were then homogenized with PCL in an organic phase of dichloromethane to form a stable emulsion system. Subsequently, the emulsion was homogenized with a PVA solution in the aqueous phase. NiMOS nanoparticles were obtained after evaporating dichloromethane in the emulsion, removing PVA by washing with distilled–deionized water, and lyophilization. The loading efficiency of siRNA in the gelatin nanoparticles was measured to be 90.2%±5.4%, and in NiMOS it was 55.2%±2.8%. The protective effects of NiMOS to siRNA were evaluated using agarose gel electrophoresis. The results suggest that NiMOS remains stable under gastric acid conditions and neutral conditions with ribonuclease. To evaluate the TNF-α gene silencing efficiency of NiMOS, TNF-α siRNA NiMOS, scramble siRNA NiMOS, and blank NiMOS were orally administered to a murine acute colitis model induced by DSS. An approximately threefold decrease of TNF-α protein was observed in the group receiving TNF-α siRNA NiMOS compared with the group that received blank NiMOS. In addition, the TNF-α siRNA NiMOS group showed the lowest TNF-α mRNA transcription compared with the other groups on day 14. However, the blank NiMOS and scramble siRNA NiMOS also exhibited a TNF-α silencing effect to some extent on day 14, which suggested that some off-target effects of NiMOS may exist. The data from ELISA indicated that TNF-α NiMOS reduced the expression of other proinflammatory cytokines, including IL-1β, interferon (IFN)-γ, and monocyte chemotactic protein-1. The therapeutic effects of TNF-α NiMOS were reflected in increased body weight and reduced levels of MPO in colitis-induced mice.81 These results demonstrated that NiMOS is a promising formulation for oral siRNA delivery in IBD treatment. NiMOS increased the loading capacity of siRNA due to the structural design of the delivery system. The multi-compartmental structures of the PCL shell and B gelatin nanoparticles also reserved the stability of the loaded siRNA in the harsh gastrointestinal environment and prolonged the exposure time to the targeted cells.
A further study of NiMOS encapsulated dual TNF-α/CyD1 siRNA.78 Combined siRNA NiMOS strongly reduced proinflammatory cytokines and relieved colonic injury of rectal bleeding, body weight loss, and colon shortening compared with TNF-α siRNA NiMOS alone. Dual TNF-α/CyD1siRNA has more reduction potency to proinflammatory cytokines than single TNF-α siRNA treatment, which is attributed to the strong CyD1 silencing effect. However, the silencing effects of dual siRNA were not as prominent as single CyD1 siRNA, which suggested that the silencing effects of dual siRNA may be diluted compared to the same overall amount of single siRNA. The histopathological assay showed that TNF-α/CyD1 NiMOS potently suppressed goblet cell destruction, ulcerations, and membrane thickening in intestinal inflammatory tissue. The MPO activities of the control group were measured to be 2–2.5 mU/mL per total protein content and were decreased to 1–1.5 mU/mL after the administration of TNF-α/CyD1 siRNA.78
Thioketal-based nanoparticles
In addition to genetic variations and immune disturbance, oxidative stress disorder is considered to be an important causative factor for IBD.82 Oxidative stress disorder in colonic tissue increases the secretion of reactive oxygen species (ROS). High levels of ROS in colonic inflammatory tissues were related to epithelial tissue injury of IBD.83
Wilson et al84 developed an exquisite delivery system of thioketal nanoparticles (TKNs) based on the characteristics of high ROS in intestinal inflammatory tissue. First, TNF-α siRNA was interacted with 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) to form siRNA–DOTAP complex particles. DOTAP is a type of cationic lipid and has preferable performance in enhancing siRNA transfection and endosomal escape.85 Subsequently, TKNs were obtained by adding these complex particles to a PPADT solution. PPADT was prepared via the acetal exchange reaction of 1,4-benzenedimethanethiol and 2,2-dimethoxypropane (Figure 7). PPADT has a special ROS-sensitive thioketal linkage and is stable in acid, base, and protease environments. The sizes of the TKNs were 600 nm to ensure that nanoparticles were localized in the inflammatory intestinal tissue and were taken up by macrophages. The cytotoxicity profile of TNF-α siRNA TKNs was similar to that of PLGA-based TNF-α siRNA nanoparticles.84 However, the nanoparticles formulated from PLGA show less silencing efficiency compared with TKNs at the same TNF-α siRNA dose (2.3 mg siRNA/kg). Moreover, the dose of 0.23 mg/kg, a tenfold lower dose, also showed silencing efficiency for TNF-α and other proinflammatory cytokines, such as IL-6, IL-1, and IFN-γ. The histopathological analysis results showed that TNF-α TKNs (2.3 mg siRNA/kg) protected intestinal inflammatory tissue from injury of epithelial cell damage and neutrophil invasion compared to the controls (Figure 8). In addition, decreased levels of MPO and increased weight were observed after treatment with TNF-α siRNA TKNs in DSS-induced mice. Thus, the clinical potential of oral TNF-α siRNA TKNs was demonstrated in colitis treatment.84
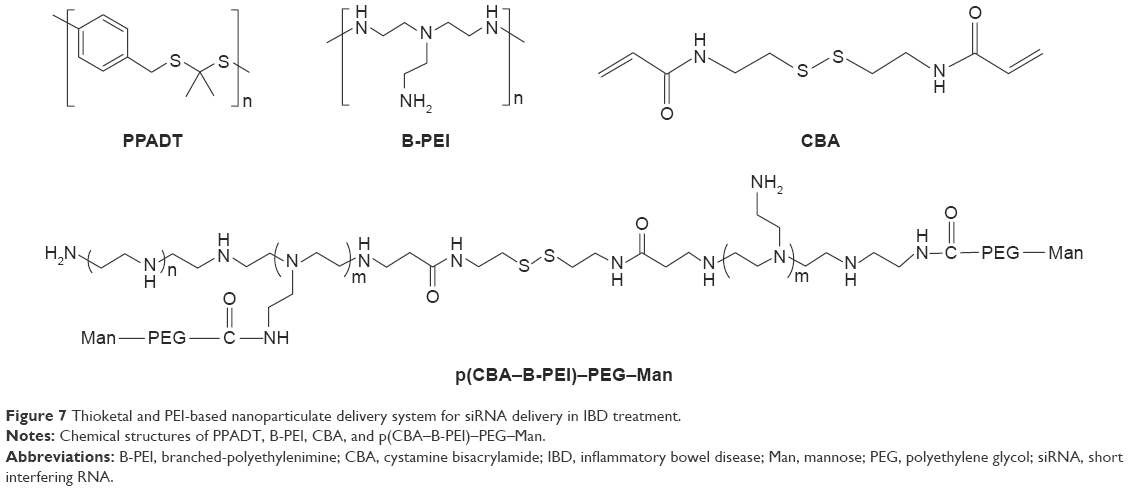 | Figure 7 Thioketal and PEI-based nanoparticulate delivery system for siRNA delivery in IBD treatment. |
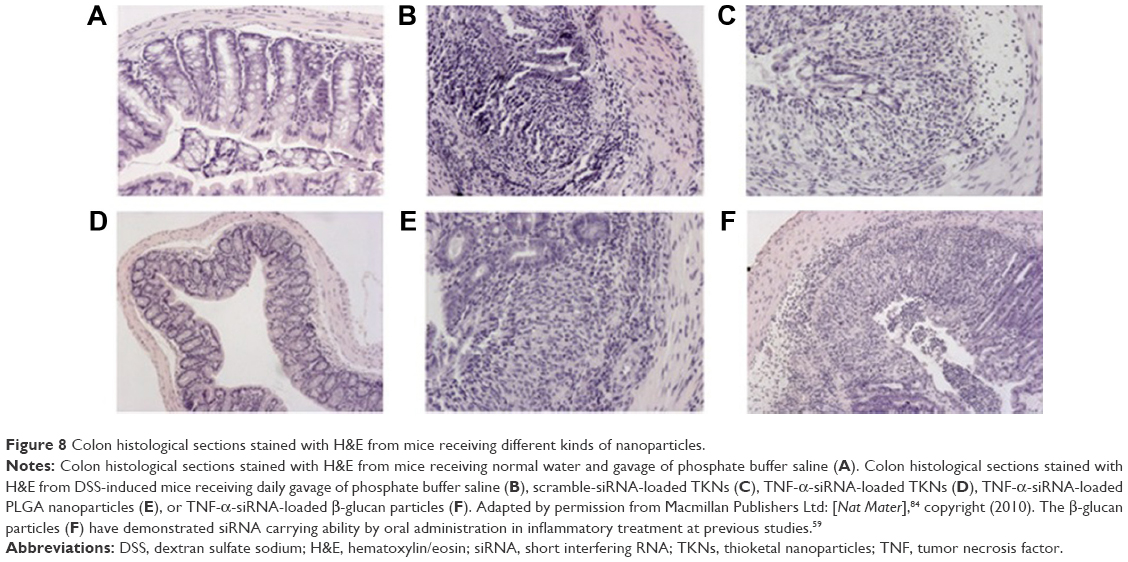 | Figure 8 Colon histological sections stained with H&E from mice receiving different kinds of nanoparticles. |
PEI-based nanoparticles
PEI is commonly used in non-viral siRNA transfection in vitro or in vivo because of the advantages of high RNA condensation and proton sponge effect to escape endosomes/lysosomes. However, the disadvantages of PEI such as non-degradability, high cytotoxicity, and aggregation in blood circulation limit its use in further clinical applications. The cytotoxicity of PEI is related to the MW, structure, and concentration.86 In the quest for a balance between cytotoxicity and high transfection efficiency, degradable PEI was developed, which consists of low MW PEI cross-linked with bioreducible molecules within disulfide linkages.87,88 The presence of disulfide bonds in the bioreducible polyplex could be reduced by intracellular glutathione, glutathione reductase, and other small molecules possessing thiol groups. Glutathione plays an antioxidant role in cells, and the intracellular concentration of glutathione was approximately threefold higher than in extracellular plasma.89 Therefore, as mentioned in the earlier section regarding the redox gradient in the intestinal lumen, the siRNA–PEI polyplex remains stable in the oxidizing extracellular space and releases siRNA in cytoplasm activated by the reduction of glutathione. Moreover, it was found that disulfide bonds increased the stability of the nanoparticles under high salt conditions.90
Xiao et al91 developed new oral nanoparticle systems, TNF-α siRNA-loaded Man-modified cationic polymeric nanoparticles (TPP–PPM/TNF-α siRNA NPs), for IBD treatment. The B-PEI (25 kDa) was connected to the end group of cystamine bisacrylamide (CBA) via a Michael addition reaction. CBA has a symmetrical acrylamide structure and contains bioreducible disulfide linkage. The amine groups of p(CBA–B-PEI) were then further reacted with HOOC–PEG–Man, and the final products were P(CBA-B-PEI)-PEG-Man (PPM) (Figure 7). The complex coacervation technique was applied to PPM package TNF-α siRNA assisted by TPP. The optimum PPM/TPP ratio was 4:1. As an anionic cross-linker, TPP has a significant effect on reducing the sizes of the nanoparticles. The sizes of TPP-free PPM/siRNA nanoparticles were 302–363 nm. However, the sizes of the TPP–PPM/siRNA nanoparticles were decreased to 211–275 nm. In addition, the quantification of nanoparticles taken up by RAW 264.7 macrophages was determined based on the mean fluorescence intensity of FITC–siRNA. The results showed that the mean fluorescence intensity of the TPP–PPM/FITC–siRNA NPs was 1–2-fold stronger than that of PPM/FITC–siRNA NPs, which indicates that TPP increases the cellular uptake of nanoparticles. Furthermore, Man was added to the culture medium of the RAW 264.7 macrophages as a competitor to TPP–PPM/FITC–siRNA NPs. It was found that the cellular uptake of TPP–PPM/FITC–siRNA NPs was decreased by adding Man, which indicated an uptake improvement caused by Man residues on the nanoparticles. In vitro, the silencing effect of TPP–PPM/TNF-α siRNA NPs is weight ratio dependent and has a maximum value of RNAi efficiency of 471.6 pg/mg protein at a TPP-PPM/siRNA weight ratio of 40:1. Furthermore, it was found that TPP–PPM/TNF-α siRNA NPs (siRNA concentration of 300 nM) reduced 61.0% of TNF-α secretion compared to the positive control in DSS-induced mouse colon tissue.91
Nanoparticle delivery systems of miRNA
MiRNA plays a critical role in the pathogenic mechanism of IBD, which includes the regulation of nuclear transcription factor, intestinal epithelial barrier function, and autophagic activity.92 Considerable attention has been devoted to miRNA as therapeutic targets or biomarkers in IBD treatment.93 At present, synthetic materials are used to condense miRNA, which mainly focus on the treatment of cancer, cardiovascular disease, and hepatitis, including lipid, PEI, PLGA, dendrimers, and so on.94,95 However, safe, effective, and stable delivery systems for miRNA are rarely reported in IBD treatment. Although commonly used as transfection reagents, liposomes are widely used in miRNA delivery in vitro.96 However, considerable unknowns still exist in miRNA nanoparticle delivery systems for IBD treatment. Currently, the design of miRNA delivery systems is normally based on the protocols of siRNA delivery systems.
PEI/miRNA complexes were prepared for miR-145 and miR-33a delivery in the miRNA replacement therapy of colon cancer.97 PEI is a positively charged polymer, and it binds with miRNA to form complexes through electrostatic interactions. Furthermore, miRNA was assisted by PEI escaping from endosomes through the proton sponge effect. MiR-145 was combined with PEI to validate the replacement therapy of PEI/miRNA complexes. This is mainly because the functions of miR-145 are analogous to tumor suppressors, which inhibit the growth and invasion of colon carcinoma cells. Decreased levels of miR-145 were detected in colon cancer models.98 The PEI/miRNA complexes increased the levels of intracellular miRNA in excess of tenfold. In addition, the cell proliferation was decreased over 60% in the group treated with PEI/miRNA complexes compared with the negative control. The jetPEI or B-PEI F25-LMW was synthesized with miRNA to investigate the delivery ability of PEI complexes. The jetPEI exhibited higher transfection efficacy than B-PEI F25-LMW in vitro.97 PEI/miR-145 complexes were intratumorally injected into athymic nude mice with a tumor xenograft. The tumor model was established by LS174T colon carcinoma cells. The results indicated clear systemic effects of tumor-suppressive function. The tumor growth decreased 50% compared with the negative controls. Subsequent studies have shown that the anti-tumor effects of PEI/miR-145 complexes were mediated by protein inhibition of ERK5 and c-Myc. To confirm the local therapeutic effects to tumors, PEI/miR-145 complexes were injected into mice with a colon cancer xenograft by the tumorigenic cell line HCT-116. Distinct tumor volume reductions of 40%–60% were observed in the PEI/miR-145 treatment group compared to the control groups.97
In addition, profound inhibition of colon cancer was similarly found in PEI/miR-33a complexes. MiR-33a shows potential suppressor activity through inhibition of the proto-oncogene Pim-1. The serine/threonine kinase Pim-1 was upregulated in colon cancer and related to early transformation and progression of tumors.99 This research first introduced PEI/miRNA complexes as miRNA delivery systems and demonstrated that PEI can be used as an efficient and biocompatible material to form miRNA delivery systems.
Atelocollagen is another promising miRNA delivery material. Atelocollagen has a positive charge and has the ability to combine with miRNA to form nanoparticles via electrostatic interactions. Atelocollagen was obtained by pepsin-treated type I collagen. Pepsin treatment decreased the immunogenicity of atelocollagen through removal of its telopeptide.100 As a nucleic acid condensation material, atelocollagen has low toxicity, nuclease resistance, and a prolonged released effect.101 Tazawa et al102 synthesized atelocollagen/miR-34a complexes to suppress colon cancer. HCT 116 and RKO cells were transplanted into nude mice to form a colon cancer model. Atelocollagen/miR-34a complexes were subcutaneously administered into mice with a colon cancer xenograft. A significant reduction in tumor volume was observed after treatment until day 6. At later time points, the suppressive effect was not desirable. The duration of the anti-tumor effect caused by atelocollagen/miR-34a complexes is ~1 week.102
Challenges of RNAi molecular delivery in IBD therapy
In general, the first preference for siRNA or miRNA delivery systems is viral vectors, such as retrovirus, lentivirus, adenovirus, adeno-associated virus, and baculovirus, due to their high transfection efficiency. However, the potential side effects of immunotoxicity, insertion of host gene, and quality problems in production have limited the applications of such systems. Non-viral nanoparticles have preferable patient compliance and are cost efficient compared with viral vectors in RNAi treatment. Thus, developing non-viral nanoparticles is an objective for scientists, and overcoming the hurdles of low transfection efficiency is the key to develop non-viral delivery systems (Table 3).
 | Table 3 Challenges of RNAi molecular delivery in IBD |
Transfection efficiency is influenced by numerous factors before siRNA or miRNA reaches the target site to elicit RNAi effects. For naked siRNA, the relatively high molecular mass (~13 kDa) and strong net negative charge (~40 negative phosphate charges) compared to small molecular drugs make siRNA difficult to be taken up by intestinal epithelial cells or macrophages. This is primarily because of the electrostatic repulsion between siRNA and the anionic cell membrane surface. However, the exorbitant positive surface charge of non-viral delivery systems may incorporate with negatively charged serum proteins to impede cellular uptake. Therefore, surface charge is an important parameter in the design of non-viral delivery systems. Another limitation of naked RNA delivery is its short half-life in vivo. This can be attributed to depurination in the low pH stomach environment and degradation by endogenous nucleases in the gut lumen or serum. The physiological conditions of patients with IBD need to be considered when designing oral RNAi molecule nanoparticles. The normal colonic pH ranges from 6.8 to 7.2. However, the colon luminal pH of patients with UC decreases to 2.3–5.5.103 The oral nanoparticles need to remain stable in the harsh gastrointestinal environment. Patients with IBD normally have diarrhea and abnormal motility of the gastrointestinal tract, which leads to accelerated transit times for drugs.104 Therefore, oral RNAi molecule nanoparticle delivery systems face the challenges of nanoparticle aggregation and physical docking/accumulation on the surface of the intestinal mucosa.105 Furthermore, in serum, RNAi molecule nanoparticles face the challenges of degradation by ribonuclease, uptake by the reticuloendothelial system, and aggregation with serum proteins.106 Moreover, it is difficult for molecules larger than 5 nm to pass through the capillary endothelium.107 Generally, delivery systems of sizes <200 μm perform the eEPR effect and have well targeted effects to immunoregulate cells of macrophages <10 μm.108 In many cases, non-viral nanoparticles of RNAi molecule are internalized into the cytoplasm by endocytosis or passive diffusion. Non-viral nanosystems via the passive diffusion process are limited to particle sizes <30 nm.109 According to the different sizes of nanoparticles, endocytosis of RNAi nanosystems mainly have clathrin-mediated endocytosis (nanoparticles: 200–500 nm) and caveolae-mediated endocytosis (nanoparticles ≤200 nm). Nanosystems via clathrin-mediated endocytosis face the challenge of gradually declining pH and hydrolytic nuclease in endosomes and lysosomes, which induce RNAi molecule degradation. In contrast, caveolae-mediated endocytosis is conducted in a neutral pH environment and without nuclease degradation by lysosomes.109
Once non-viral nanosystems enter the cytoplasm, they need to escape from endosomes/lysosomes and release their siRNA or miRNA cargo. There are several strategies that have been employed to escape endosomes/lysosomes in nanosystem delivery at the time of writing. The common strategy achieves endosomes/lysosomes escape by applying “proton sponge”, which are types of protonable molecules with abundant primary and tertiary amine groups, including PEI and imidazole groups. The “proton sponge” captures abundant protons in low pH environments in endosomes/lysosomes to cause chloride ions to flood into cells via passive transport, leading to osmotic swelling. An abnormal increase in osmotic pressure eventually leads to the rupture of the membranes of endosomes/lysosomes and the entry of nanosystems into the cytoplasm. Another strategy for endosome/lysosome escape is adding endosomolytic agents into delivery systems, such as 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine (DOPE) and chloroquine. According to reports, DOPE can incorporate with neutral liposomes to form lipid-based nanoparticles.110 In addition, dendrimers have no ability to transfer nucleic acid into cytoplasm; however, this situation will change by adding chloroquine to facilitate dendrimers escaping from endosomes. Chloroquine normally promotes the transfection efficiency of delivery systems with a low pH value in the vesicular stage.
Conclusion
Over the past decades, RNAi therapeutic studies have developed rapidly in the fields of cancer, immunological diseases, infectious diseases, and genetic diseases.111–113 In theory, any genetic target can be regulated by siRNA or miRNA technology. The sequence design and chemical modification of siRNA or miRNA are no longer the development bottlenecks of RNAi. Currently, the critical factors in RNAi treatment for IBD are the development of appropriate delivery systems for special targets and effectively regulating the target gene without side effects. In addition, in-depth studies of IBD pathology and new nanotechnology continuously provide new nanoparticle mechanisms for IBD therapy. For example, iron deficiency anemia, which is often associated with IBD, occurs due to both low intestinal intake of iron and blood loss.114 Furthermore, superparamagnetic iron oxide nanoparticles have been confirmed as a theranostic system in siRNA delivery.115 Iron oxide nanoparticles with siRNA may be an option in future IBD treatment. Although there are various complex problems associated with RNAi-based nanoparticles, they are regarded as a type of potent alternative to conventional IBD medicinal treatments. Herein, we analyzed the advantages and disadvantages of different targets and mechanisms of siRNA and miRNA nanoparticles in vivo or invitro, with hopes of promoting the innovation and creativity in nanoparticle formulations or preparations. To apply RNAi to clinical IBD therapy in the future, a substantial improvement of RNAi-based nanosystems is still necessary.
Abbreviations
Ab, antibody; B-PEI, branched-polyethylenimine; CaP, calcium phosphate; CBA, cystamine bisacrylamide; CD, Crohn’s disease; cKGM, cationic konjac glucomannan; CyD1, cyclin D1; DOPE, 1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine; DOTAP, 1,2-dioleoyl-3-trimethylammonium-propane; DPPE, dipalmitoyl phosphatidylethanolamine; DSS, dextran sulfate sodium; eEPR, epithelial enhanced permeability and retention; FAE, Peyer’s patches; FAM, carboxyfluorescein; FDA, Food and Drug Administration; FITC, fluorescein isothiocyanate; GeRPs, β-1,3-D-glucan-encapsulated siRNA particles; GTC, galactosylated-trimethyl chitosan; HA, hyaluronic acid; H&E, hematoxylin/eosin; IBD, inflammatory bowel disease; IFN, interferon; IL, interleukin; Klf4, Krüppel-like factor 4; L-PEI, linear-polyethylenimine; LPS, lipopolysaccharides; mAb, monoclonal antibody; Mal, maleimide; Man, mannose; Map4k4, mitogen-activated protein kinase kinase kinase kinase 4; MGL, macrophage galactose-type lectin; miRNA, micro RNA; MPO, myeloperoxidase; MPs, microspheres; MR, mannose receptor; MTC, mannose-modified trimethyl chitosan-cysteine TNF-α siRNA nanoparticles; MW, molecular weight; NF-κB, nuclear factor-κB; NiMOS, nanoparticles-in-microsphere oral system; NPs, nanoparticles; Papp, apparent permeability coefficients; PCL, poly-ε-caprolactone; PEG, polyethylene glycol; PEI, polyethylenimine; PLA, polylactide; PLGA, poly(D,L-lactide-co-glycolide acid); PVA, polyvinyl alcohol; RISC, RNA-induced silencing complex; RNAi, RNA interference; ROS, reactive oxygen species; ScCD98, single-chain CD98; siRNA, short interfering RNA; SPR, surface plasmon resonance; TC, trimethyl chitosan; TH, T-helper cell; TKNs, thioketal nanoparticles; TNF, tumor necrosis factor; TPP, tripolyphosphate; UAC, urocanic acid; UC, ulcerative colitis; W/O, water/oil; W/O/W, water/oil/water; β7 I-tsNPs, β7 antibody-equipped liposome-siRNA complexes.
Acknowledgments
The authors wish to express their gratitude to Dr Ping Ma for his useful discussions and constructive suggestions during the progression of the work. This work was supported by the National Natural Science Foundation of China (81274099).
Disclosure
The authors report no conflicts of interest in this work.
References
Baumgart DC, Sandborn WJ. Crohn’s disease. The Lancet. 2012;380(9853):1590–1605. | ||
Talley NJ, Abreu MT, Achkar JP, et al. An evidence-based systematic review on medical therapies for inflammatory bowel disease. Am J Gastroenterol. 2011;106(Suppl 1):S2–S25. | ||
Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015;12(12):720–727. | ||
Ng SC. Emerging leadership lecture: inflammatory bowel disease in Asia: emergence of a “Western” disease. J Gastroenterol Hepatol. 2015;30(3):440–445. | ||
Singh D, Srivastava S, Pradhan M, Kanwar JR, Singh MR. Inflammatory bowel disease: pathogenesis, causative factors, issues, drug treatment strategies, and delivery approaches. Crit Rev Ther Drug Carrier Syst. 2015;32(3):181–214. | ||
van der Have M, Mangen MJ, van der Valk ME, et al. Effect of aging on healthcare costs of inflammatory bowel disease: a glimpse into the future. Inflamm Bowel Dis. 2014;20(4):637–645. | ||
Kappelman MD, Rifas-Shiman SL, Porter CQ, et al. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology. 2008;135(6):1907–1913. | ||
Triantafillidis JK, Merikas E, Georgopoulos F. Current and emerging drugs for the treatment of inflammatory bowel disease. Drug Des Devel Ther. 2011;5:185–210. | ||
Olesen MT, Ballarin-Gonzalez B, Howard KA. The application of RNAi-based treatments for inflammatory bowel disease. Drug Deliv Transl Res. 2014;4(1):4–18. | ||
Magro F, Portela F. Management of inflammatory bowel disease with infliximab and other anti-tumor necrosis factor alpha therapies. BioDrugs. 2010;24(suppl 1):3–14. | ||
Zamore PD, Tuschl T, Sharp PA, Bartel DP. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell. 2000;101(1):25–33. | ||
Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123(4):631–640. | ||
Piatek MJ, Werner A. Endogenous siRNAs: regulators of internal affairs. Biochem Soc Trans. 2014;42(4):1174–1179. | ||
Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23(2):222–226. | ||
Behlke MA. Progress towards in vivo use of siRNAs. Mol Ther. 2006;13(4):644–670. | ||
Kriegel C, Attarwala H, Amiji M. Multi-compartmental oral delivery systems for nucleic acid therapy in the gastrointestinal tract. Adv Drug Deliv Rev. 2013;65(6):891–901. | ||
Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5(4):505–515. | ||
Akhtar S, Benter IF. Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest. 2007;117(12):3623–3632. | ||
Chiu YL, Rana TM. siRNA function in RNAi: a chemical modification analysis. RNA. 2003;9(9):1034–1048. | ||
Chapman CG, Pekow J. The emerging role of miRNAs in inflammatory bowel disease: a review. Therap Adv Gastroenterol. 2015;8(1):4–22. | ||
Lennox KA, Behlke MA. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011;18(12):1111–1120. | ||
Kim WJ, Kim SW. Efficient siRNA delivery with non-viral polymeric vehicles. Pharm Res. 2009;26(3):657–666. | ||
Jackson AL, Burchard J, Leake D, et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA. 2006;12(7):1197–1205. | ||
Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7(1):33–40. | ||
Beloqui A, Coco R, Alhouayek M, et al. Budesonide-loaded nanostructured lipid carriers reduce inflammation in murine DSS-induced colitis. Int J Pharm. 2013;454(2):775–783. | ||
Talaei F, Atyabi F, Azhdarzadeh M, Dinarvand R, Saadatzadeh A. Overcoming therapeutic obstacles in inflammatory bowel diseases: a comprehensive review on novel drug delivery strategies. Eur J Pharm Sci. 2013;49(4):712–722. | ||
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. | ||
Myers KJ, Murthy S, Flanigan A, et al. Antisense oligonucleotide blockade of tumor necrosis factor-alpha in two murine models of colitis. J Pharmacol Exp Ther. 2003;304(1):411–424. | ||
Zhang Y, Cristofaro P, Silbermann R, et al. Engineering mucosal RNA interference in vivo. Mol Ther. 2006;14(3):336–342. | ||
Peer D, Park EJ, Morishita Y, Carman CV, Shimaoka M. Systemic leukocyte-directed siRNA delivery revealing cyclin D1 as an anti-inflammatory target. Science. 2008;319(5863):627–630. | ||
Peer D, Florentin A, Margalit R. Hyaluronan is a key component in cryoprotection and formulation of targeted unilamellar liposomes. Biochim Biophys Acta. 2003;1612(1):76–82. | ||
Zheng X, Vladau C, Zhang X, et al. A novel in vivo siRNA delivery system specifically targeting dendritic cells and silencing CD40 genes for immunomodulation. Blood. 2009;113(12):2646–2654. | ||
Xue HY, Guo P, Wen WC, Wong HL. Lipid-based nanocarriers for RNA delivery. Curr Pharm Des. 2015;21(22):3140–3147. | ||
Hua S. Orally administered liposomal formulations for colon targeted drug delivery. Front Pharmacol. 2014;5:138. | ||
Mizrahy S, Peer D. Polysaccharides as building blocks for nanotherapeutics. Chem Soc Rev. 2012;41(7):2623–2640. | ||
Raemdonck K, Martens TF, Braeckmans K, Demeester J, De Smedt SC. Polysaccharide-based nucleic acid nanoformulations. Adv Drug Deliv Rev. 2013;65(9):1123–1147. | ||
Lee JW, Park JH, Robinson JR. Bioadhesive-based dosage forms: the next generation. J Pharm Sci. 2000;89(7):850–866. | ||
Chourasia MK, Jain SK. Polysaccharides for colon targeted drug delivery. Drug Deliv. 2004;11(2):129–148. | ||
van der Lubben IM, Verhoef JC, Borchard G, Junginger HE. Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur J Pharm Sci. 2001;14(3):201–207. | ||
Rudzinski WE, Aminabhavi TM. Chitosan as a carrier for targeted delivery of small interfering RNA. Int J Pharm. 2010;399(1–2):1–11. | ||
Xiao B, Laroui H, Viennois E, et al. Nanoparticles with surface antibody against CD98 and carrying CD98 small interfering RNA reduce colitis in mice. Gastroenterology. 2014;146(5):1289–1300.e1281–e1219. | ||
Wang YY, Lai SK, Suk JS, Pace A, Cone R, Hanes J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “slip” through the human mucus barrier. Angew Chem Int Ed Engl. 2008;47(50):9726–9729. | ||
Kean T, Roth S, Thanou M. Trimethylated chitosans as non-viral gene delivery vectors: cytotoxicity and transfection efficiency. J Control Release. 2005;103(3):643–653. | ||
Mao HQ, Roy K, Troung-Le VL, et al. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterization and transfection efficiency. J Control Release. 2001;70(3):399–421. | ||
Dehousse V, Garbacki N, Jaspart S, et al. Comparison of chitosan/siRNA and trimethylchitosan/siRNA complexes behaviour in vitro. Int J Biol Macromol. 2010;46(3):342–349. | ||
Zhang J, Tang C, Yin C. Galactosylated trimethyl chitosan-cysteine nanoparticles loaded with Map4k4 siRNA for targeting activated macrophages. Biomaterials. 2013;34(14):3667–3677. | ||
Wang X, Zheng C, Wu Z, et al. Chitosan-NAC nanoparticles as a vehicle for nasal absorption enhancement of insulin. J Biomed Mater Res B Appl Biomater. 2009;88(1):150–161. | ||
Palmeira-de-Oliveira R, Palmeira-de-Oliveira A, Gaspar C, et al. Sodium tripolyphosphate: an excipient with intrinsic in vitro anti-Candida activity. Int J Pharm. 2011;421(1):130–134. | ||
Katas H, Alpar HO. Development and characterisation of chitosan nanoparticles for siRNA delivery. J Control Release. 2006;115(2):216–225. | ||
Sun Y, Wan A. Preparation of nanoparticles composed of chitosan and its derivatives as delivery systems for macromolecules. J Appl Polymer Sci. 2007;105(2):552–561. | ||
van Vliet SJ, Saeland E, van Kooyk Y. Sweet preferences of MGL: carbohydrate specificity and function. Trends Immunol. 2008;29(2):83–90. | ||
He C, Yin L, Song Y, Tang C, Yin C. Optimization of multifunctional chitosan-siRNA nanoparticles for oral delivery applications, targeting TNF-alpha silencing in rats. Acta Biomater. 2015;17:98–106. | ||
He C, Yin L, Tang C, Yin C. Multifunctional polymeric nanoparticles for oral delivery of TNF-alpha siRNA to macrophages. Biomaterials. 2013;34(11):2843–2854. | ||
Chu S, Tang C, Yin C. Effects of mannose density on in vitro and in vivo cellular uptake and RNAi efficiency of polymeric nanoparticles. Biomaterials. 2015;52:229–239. | ||
Chaturvedi K, Ganguly K, Kulkarni AR, et al. Cyclodextrin-based siRNA delivery nanocarriers: a state-of-the-art review. Expert Opin Drug Deliv. 2011;8(11):1455–1468. | ||
Davis ME, Brewster ME. Cyclodextrin-based pharmaceutics: past, present and future. Nat Rev Drug Discov. 2004;3(12):1023–1035. | ||
McCarthy J, O’Neill MJ, Bourre L, et al. Gene silencing of TNF-alpha in a murine model of acute colitis using a modified cyclodextrin delivery system. J Control Release. 2013;168(1):28–34. | ||
O’Mahony AM, Ogier J, Desgranges S, Cryan JF, Darcy R, O’Driscoll CM. A click chemistry route to 2-functionalised PEGylated and cationic beta-cyclodextrins: co-formulation opportunities for siRNA delivery. Org Biomol Chem. 2012;10(25):4954–4960. | ||
Aouadi M, Tesz GJ, Nicoloro SM, et al. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458(7242):1180–1184. | ||
Huang Z, Gan J, Jia L, et al. An orally administrated nucleotide-delivery vehicle targeting colonic macrophages for the treatment of inflammatory bowel disease. Biomaterials. 2015;48:26–36. | ||
Singh S, Ray SS. Polylactide based nanostructured biomaterials and their applications. J Nanosci Nanotechnol. 2007;7(8):2596–2615. | ||
Kumari A, Yadav SK, Yadav SC. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf B Biointerfaces. 2010;75(1):1–18. | ||
Laroui H, Theiss AL, Yan Y, et al. Functional TNFalpha gene silencing mediated by polyethyleneimine/TNFalpha siRNA nanocomplexes in inflamed colon. Biomaterials. 2011;32(4):1218–1228. | ||
Theiss AL, Laroui H, Obertone TS, et al. Nanoparticle-based therapeutic delivery of prohibitin to the colonic epithelial cells ameliorates acute murine colitis. Inflamm Bowel Dis. 2011;17(5):1163–1176. | ||
Ghaleb AM, Laroui H, Merlin D, Yang VW. Genetic deletion of Klf4 in the mouse intestinal epithelium ameliorates dextran sodium sulfate-induced colitis by modulating the NF-kappaB pathway inflammatory response. Inflamm Bowel Dis. 2014;20(5):811–820. | ||
Laroui H, Viennois E, Xiao B, et al. Fab’-bearing siRNA TNFalpha-loaded nanoparticles targeted to colonic macrophages offer an effective therapy for experimental colitis. J Control Release. 2014;186:41–53. | ||
Liberman EA, Topaly VP, Tsofina LM, Jasaitis AA, Skulachev VP. Mechanism of coupling of oxidative phosphorylation and the membrane potential of mitochondria. Nature. 1969;222(5198):1076–1078. | ||
Laroui H, Dalmasso G, Nguyen HT, Yan Y, Sitaraman SV, Merlin D. Drug-loaded nanoparticles targeted to the colon with polysaccharide hydrogel reduce colitis in a mouse model. Gastroenterology. 2010;138(3):843–853.e841–e842. | ||
Theiss AL, Jenkins AK, Okoro NI, Klapproth JM, Merlin D, Sitaraman SV. Prohibitin inhibits tumor necrosis factor alpha-induced nuclear factor-kappa B nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol Biol Cell. 2009;20(20):4412–4423. | ||
Bouillot P, Ubrich N, Sommer F, Duc TM, Loeffler JP, Dellacherie E. Protein encapsulation in biodegradable amphiphilic microspheres. Int J Pharm. 1999;181(2):159–172. | ||
Uskokovic V, Uskokovic DP. Nanosized hydroxyapatite and other calcium phosphates: chemistry of formation and application as drug and gene delivery agents. J Biomed Mater Res B Appl Biomater. 2011;96(1):152–191. | ||
Frede A, Neuhaus B, Klopfleisch R, et al. Colonic gene silencing using siRNA-loaded calcium phosphate/PLGA nanoparticles ameliorates intestinal inflammation in vivo. J Control Release. 2016;222:86–96. | ||
Sokolova V, Epple M. Inorganic nanoparticles as carriers of nucleic acids into cells. Angew Chem Int Ed Engl. 2008;47(8):1382–1395. | ||
Dordelmann G, Kozlova D, Karczewski S, Lizio R, Knauer S, Epple M. Calcium phosphate increases the encapsulation efficiency of hydrophilic drugs (proteins, nucleic acids) into poly(d,l-lactide-co-glycolide acid) nanoparticles for intracellular delivery. J Mat Chem B. 2014;2(41):7250–7259. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Cun D, Foged C, Yang M, Frokjaer S, Nielsen HM. Preparation and characterization of poly(DL-lactide-co-glycolide) nanoparticles for siRNA delivery. Int J Pharm. 2010;390(1):70–75. | ||
Ali H, Weigmann B, Collnot EM, Khan SA, Windbergs M, Lehr CM. Budesonide loaded PLGA nanoparticles for targeting the inflamed intestinal mucosa – pharmaceutical characterization and fluorescence imaging. Pharm Res. 2016;33(5):1085–1092. | ||
Kriegel C, Amiji MM. Dual TNF-alpha/Cyclin D1 gene silencing with an oral polymeric microparticle system as a novel strategy for the treatment of inflammatory bowel disease. Clin Transl Gastroenterol. 2011;2:e2. | ||
Xiao B, Merlin D. Oral colon-specific therapeutic approaches toward treatment of inflammatory bowel disease. Expert Opin Drug Deliv. 2012;9(11):1393–1407. | ||
Bhavsar MD, Tiwari SB, Amiji MM. Formulation optimization for the nanoparticles-in-microsphere hybrid oral delivery system using factorial design. J Control Release. 2006;110(2):422–430. | ||
Kriegel C, Amiji M. Oral TNF-alpha gene silencing using a polymeric microsphere-based delivery system for the treatment of inflammatory bowel disease. J Control Release. 2011;150(1):77–86. | ||
Rezaie A, Parker RD, Abdollahi M. Oxidative stress and pathogenesis of inflammatory bowel disease: an epiphenomenon or the cause? Dig Dis Sci. 2007;52(9):2015–2021. | ||
Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003;201(1):28–36. | ||
Wilson DS, Dalmasso G, Wang L, Sitaraman SV, Merlin D, Murthy N. Orally delivered thioketal nanoparticles loaded with TNF-alpha-siRNA target inflammation and inhibit gene expression in the intestines. Nat Mater. 2010;9(11):923–928. | ||
Zhang S, Zhao B, Jiang H, Wang B, Ma B. Cationic lipids and polymers mediated vectors for delivery of siRNA. J Control Release. 2007;123(1):1–10. | ||
Islam MA, Park TE, Singh B, et al. Major degradable polycations as carriers for DNA and siRNA. J Control Release. 2014;193:74–89. | ||
Ou M, Xu R, Kim SH, Bull DA, Kim SW. A family of bioreducible poly(disulfide amine)s for gene delivery. Biomaterials. 2009;30(29):5804–5814. | ||
Breunig M, Lungwitz U, Liebl R, Goepferich A. Breaking up the correlation between efficacy and toxicity for nonviral gene delivery. Proc Natl Acad Sci U S A. 2007;104(36):14454–14459. | ||
Son S, Namgung R, Kim J, Singha K, Kim WJ. Bioreducible polymers for gene silencing and delivery. Acc Chem Res. 2012;45(7):1100–1112. | ||
Varkouhi AK, Verheul RJ, Schiffelers RM, Lammers T, Storm G, Hennink WE. Gene silencing activity of siRNA polyplexes based on thiolated N,N,N-trimethylated chitosan. Bioconjug Chem. 2010;21(12):2339–2346. | ||
Xiao B, Laroui H, Ayyadurai S, et al. Mannosylated bioreducible nanoparticle-mediated macrophage-specific TNF-alpha RNA interference for IBD therapy. Biomaterials. 2013;34(30):7471–7482. | ||
Coskun M, Bjerrum JT, Seidelin JB, Nielsen OH. MicroRNAs in inflammatory bowel disease – pathogenesis, diagnostics and therapeutics. World J Gastroenterol. 2012;18(34):4629–4634. | ||
Chen WX, Ren LH, Shi RH. Implication of miRNAs for inflammatory bowel disease treatment: systematic review. World J Gastrointest Pathophysiol. 2014;5(2):63–70. | ||
Zhang Y, Wang Z, Gemeinhart RA. Progress in microRNA delivery. J Control Release. 2013;172(3):962–974. | ||
van Rooij E, Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Mol Med. 2014;6(7):851–864. | ||
Pathak S, Grillo AR, Scarpa M, et al. MiR-155 modulates the inflammatory phenotype of intestinal myofibroblasts by targeting SOCS1 in ulcerative colitis. Exp Mol Med. 2015;47:e164. | ||
Ibrahim AF, Weirauch U, Thomas M, Grunweller A, Hartmann RK, Aigner A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011;71(15):5214–5224. | ||
Sachdeva M, Mo YY. miR-145-mediated suppression of cell growth, invasion and metastasis. Am J Transl Res. 2010;2(2):170–180. | ||
Thomas M, Lange-Grunweller K, Weirauch U, et al. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene. 2012;31(7):918–928. | ||
Minakuchi Y, Takeshita F, Kosaka N, et al. Atelocollagen-mediated synthetic small interfering RNA delivery for effective gene silencing in vitro and in vivo. Nucleic Acids Res. 2004;32(13):e109. | ||
Ochiya T, Nagahara S, Sano A, Itoh H, Terada M. Biomaterials for gene delivery: atelocollagen-mediated controlled release of molecular medicines. Curr Gene Ther. 2001;1(1):31–52. | ||
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A. 2007;104(39):15472–15477. | ||
Fallingborg J, Christensen LA, Jacobsen BA, Rasmussen SN. Very low intraluminal colonic pH in patients with active ulcerative colitis. Dig Dis Sci. 1993;38(11):1989–1993. | ||
Hua S, Marks E, Schneider JJ, Keely S. Advances in oral nano-delivery systems for colon targeted drug delivery in inflammatory bowel disease: selective targeting to diseased versus healthy tissue. Nanomedicine. 2015;11(5):1117–1132. | ||
Pouton CW, Seymour LW. Key issues in non-viral gene delivery. Adv Drug Deliv Rev. 2001;46(1–3):187–203. | ||
Dykxhoorn DM, Palliser D, Lieberman J. The silent treatment: siRNAs as small molecule drugs. Gene Ther. 2006;13(6):541–552. | ||
Rippe B, Rosengren BI, Carlsson O, Venturoli D. Transendothelial transport: the vesicle controversy. J Vasc Res. 2002;39(5):375–390. | ||
Lamprecht A. IBD: selective nanoparticle adhesion can enhance colitis therapy. Nat Rev Gastroenterol Hepatol. 2010;7(6):311–312. | ||
Daka A, Peer D. RNAi-based nanomedicines for targeted personalized therapy. Adv Drug Deliv Rev. 2012;64(13):1508–1521. | ||
Farhood H, Serbina N, Huang L. The role of dioleoyl phosphatidylethanolamine in cationic liposome mediated gene transfer. Biochim Biophys Acta. 1995;1235(2):289–295. | ||
Dyawanapelly S, Ghodke SB, Vishwanathan R, Dandekar P, Jain R. RNA interference-based therapeutics: molecular platforms for infectious diseases. J Biomed Nanotechnol. 2014;10(9):1998–2037. | ||
Xu H, Li Z, Si J. Nanocarriers in gene therapy: a review. J Biomed Nanotechnol. 2014;10(12):3483–3507. | ||
Seyhan AA. RNAi: a potential new class of therapeutic for human genetic disease. Hum Genet. 2011;130(5):583–605. | ||
Wells CW, Lewis S, Barton JR, Corbett S. Effects of changes in hemoglobin level on quality of life and cognitive function in inflammatory bowel disease patients. Inflamm Bowel Dis. 2006;12(2):123–130. | ||
Mahajan UM, Teller S, Sendler M, et al. Tumour-specific delivery of siRNA-coupled superparamagnetic iron oxide nanoparticles, targeted against PLK1, stops progression of pancreatic cancer. Gut. Epub 2016 May 12. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.