")
Back to Journals » Clinical Interventions in Aging » Volume 12
Rivastigmine: the advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia
Authors Kandiah N, Pai MC , Senanarong V, Looi I, Ampil E, Park KW , Karanam AK, Christopher S
Received 1 December 2016
Accepted for publication 21 February 2017
Published 18 April 2017 Volume 2017:12 Pages 697—707
DOI https://doi.org/10.2147/CIA.S129145
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
Nagaendran Kandiah,1,2 Ming-Chyi Pai,3,4 Vorapun Senanarong,5 Irene Looi,6,7 Encarnita Ampil,8 Kyung Won Park,9 Ananda Krishna Karanam,10 Stephen Christopher11
1Department of Neurology, National Neuroscience Institute, Tan Tock Seng Hospital, 2Duke-NUS, Graduate Medical School, Singapore; 3Division of Behavioral Neurology, Department of Neurology, 4Alzheimer’s Disease Research Center, National Cheng Kung University Hospital, College of Medicine, National Cheng Kung University, Tainan City, Taiwan; 5Division of Neurology, Faculty of Medicine, Siriraj Hospital, Mahidol University, Bangkok, Thailand; 6Clinical Research Centre, 7Department of Medicine, Hospital Seberang Jaya, Penang, Malaysia; 8Department of Neurology and Psychiatry, Faculty of Medicine and Surgery, University of Santo Tomas, Manila, Philippines; 9Department of Neurology and Cognitive Disorders and Dementia Center, Institute of Convergence Bio-Health, Dong-A University College of Medicine, Busan, Republic of Korea; 10Novartis Healthcare Private Limited, Hyderabad, India; 11Novartis (Singapore) Pte. Ltd., Singapore
Abstract: Several studies have demonstrated clinical benefits of sustained cholinesterase inhibition with rivastigmine in Alzheimer’s disease (AD) and Parkinson’s disease dementia (PDD). Unlike donepezil and galantamine that selectively inhibit acetylcholinesterase (AChE; EC 3.1.1.7), rivastigmine is a unique cholinesterase inhibitor with both AChE and butyrylcholinesterase (BuChE; EC 3.1.1.8) inhibitory activity. Rivastigmine is also available as transdermal patch that has been approved by the US Food and Drug Administration for the treatment of mild, moderate, and severe AD as well as mild-to-moderate PDD. In this review, we explore the role of BuChE inhibition in addition to AChE inhibition with rivastigmine in the outcomes of cognition, global function, behavioral symptoms, and activities of daily living. Additionally, we review the evidence supporting the use of dual AChE-BuChE inhibitory activity of rivastigmine as a therapeutic strategy in the treatment of neurological disorders, with a focus on the role of rivastigmine in subcortical dementias such as vascular dementia (VaD) and PDD. Toward this objective, we performed a literature search in PubMed and Ovid with limits to articles published in the English language before June 2016. The available evidence from the literature suggests that the dual inhibition of AChE and BuChE may afford additional therapeutic potential of rivastigmine in subcortical dementias (subcortical VaD and PDD) with benefits on cognition and behavioral symptoms. Rivastigmine was found to specifically benefit executive dysfunction frequently observed in subcortical dementias; however, large randomized clinical studies are warranted to support these observations.
Keywords: acetylcholinesterase, BuChE genotype, butyrylcholinesterase, Parkinson’s disease dementia, rivastigmine, subcortical vascular dementia
Introduction
Cholinesterase inhibitors were developed based on the cholinergic hypothesis of Alzheimer’s disease (AD), and in this condition, degeneration of cholinergic neurons in the brain leads to reduction in the levels of acetylcholine and cholinergic function, resulting in cognitive deficits.1–3 Cholinesterase inhibitors reduce degradation of synaptic acetylcholine, improve brain acetylcholine levels in a dose-dependent manner, and thereby enhance cholinergic transmission in patients with Alzheimer’s and other dementias.4,5 Donepezil and galantamine are cholinesterase inhibitors with acetylcholinesterase (AChE; EC 3.1.1.7)-inhibiting activity, whereas rivastigmine inhibits both AChE and butyrylcholinesterase (BuChE; EC 3.1.1.8).6 Evidence suggests that cholinesterase inhibitors (donepezil, rivastigmine, and galantamine) and memantine provide symptomatic pharmacological treatment, improving cognition and global function in patients with dementia.7,8 Currently, the cholinesterase inhibitors approved by the US Food and Drug Administration (FDA) for the treatment of mild-to-moderate AD include donepezil,9 rivastigmine,10,11 and galantamine.12 The US FDA also approved memantine, an N-methyl-D-aspartate receptor antagonist, for the treatment of moderate-to-severe AD.13
Rivastigmine is a pseudo-irreversible, carbamate-type, brain-selective, dual AChE-BuChE inhibitor (the structure and properties are shown in Table 1). The pharmacokinetic profile showed that compared with the oral formulation, rivastigmine transdermal patch provides smoother and continuous as well as controlled drug delivery over 24 h, thereby resulting in fewer side effects.14–16 The transdermal patch formulation of rivastigmine has been approved in the US (FDA) for the treatment of mild, moderate, and severe AD and mild-to-moderate Parkinson’s disease dementia (PDD),11 and in the European Union, it is approved for the treatment of mild-to-moderately severe AD.17 Both AD and PDD are associated with cortical cholinergic deficits18 and therefore form the rationale for the use of pharmacological symptomatic treatment. The findings from an in vivo positron emission tomography (PET) imaging study showed greater cortical AChE deficit in patients with PDD than in those with AD.18 Rivastigmine exerts symptomatic therapeutic effects through increasing acetylcholine levels in the brain, thereby making more acetylcholine available for synaptic transmission.10,11,19 This increase in brain acetylcholine levels is believed to be responsible for the clinical improvements in AD and PDD.
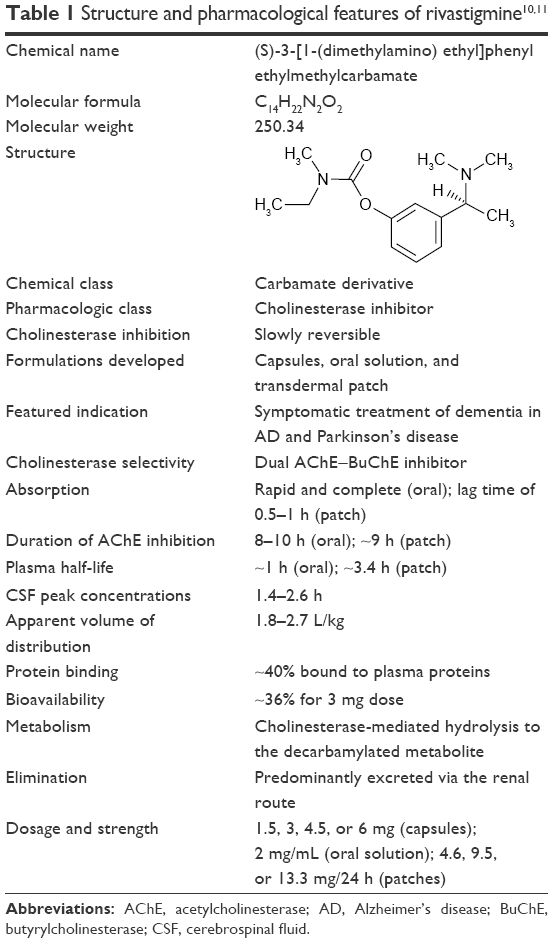 | Table 1 Structure and pharmacological features of rivastigmine10,11 |
In this review, we discuss the advantage of BuChE inhibition in addition to AChE inhibition with rivastigmine on the outcomes of cognition, global function, behavioral symptoms, and activities of daily living (ADL). We also focus on the role of rivastigmine in subcortical vascular dementia (VaD) and PDD. Based on this context, we performed a literature search of English language articles on rivastigmine published in PubMed and Ovid before June 2016. Studies identified during the search were assessed for relevance based on the titles, abstracts, and/or the full text of the retrieved articles.
Rivastigmine: dual inhibitor AChE/BuChE
AChE and BuChE are two different cholinesterase enzymes located in the brain that are responsible for acetylcholine hydrolysis.20,21 Of these, AChE is the primary cholinesterase mostly found at the nerve synaptic junctions and the areas that express intense activity in the adult human cerebral cortex,22 whereas BuChE is mainly located in the glial cells of the brain23 and plays an important role in cholinergic mediation.24 As AD progresses, BuChE activity increases in the hippocampus and temporal cortex in contrast to the AChE activity, which decreases in these specific regions of the brain, thereby supporting the key role of BuChE in regulating brain acetylcholine levels.25,26 Moreover, while BuChE activity increased with age in the AD brain, no correlation was observed in AChE activity with increasing age.26 Studies have also shown that BuChE inhibition with rivastigmine correlated with improved cognition in patients with AD.25,27,28 Initially, treatment efforts were focused on the inhibition of AChE; however, several studies have demonstrated the importance of both AChE and BuChE inhibition in the pathophysiology and pharmacological treatment of AD.21 Further to the evidence of BuChE involvement in cholinergic regulation20 and the suggested role of BuChE (K-variant allele) in the progression of AD,29 dual inhibition may offer additional benefits,30,31 especially over a long term.32
Rivastigmine has been shown to inhibit AChE in the brain in preclinical33 and clinical34 studies. Several large clinical studies35–37 and meta-analyses38–41 have demonstrated that rivastigmine improves cognitive function in AD. Studies in patients with AD have shown that rivastigmine exhibited dose-dependent efficacy,42,43 and further investigations suggested that a higher dose of rivastigmine patch 13.3 mg/24 h (15 cm2) conferred greater benefits on cognition, ADL, and global function than the 4.6 mg/24 h patch (5 cm2) in patients with severe AD.44,45 In the following sections, we provide evidence supporting the use of dual AChE-BuChE inhibitory activity of rivastigmine and summarize relevant findings in Table 2.
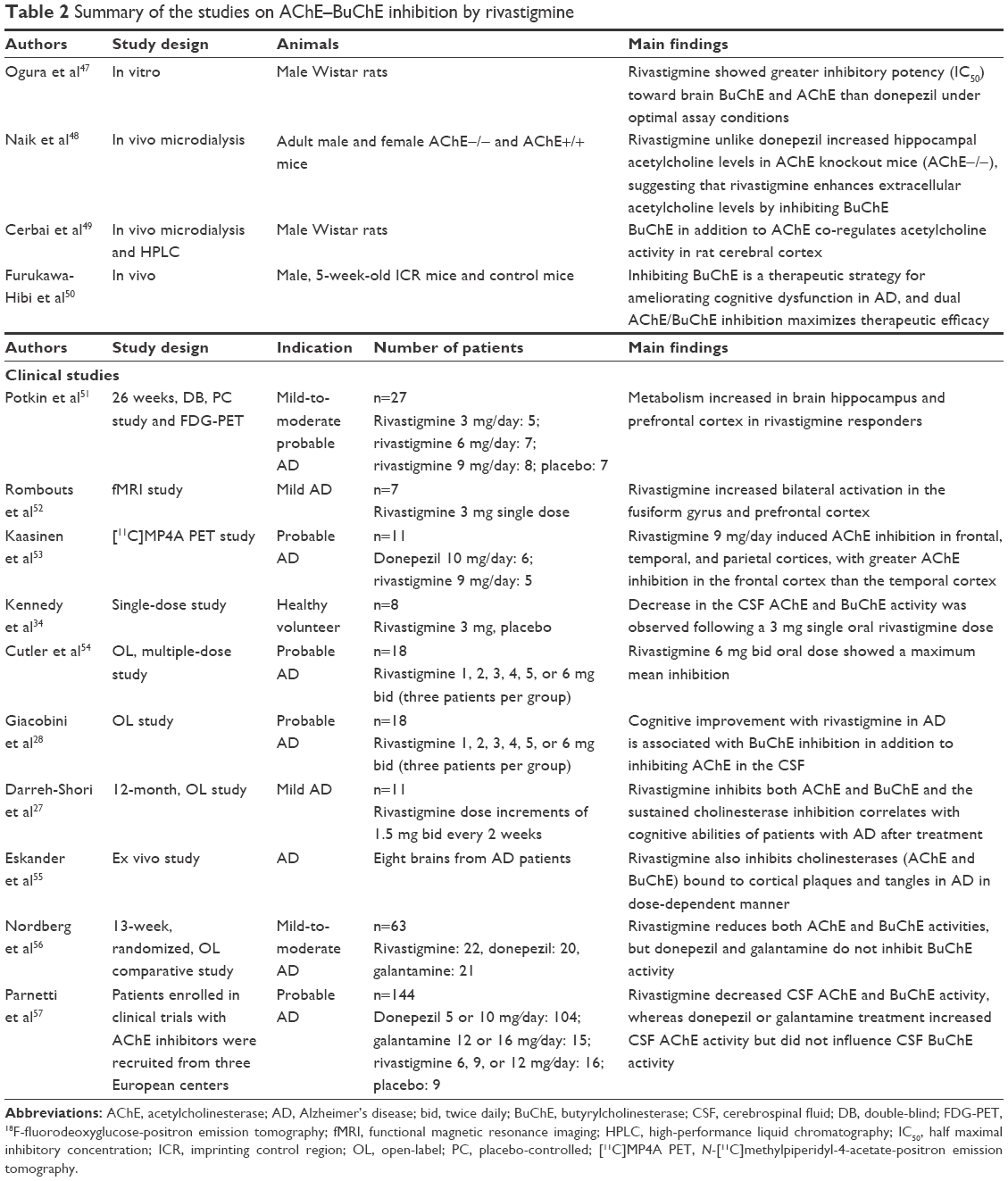 | Table 2 Summary of the studies on AChE–BuChE inhibition by rivastigmine |
Preclinical studies of rivastigmine
Several preclinical studies have been conducted to investigate the effect of AChE and BuChE inhibition on the hydrolysis of brain acetylcholine. Preclinical studies24,46 in AChE knockout mice have shown that BuChE was functional in the brain and had a role in the hydrolysis of acetylcholine, suggesting that inhibition of BuChE may enhance cholinergic transmission and is therefore desirable for cholinergic therapies. An in vitro study in male Wistar rats showed greater inhibitory potency (half maximal inhibitory concentration [IC50]) of rivastigmine toward brain BuChE and AChE compared with donepezil under optimal assay conditions (BuChE: 31 vs 7,400 nM; AChE: 4.3 vs 6.7 nM).47 The results from a preclinical study48 using AChE knockout mice showed that infusion of rivastigmine increased hippocampal acetylcholine levels but not donepezil, suggesting that rivastigmine enhances acetylcholine levels by inhibiting BuChE. A 30-fold increase in the extracellular acetylcholine levels was observed in the hippocampus of untreated AChE knockout mice compared with the wild-type mice. The infusion of rivastigmine (1 and 10 μM) further doubled (30%–50%) acetylcholine levels in AChE knockout mice, whereas no effect was observed with donepezil (1 μM); however, following infusion of donepezil (1 μM) in wild-type mice, the hippocampal acetylcholine levels increased by more than 2-fold (150%).48 A microdialysis study49 was conducted to clarify the importance of BuChE in regulating brain cholinergic function in rat cerebral cortex. The results showed that rivastigmine (0.6 mg/kg intraperitoneally [i.p.]) inhibited AChE and BuChE by 40% and 25%, respectively, in the rat cerebral cortex, whereas administration of donepezil (1 mg/kg i.p.) was associated with a 27% inhibition of AChE, but no BuChE inhibition was observed. A preclinical study50 in 5-week-old imprinting control region mice with cognitive dysfunction induced by amyloid-β peptide (Aβ1–40) showed that repeated daily administration of a BuChE-selective inhibitor, N1-phenethyl-norcymserine (1 and 3 mg/kg); a dual AChE/BuChE inhibitor, rivastigmine (0.03, 0.1, and 0.3 mg/kg), or an AChE-selective inhibitor, donepezil (1 mg/kg), on days 0–3 significantly ameliorated cognitive dysfunction compared to the control mice (P<0.001 at all doses; rivastigmine 0.3 mg/kg: P<0.05). The finding from this study supports the role of BuChE inhibition as a therapeutic strategy in ameliorating cognitive dysfunction induced by AD pathology; further, the dual AChE/BuChE inhibition maximizes therapeutic efficacy.
Clinical studies of rivastigmine
The effect of rivastigmine in the treatment of patients with AD has been studied using medical imaging methods such as PET and magnetic resonance imaging (MRI). The findings from a double-blind, placebo-controlled study51 using 18F-fluorodeoxyglucose-PET demonstrated that treatment with rivastigmine (3, 6, or 9 mg/day) over 6 months in patients with mild-to-moderate probable AD significantly increased brain hippocampal metabolism by 32.5% in rivastigmine responders (P<0.03) compared with nonsignificant decrease in rivastigmine nonresponders and those treated with placebo (6.4% and 4.1%, respectively). Rivastigmine prevented clinical progression of symptoms and showed marked metabolic increase in memory-related cortices and the prefrontal system in rivastigmine responders.51 A pilot study52 in patients with mild AD using functional MRI showed that a single 3 mg dose of rivastigmine increased bilateral activation in the fusiform gyrus during the face-encoding task and in the prefrontal cortex during a simple working memory task. Results from the N-[11C]methylpiperidyl-4-acetate-PET study53 showed that in patients with probable AD (n=11), rivastigmine at a dose of 9 mg/day (for 3–5 months) reduced AChE activity by 29%–37% in various cortical regions (frontal, temporal, and parietal cortices), with greater AChE inhibition in the frontal cortex (P=0.003) than the temporal cortex (P=0.05), whereas donepezil 10 mg/day reduced the brain AChE activity by 38.5% in the frontal cortex (P=0.0006), by 28.7% in the temporal cortex (P=0.02), and by 28.1% in the parietal cortex (P=0.05). The findings from this study showed that AChE inhibitors (rivastigmine and donepezil) induce greater AChE inhibition in the frontal cortex in the AD brain.
Several clinical studies have also demonstrated the role of rivastigmine in dual AChE–BuChE inhibition. A clinical study34 in healthy young male volunteers showed that a single 3 mg oral dose of rivastigmine produced a maximum cerebrospinal fluid (CSF) AChE inhibition of 38.9% and CSF BuChE inhibition of 9.7% at 2.4 h. Furthermore, an open-label, multiple-dose study54 in patients with probable AD showed the therapeutic potential of rivastigmine, with the 6 mg twice daily (bid) group showing a maximum mean inhibition of 61.7% of AChE activity at 5.6 h post-dose. Results from a single-center, open-label study28 supported the role of BuChE and its inhibition with rivastigmine in the regulation of cognitive functions in patients with probable AD. A significant correlation was observed between the change in the Computerized Neuropsychological Test Battery summary score and the CSF AChE (r=−0.56, P<0.05) and CSF BuChE (r=−0.65, P<0.01) inhibition activity. The important role of BuChE in cognitive processing was further supported by the result of this study because improvement in speed-, attention-, and memory-related performance was significantly correlated with the inhibition of BuChE activity but not with AChE activity in the CSF. A 12-month, open-label study27 in patients with mild AD showed sustained AChE–BuChE inhibition of rivastigmine in the CSF (3 months: AChE: r=0.87, P=0.0002; BuChE; r=0.95, P<0.0001; 12 months: AChE: r=−0.67, P=0.02; BuChE: r=−0.86, P=0.0003). The findings showed that at 12 months, the AChE and BuChE activities in the CSF were lower compared with baseline values by 36% and 45%, respectively. In addition, a positive cognitive effect, particularly in memory and attention, was noted for up to 6 months of treatment.
According to the results from an ex vivo study,55 in patients with a clinical history of AD, rivastigmine could inhibit AChE activity in neurons and axons in a dose-dependent manner at concentrations of 10−6–10−4 M, whereas rivastigmine inhibited BuChE activity in the cerebral cortical neurons at a concentration of 10−5 M. The results from this study suggest that rivastigmine is a more potent inhibitor of BuChE. Moreover, a 13-week, randomized, open-label study56 in patients with mild-to-moderate AD showed that rivastigmine significantly decreased activity of both AChE and BuChE in the CSF by 42.6% (P<0.001 vs baseline; AChE protein levels decreased by 21.8%) and 45.4% (P<0.001 vs baseline; BuChE protein levels decreased by 9.3%), respectively, whereas donepezil and galantamine did not significantly decrease BuChE activity, and these enzymes were associated with increased levels of cholinesterases in the CSF. Similarly, results from a clinical study57 in patients with probable AD showed that rivastigmine treatment decreased CSF AChE and BuChE activity and was statistically significant in only rivastigmine 12 mg/day group (P<0.01) as there were fewer patients in the 6 and 9 mg/day groups; however, treatment with donepezil (P<0.0001) and galantamine (P<0.001) showed significant increase in CSF AChE activity, but no change in BuChE activity was observed. The change in CSF AChE activity with donepezil treatment was dose related (mean percentage change with 5 and 10 mg/day: +88% and +116%, respectively), and the percentage change in CSF AChE activity was significantly different between the two doses (P<0.01), whereas the change in CSF AChE activity with galantamine was not dose related (8 and 12 mg/day: +53% and +52%, respectively).
BuChE genotype
The most common BuChE variant is the K-variant that develops due to a point mutation in the DNA that results in alanine-to-threonine substitution at the 539 amino acid position (Ala539 → Thr). This polymorphism is associated with a 30% reduction of serum BuChE activity due to lower expression of the enzyme.58,59 Several studies have been conducted to determine the association between the K allele of BuChE and the risk of developing AD, and the association of BuChE-K variant and risk of developing AD still remains unclear.58 Results from a study suggested a protective role of the BuChE-K allele in disease progression.29 Further, a retrospective analysis60 of patients with AD (n=994) investigated the influence of age on response to cholinesterase inhibitor therapy. An analysis of response according to the BuChE genotype suggested that the differential effect might be related to the ability of rivastigmine to inhibit BuChE in addition to AChE.60 Furthermore, another retrospective analysis32 showed differential responses to cholinesterase inhibitors in patients with AD (aged <75 years) (n=994) who were grouped according to their BuChE genotype. The results demonstrated significantly greater responses to rivastigmine compared with donepezil on the Severe Impairment Battery, Alzheimer’s Disease Cooperative Study-Activities of Daily Living (ADCS-ADL) scale, Global Deterioration Scale, and Neuropsychiatric Inventory (NPI) in BuChE wild-type carriers but not in carriers of BuChE-K. A retrospective exploratory analysis61 of a randomized, placebo-controlled study (Investigation in Delay to Diagnosis of Alzheimer’s disease with Exelon [InDDEx];62 n=1,018) of patients with amnestic mild cognitive impairment showed that rivastigmine compared with placebo reduced progression to AD (12.9% vs 30%; hazard ratio: 0.369; χ2 test, P=0.0745) and functional decline on ADCS-ADL (−6.08 vs −10.9, respectively; P=0.046) in women with the BuChE wild-type genotype, and rivastigmine reduced cognitive decline in men with the BuChE-K allele compared with those receiving placebo (−2.23 vs −4.71, respectively; P=0.037). The results from a community-based study of patients with severe AD29 showed that individuals (with baseline Mini-Mental State Examination [MMSE] scores ≤8 points) carrying the BuChE-K allele had slower rate of cognitive decline. A multicenter outpatient study63 (n=152) on the prevalence of wild-type BuChE genotype showed that most of the patients (n=146 [96.1%]) with AD were wild-type BuChE carriers, and those with AD with wild-type BuChE were associated with greater cognitive decline, unlike patients with the BuChE-K allele that is associated with lower BuChE enzyme expression, thereby leading to less rapid cognitive decline. Moreover, various studies58,64–67 have shown the protective role of the BuChE K-variant alleles in reducing the risk of AD in apolipoprotein E ε4 (APOE ε4) noncarriers and delaying the onset and progression of AD.
Although APOE ε4 is the main genetic risk factor for AD, it has been understudied in the literature.68 The results from a systematic review69 showed that the Northern Europe had the highest regional prevalence estimates for APOE e4 carriers in AD populations (e4/−: 61.3% [95% CI 55.9–66.7]; e4/4: 14.1% [95% CI 12.2–16.0]), whereas Asia (e4/−: 41.9% [95% CI 38.5–45.3]; e4/4: 7.7% [95% CI 5.8–9.6]) or Southern Europe/Mediterranean (e4/−: 40.5% [95% CI: 36.8–44.1]; e4/4 prevalence: 4.6% [95% CI: 2.7–6.4]) had the lowest regional prevalence estimates. The findings from a meta-analysis70 in Chinese population showed statistically significant positive association between the APOE ε4 allele carriers and AD (odds ratio [OR] =3.93, 95% CI 3.37–4.58, P<0.00001); moreover, carriers of homozygotes APOE ε4/ε4 and heterozygous APOE ε4/ε3 alleles have statistically significant association with AD (OR =11.76, 95% CI 6.38–21.47, and OR =3.08, 95% CI 2.57–3.69, respectively; P<0.00001). A retrospective analysis71 in mild-to-moderately severe AD patients showed inconsistency with carriers of APOE ε4 and drug response. In this study, rivastigmine 6–12 mg/day improved ADAS-cog by 1.67 points in APOE ε4 carriers (n=246) and declined ADAS-cog by 0.48 points in the APOE ε4 noncarriers (n=121). Moreover, an open-label, nonrandomized, multicenter study72 in patients with mild-to-moderately severe AD (n=214) showed no difference in response to rivastigmine (1.5–6 mg bid) for APOE ε4 carriers and noncarriers on cognition, indicating that at least one APOE ε4 allele does not determine difference in the response to rivastigmine.
BuChE progressively replaces AChE in acetylcholine hydrolysis in patients with advanced stages of AD, and particularly in the BuChE wild-type carriers, the rate of hydrolysis is higher compared with the BuChE K-variant. Rivastigmine has an added advantage of inhibiting BuChE in addition to AChE, thereby showing advantages over other AChE inhibitors, and particularly in individuals with BuChE wild-type genotype.
Rivastigmine in subcortical VaD
VaD is associated with cholinergic deficit, and cholinesterase inhibitors may be beneficial in the treatment of VaD.73 The commonest type of VaD is subcortical VaD, which develops due to interruption or reduction in the blood flow in subcortical region of brain.74 A range of behavioral and cognitive changes, particularly executive dysfunction (main characteristic of subcortical dementia), accompany subcortical VaD and are attributed to disruption of the frontal subcortical circuits.75 Rivastigmine has shown beneficial effects of improvement in global function, ADL, behavioral symptoms, and/or cognition in patients with AD and PDD.76 Data from the open-label studies77 have shown similar benefits of improved cognition and behavioral symptoms in patients with subcortical VaD. The results from a 12-month, follow-up study78 showed that rivastigmine (3–6 mg/day) improved cognition (ten-point clock [TPC] drawing test) (P<0.05) and reduced both caregiver stress (relative stress scale [RSS]) (P<0.05) and behavioral discharge (P=0.09) in patients with subcortical VaD. Moreover, an open-label follow-up study79 in patients with subcortical VaD (n=16) showed significant improvements with rivastigmine (3–6 mg/day) at 22 months in the TPC drawing test scores of 2.1 points over baseline (P<0.01) and 3 points over aspirin (100 mg/day) (P<0.001); NPI scores by 3.3 points over baseline (P<0.05) and 4.2 points over aspirin (100 mg/day) (P<0.01); and reduced caregiver stress by 8.5 points on the RSS over baseline (P<0.05) and a mean treatment effect of 11.8 points (P<0.01). A 12-month, randomized, controlled, open-label study80 (n=208) suggested that rivastigmine (3–6 mg/day) is useful in probable subcortical VaD. The results showed significant improvement in the total scores of the Behavioral Pathology in Alzheimer’s Disease (BEHAVE-AD) rating scale compared with baseline and in those treated with aspirin (100 mg/day) at month 12 (P<0.001). Further analysis of the individual items on the BEHAVE-AD showed that rivastigmine treatment provided benefits on all items of the scale, except for delusions (not significant). Rivastigmine was also shown to specifically address the impairment in executive function commonly observed in subcortical VaD. The specific improvement in executive function provided by rivastigmine was also supported by results from a 22-month, open-label study81 in patients with subcortical VaD (n=16). There was significant improvement on the total BEHAVE-AD score in patients receiving rivastigmine (3–6 mg/day) compared to those on aspirin (100 mg/day) (P=0.001), the other treatment arm. Analysis of the individual items on the BEHAVE-AD indicated that rivastigmine therapy was associated with improvements on all items of the scale, except for diurnal rhythm disturbance. Moreover, the sub-analysis of individual NPI items indicated significant benefits in terms of anxiety, hallucinations, and wandering in patients treated with rivastigmine compared to those treated with aspirin (P=0.001, P=0.005, and P=0.014, respectively). Rivastigmine (3–6 mg/day) was also shown to be superior to aspirin (100 mg/day) plus nimodipine (60 mg/day) in cognition and functional and behavioral outcome measures in a 16-month open-label study82 in subcortical VaD (n=64). In particular, benefits in executive function and attention that are the core cognitive features of subcortical VaD were observed in the rivastigmine group compared to the aspirin plus nimodipine group. An overall improvement was observed in the behavioral symptoms with rivastigmine as assessed using BEHAVE-AD. In patients treated with rivastigmine, the total BEHAVE-AD scores at 16 months significantly improved compared with baseline and with that of the aspirin plus nimodipine group (P<0.001); conversely, a significant deterioration in BEHAVE-AD scores compared with baseline (P<0.05) was observed in patients treated with aspirin plus nimodipine. Long-term treatment with rivastigmine in a prospective study83 (n=245) consistently showed improvement of behavioral symptoms (BEHAVE-AD) on both total score and individual items in patients with subcortical VaD (P<0.0001); only delusion was found to not have significant improvement in this study. However, a 26-week, randomized, placebo-controlled study84 of rivastigmine (6 mg/day) in patients from China with subcortical VaD (n=40) showed no apparent cognitive benefit (on MMSE and Frontal Assessment Battery) over 6 months, whereas the mean total NPI score (standard deviation [SD]) decreased from 15.0 (14.6) to 11.4 (9.4) with rivastigmine treatment, indicating a positive treatment effect of ~4 points in behavioral symptoms (on the NPI). However, large randomized clinical studies with longer duration are warranted to investigate treatment benefits of rivastigmine in subcortical VaD.
Rivastigmine in PDD
Studies have shown that rivastigmine provides significant benefits on cognition, particularly in executive function and attention, ADL, and neuropsychiatric symptoms, especially hallucinations and delusions, in patients with PDD. In an open-label study85 in patients with PDD (n=28), rivastigmine improved the cognitive function. At week 26, the cognition as assessed by ADAS-cog (total score) and the motor function as assessed by Unified Parkinson’s Disease Rating Scale (UPDRS; total score) improved significantly from baseline by 7.3 points (P<0.002) and 3.2 points (P<0.06), respectively. The specific items that significantly improved in the ADAS-cog at week 26 were recognition (P=0.02), word finding (P=0.05), remembering instructions (P=0.005), and concentration (P=0.003). Further, the MMSE subscore of attention improved significantly by 1.1 points at week 26 (P<0.002); however, rivastigmine did not cause worsening of motor function (a subscore of UPDRS) during the study. Rivastigmine targets the cognitive domain that is frequently impaired in PDD. A randomized, double-blind, placebo-controlled, multicenter study86 in patients with PDD (n=487) showed significant improvements with rivastigmine over placebo on the power of attention at week 24 (P<0.01), continuity of attention at both week 16 (P=0.001) and week 24 (P=0.0001), cognitive reaction time at week 24 (P<0.0001), and reaction time variability at both week 16 (P<0.05) and week 24 (P<0.001). The findings from this study showed that rivastigmine treatment was associated with significant benefits in all aspects of attention (sustained attention, focused attention, consistence of responding, and central processing speed). A randomized, double-blind, placebo-controlled study87 in PD patients with cognitive impairment (ie, mild cognitive impairment and PDD) (n=176) showed that rivastigmine delayed the deterioration of cognitive function and lowered the incidence of falls. In the rivastigmine group, the Montreal Cognitive Assessment scores were significantly higher compared with the placebo group (P=0.002) at 12 months; and the average number of falls per person and the incidence of falls of patients were significantly lower in the rivastigmine group compared with the placebo group (P<0.01) at 12 months. A retrospective ADAS-cog factor analysis study88 including data from patients with PDD (n=969) showed significant improvements on both “memory” (P<0.0001) and “language” (P=0.0033) with rivastigmine (6–12 mg/day) treatment compared with placebo at week 24/26. Results from a multicenter, double-blind, placebo-controlled, parallel-group study89 in patients with PDD (n=541) showed that rivastigmine was associated with significant improvements over placebo on executive function tests and had an impact on flexibility of thinking, problem solving, and planning skills. With regard to letter fluency, rivastigmine was associated with improvements in correct responses, set loss errors, and responses made (P<0.05) at week 24; more correct substitutions on the symbol digit modalities test (P=0.02) and significantly higher card sorting recognition errors (P=0.03) were recorded. Several neuropsychiatric disorders are associated with dysfunction in the frontal–subcortical circuit at cortical and/or subcortical levels,90,91 and the findings from this study89 support the hypothesis that rivastigmine may affect the frontal subcortical circuits contributing to improvements in executive function.
Visual hallucinations frequently complicate the clinical course of PDD. They predict more rapid decline in the clinical course of the illness. A specific benefit was found with rivastigmine treatment in patients with PDD who manifest with visual hallucinations. A double-blind placebo-controlled study92 in patients with PDD (n=541) showed significantly greater rivastigmine–placebo treatment difference at week 24 on the ADAS-cog in visual hallucinators (4.27 points; P=0.002) than nonhallucinators (2.09 points; P=0.015) at week 24. The rivastigmine–placebo treatment difference on the Alzheimer’s Disease Cooperative Society-Clinician’s Global Impression of Change (ADCS-CGIC) was 0.5 among the visual hallucinators (P=0.030) and 0.3 among the nonhallucinators (P=0.111). This supported a greater benefit of rivastigmine in visual hallucinators. A 48-week active treatment extension study showed sustained benefits of rivastigmine over baseline in patients with PDD with no new safety concerns.93 The results from a 6-month, prospective, longitudinal, open-label, observational, single-center study in South Korea94 in patients with PDD (n=23) showed that rivastigmine is useful in controlling several neuropsychiatric symptoms and is beneficial for caregiver distress in patients with PDD. Over 6 months, the NPI total score significantly improved with rivastigmine treatment by −5.4 points (P=0.049), with improvements in the domains of hallucination (P=0.048), depression and dysphoria (P=0.001), and appetite (P=0.024); moreover, total caregiver distress scores decreased significantly (P=0.020).
Approval of rivastigmine for the treatment of mild-to-moderate PDD10 was based on EXPRESS (n=541), a large, 24-week, double-blind, randomized, placebo-controlled study.95 The study showed significant improvements with rivastigmine in ADAS-cog scores compared with placebo at week 16 (P=0.002) and week 24 (P<0.001) (mean −2.1 points improvement from baseline with rivastigmine vs a 0.7 point decline from baseline with placebo at week 24). There were clinically meaningful improvements (moderate or marked) observed in the ADCS-CGIC scores in patients treated with rivastigmine compared with those treated with placebo (19.8% vs 14.5%, respectively) at week 24. Compared with placebo, rivastigmine also demonstrated significant benefits on ADCS-ADL (P=0.02), NPI-10 (P=0.02), MMSE (P=0.03), cognitive drug research power of attention tests (P=0.009), Delis–Kaplan executive function system verbal fluency test (P<0.001), and the TPC drawing test (P=0.02) at week 24. In the retrospective analysis of the EXPRESS study,96 ADAS-cog items were further examined to determine the impact of rivastigmine on various cognitive domains. Patients receiving rivastigmine (3–12 mg/day) improved significantly on all three key symptom domains (memory [P<0.001], language [P=0.003] and praxis [P=0.003]) compared with placebo at week 24; moreover, significant treatment differences with rivastigmine (3–12 mg/day) were observed on the following cognitive items: word recall, following commands, ideational praxis, remembering test instructions, and comprehension of spoken language (P<0.05).
The long-term safety data from a 76-week, prospective, open-label, randomized study97 in patients with mild-to-moderate PDD (n=583) showed that treatment with rivastigmine capsule (12 mg/day) or patch (9.5 mg/24 h patch) was well tolerated. The commonest adverse events (AEs) with rivastigmine capsules and patch were nausea (40.5% and 8.3%, respectively), tremor (24.5% and 9.7%, respectively), fall (17.0% and 20.1%, respectively), vomiting (15.3% and 2.8%, respectively), and application site erythema (0% and 13.9%, respectively). The motor symptoms of PD were not found to worsen with treatment as the rate of progression was in the expected range due to the natural progression of the disease. The results from a 24-week, randomized, double-blind, placebo-controlled study98 on rivastigmine showed that more patients receiving rivastigmine (10.2%) compared with those receiving placebo (3.9%) reported tremor, although this did not reflect in the total UPDRS part III score and there was no significant difference between rivastigmine and placebo (P=0.83) (mean baseline UPDRS part III motor scores [SD]: 34.0 [14.6] [rivastigmine] and 32.2 [13.2] [placebo]). The incidence of worsening Parkinsonism, bradykinesia, and rigidity were all <5% in both treatment groups. The positive impact of rivastigmine on the ability of patients to perform various ADLs was observed in a post hoc analysis of a prospective, multicenter, randomized, double-blind, placebo-controlled study.99 At week 24, compared with placebo, rivastigmine was associated with significantly better outcomes in both basic ADLs (−0.5±6.19 vs −1.7±5.46; P=0.025; effect size 22.1%) and high-level function ADLs (0.1±4.95 vs −1.0±4.49; P=0.017; effect size 22.9%).
Conclusion
Dual inhibition of AChE and BuChE by rivastigmine may be considered as a potential therapeutic advantage for subcortical dementias (PDD and subcortical VaD) to benefit in cognition, global function, and behavioral symptoms. Rivastigmine was found to specifically benefit executive dysfunction that is frequently observed in subcortical dementias; however, large randomized clinical studies are warranted to support these observations.
Acknowledgments
This review article is sponsored by Novartis (Singapore) Pte. Ltd. The publication processing fees were funded by Novartis (Singapore) Pte. Ltd. All authors of this review article met the International Committee of Medical Journal Editors (ICMJE) criteria for authorship and gave their approvals for this article to be published.
Disclosure
Nagaendran Kandiah has received honorarium and CME sponsorship from Lundbeck, Novartis, Pfizer, Schwabbe, and Eisai. He has also received research funding from Singhealth Foundation, Media Development Authority of Singapore, Biomedical Research Council, and the National Medical Research Council of Singapore. Ming-Chyi Pai has received grant/clinical trial support from AB Science, Lundbeck, Biogen, BAC, Lilly and Axovant and has served as a consultant and/or has received lecture fees from Janssen, Lilly, Novartis, Lotus, and Eisai. Vorapun Senanarong has received honorarium from Boehringer Ingelheim, Eisai, Janssen, Lundbeck, Novartis, Otsuka, and Pfizer as a speaker. Irene Looi has received lectures fees and grants from Novartis and Eisai. Encarnita Ampil is on the Advisory Board of Lundbeck Philippines and previously Novartis and Hi Eisai. She has been speaker for and has received honoraria from Lundbeck, Hi Eisai, Menarini, Novartis, Janssen, Otsuka, Astra Zeneca, Pharmalink, and Torrent Pharma. She is currently involved in clinical trials with E Chimes and Amylex Pharmaceuticals, Inc. Kyung Won Park has received research funding, lecture fees, or honoraria from Eisai, Lundbeck, and Novartis. Stephen Christopher and Ananda Krishna Karanam are employees of Novartis. The authors report no other conflicts of interest in this work.
References
Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2(8000):1403. | ||
Martorana A, Esposito Z, Koch G. Beyond the cholinergic hypothesis: do current drugs work in Alzheimer’s disease? CNS Neurosci Ther. 2010;16(4):235–245. | ||
Bartus RT. On neurodegenerative diseases, models, and treatment strategies: lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp Neurol. 2000;163(2):495–529. | ||
Rogan S, Lippa CF. Alzheimer’s disease and other dementias: a review. Am J Alzheimers Dis Other Demen. 2002;17(1):11–17. | ||
Nordberg A, Ballard C, Bullock R, Darreh-Shori T, Somogyi M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim Care Companion CNS Disord. 2013;15(2):ii:CC.12r01412. | ||
Weinstock M. Selectivity of cholinesterase inhibition: clinical implications for the treatment of Alzheimer’s disease. CNS Drugs. 1999;12(4):307–323. | ||
Rodda J, Carter J. Cholinesterase inhibitors and memantine for symptomatic treatment of dementia. BMJ. 2012;344:e2986. | ||
Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med. 2008;148(5):379–397. | ||
Donepezil (Aricept®) [US prescribing information]. Available from: http://www.aricept.com/docs/pdf/aricept_PI.pdf. Accessed November 17, 2016. | ||
Rivastigmine (Exelon®) [US prescribing information]. Available from: https://www.pharma.us.novartis.com/product/pi/pdf/exelon.pdf. Accessed November 17, 2016. | ||
Rivastigmine patch (Exelon Patch®) [US prescribing information]. Available from: https://www.pharma.us.novartis.com/product/pi/pdf/exelonpatch.pdf. Accessed November 21, 2016. | ||
Galantamine (Razadyne ER® and Razadyne®) [US prescribing information]. Available from: https://www.janssenmd.com/pdf/razadyne/PI-Razadyne-RazadyneER.pdf. Accessed November 21, 2016. | ||
Memantine (Namenda®) [US prescribing information]. Available from: https://www.allergan.com/assets/pdf/namenda_pi. Accessed November 21, 2016. | ||
Cummings J, Lefèvre G, Small G, Appel-Dingemanse S. Pharmacokinetic rationale for the rivastigmine patch. Neurology. 2007;69(4 suppl 1):S10–S13. | ||
Mercier F, Lefèvre G, Huang HL, Schmidli H, Amzal B, Appel-Dingemanse S. Rivastigmine exposure provided by a transdermal patch versus capsules. Curr Med Res Opin. 2007;23(12):3199–3204. | ||
Winblad B, Machado JC. Use of rivastigmine transdermal patch in the treatment of Alzheimer’s disease. Expert Opin Drug Deliv. 2008;5(12):1377–1386. | ||
Rivastigmine (Exelon®) summary of product characteristics. Available from: http://www.ema.europa.eu/ema/. Accessed November 21, 2016. | ||
Bohnen NI, Kaufer DI, Ivanco LS, et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol. 2003;60(12):1745–1748. | ||
Gottwald MD, Rozanski RI. Rivastigmine, a brain-region selective acetylcholinesterase inhibitor for treating Alzheimer’s disease: review and current status. Expert Opin Investig Drugs. 1999;8(10):1673–1682. | ||
Mesulam M, Guillozet A, Shaw P, Quinn B. Widely spread butyrylcholinesterase can hydrolyze acetylcholine in the normal and Alzheimer brain. Neurobiol Dis. 2002;9(1):88–93. | ||
Lane RM, Potkin SG, Enz A. Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int J Neuropsychopharmacol. 2006;9(1):101–124. | ||
Mesulam MM, Geula C. Acetylcholinesterase-rich neurons of the human cerebral cortex: cytoarchitectonic and ontogenetic patterns of distribution. J Comp Neurol. 1991;306(2):193–220. | ||
Wright CI, Geula C, Mesulam MM. Neurological cholinesterases in the normal brain and in Alzheimer’s disease: relationship to plaques, tangles, and patterns of selective vulnerability. Ann Neurol. 1993;34(3):373–384. | ||
Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O. Acetylcholinesterase knockouts establish central cholinergic pathways and can use butyrylcholinesterase to hydrolyze acetylcholine. Neuroscience. 2002;110(4):627–639. | ||
Greig NH, Utsuki T, Yu Q, et al. A new therapeutic target in Alzheimer’s disease treatment: attention to butyrylcholinesterase. Curr Med Res Opin. 2001;17(3):159–165. | ||
Perry EK, Perry RH, Blessed G, Tomlinson BE. Changes in brain cholinesterases in senile dementia of Alzheimer type. Neuropathol Appl Neurobiol. 1978;4(4):273–277. | ||
Darreh-Shori T, Almkvist O, Guan ZZ, et al. Sustained cholinesterase inhibition in AD patients receiving rivastigmine for 12 months. Neurology. 2002;59(4):563–572. | ||
Giacobini E, Spiegel R, Enz A, Veroff AE, Cutler NR. Inhibition of acetyl- and butyryl-cholinesterase in the cerebrospinal fluid of patients with Alzheimer’s disease by rivastigmine: correlation with cognitive benefit. J Neural Transm (Vienna). 2002;109(7–8):1053–1065. | ||
Holmes C, Ballard C, Lehmann D, et al. Rate of progression of cognitive decline in Alzheimer’s disease: effect of butyrylcholinesterase K gene variation. J Neurol Neurosurg Psychiatry. 2005;76(5):640–643. | ||
Poirier J. Evidence that the clinical effects of cholinesterase inhibitors are related to potency and targeting of action. Int J Clin Pract Suppl. 2002;(127):6–19. | ||
Bullock R, Lane R. Executive dyscontrol in dementia, with emphasis on subcortical pathology and the role of butyrylcholinesterase. Curr Alzheimer Res. 2007;4(3):277–293. | ||
Blesa R, Bullock R, He Y, et al. Effect of butyrylcholinesterase genotype on the response to rivastigmine or donepezil in younger patients with Alzheimer’s disease. Pharmacogenet Genomics. 2006;16(11):771–774. | ||
Enz A, Amstutz R, Boddeke H, Gmelin G, Malanowski J. Brain selective inhibition of acetylcholinesterase: a novel approach to therapy for Alzheimer’s disease. Prog Brain Res. 1993;98:431–438. | ||
Kennedy JS, Polinsky RJ, Johnson B, et al. Preferential cerebrospinal fluid acetylcholinesterase inhibition by rivastigmine in humans. J Clin Psychopharmacol. 1999;19(6):513–521. | ||
Corey-Bloom J, Anand R, Veach J; ENA 713 B352 Study. A randomised trial evaluating the efficacy and safety of ENA 713 (rivastigmine tartrate), a new acetylcholinesterase inhibitor, in patients with mild to moderately severe Alzheimer’s disease. Int J Ger Psychopharmacol. 1998;1:55–65. | ||
Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318(7184):633–638. | ||
Winblad B, Cummings J, Andreasen N, et al. A six-month double-blind, randomized, placebo – controlled study of a transdermal patch in Alzheimer’s disease – rivastigmine patch versus capsule. Int J Geriatr Psychiatry. 2007;22(5):456–467. | ||
Birks JS, Chong LY, Grimley Evans J. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev. 2015;9:CD001191. | ||
Farlow MR, Small GW, Quarg P, Krause A. Efficacy of rivastigmine in Alzheimer’s disease patients with rapid disease progression: results of a meta-analysis. Dement Geriatr Cogn Disord. 2005;20(2–3):192–197. | ||
Su J, Liu Y, Liu Y, Ren L. Long-term effectiveness of rivastigmine patch or capsule for mild-to-severe Alzheimer’s disease: a meta-analysis. Expert Rev Neurother. 2015;15(9):1093–1103. | ||
Birks JS, Grimley Evans J. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev. 2015;4:CD001191. | ||
Anand R, Messina J, Hartman R. Dose-response effect of rivastigmine in the treatment of Alzheimer’s disease. Int J Geriatr Psychopharmacol. 2000;2:68–72. | ||
Grossberg GT, Olin JT, Somogyi M, Meng X. Dose effects associated with rivastigmine transdermal patch in patients with mild-to-moderate Alzheimer’s disease. Int J Clin Pract. 2011;65(4):465–471. | ||
Farlow MR, Grossberg GT, Sadowsky CH, Meng X, Somogyi M. A 24-week, randomized, controlled trial of rivastigmine patch 13.3 mg/24 h versus 4.6 mg/24 h in severe Alzheimer’s dementia. CNS Neurosci Ther. 2013;19(10):745–752. | ||
Isaacson RS, Ferris S, Velting DM, Meng X. Cognitive efficacy (SIB) of 13.3 versus 4.6 mg/24 h rivastigmine patch in severe Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2016;31(3):270–277. | ||
Xie W, Stribley JA, Chatonnet A, et al. Postnatal developmental delay and supersensitivity to organophosphate in gene-targeted mice lacking acetylcholinesterase. J Pharmacol Exp Ther. 2000;293(3):896–902. | ||
Ogura H, Kosasa T, Kuriya Y, Yamanishi Y. Comparison of inhibitory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butyrylcholinesterase in vitro. Methods Find Exp Clin Pharmacol. 2000;22(8):609–613. | ||
Naik RS, Hartmann J, Kiewert C, Duysen EG, Lockridge O, Klein J. Effects of rivastigmine and donepezil on brain acetylcholine levels in acetylcholinesterase-deficient mice. J Pharm Pharm Sci. 2009;12(1):79–85. | ||
Cerbai F, Giovannini MG, Melani C, Enz A, Pepeu G. N1phenethyl-norcymserine, a selective butyrylcholinesterase inhibitor, increases acetylcholine release in rat cerebral cortex: a comparison with donepezil and rivastigmine. Eur J Pharmacol. 2007;572(2–3):142–150. | ||
Furukawa-Hibi Y, Alkam T, Nitta A, et al. Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-β peptide in mice. Behav Brain Res. 2011;225(1):222–229. | ||
Potkin SG, Anand R, Fleming K, et al. Brain metabolic and clinical effects of rivastigmine in Alzheimer’s disease. Int J Neuropsychopharmacol. 2001;4(3):223–230. | ||
Rombouts SA, Barkhof F, van Meel CS, Scheltens P. Alterations in brain activation during cholinergic enhancement with rivastigmine in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2002;73(6):665–671. | ||
Kaasinen V, Någren K, Järvenpää T, et al. Regional effects of donepezil and rivastigmine on cortical acetylcholinesterase activity in Alzheimer’s disease. J Clin Psychopharmacol. 2002;22(6):615–620. | ||
Cutler NR, Polinsky RJ, Sramek JJ, et al. Dose-dependent CSF acetylcholinesterase inhibition by SDZ ENA 713 in Alzheimer’s disease. Acta Neurol Scand. 1998;97(4):244–250. | ||
Eskander MF, Nagykery NG, Leung EY, Khelghati B, Geula C. Rivastigmine is a potent inhibitor of acetyl- and butyrylcholinesterase in Alzheimer’s plaques and tangles. Brain Res. 2005;1060(1–2):144–152. | ||
Nordberg A, Darreh-Shori T, Peskind E, et al. Different cholinesterase inhibitor effects on CSF cholinesterases in Alzheimer patients. Curr Alzheimer Res. 2009;6(1):4–14. | ||
Parnetti L, Chiasserini D, Andreasson U, et al. Changes in CSF acetyl- and butyrylcholinesterase activity after long-term treatment with AChE inhibitors in Alzheimer’s disease. Acta Neurol Scand. 2011;124(2):122–129. | ||
Wang Z, Jiang Y, Wang X, et al. Butyrylcholinesterase K variant and Alzheimer’s disease risk: a meta-analysis. Med Sci Monit. 2015;21:1408–1413. | ||
Bartels CF, Jensen FS, Lockridge O, et al. DNA mutation associated with the human butyrylcholinesterase K-variant and its linkage to the atypical variant mutation and other polymorphic sites. Am J Hum Genet. 1992;50(5):1086–1103. | ||
Bullock R, Bergman H, Touchon J, et al. Effect of age on response to rivastigmine or donepezil in patients with Alzheimer’s disease. Curr Med Res Opin. 2006;22(3):483–494. | ||
Ferris S, Nordberg A, Soininen H, Darreh-Shori T, Lane R. Progression from mild cognitive impairment to Alzheimer’s disease: effects of sex, butyrylcholinesterase genotype, and rivastigmine treatment. Pharmacogenet Genomics. 2009;19(8):635–646. | ||
Feldman HH, Ferris S, Winblad B, et al. Effect of rivastigmine on delay to diagnosis of Alzheimer’s disease from mild cognitive impairment: the InDDEx study. Lancet Neurol. 2007;6(6):501–512. | ||
Mueller B, Adler G. Prevalence of wild-type butyrylcholinesterase genotype in patients with Alzheimer’s dementia. World J Neurosci. 2015;5:175–179. | ||
Alvarez-Arcaya A, Combarros O, Llorca J, et al. The butyrylcholinesterase K variant is a protective factor for sporadic Alzheimer’s disease in women. Acta Neurol Scand. 2000;102(6):350–353. | ||
Darreh-Shori T, Siawesh M, Mousavi M, Andreasen N, Nordberg A. Apolipoprotein ε4 modulates phenotype of butyrylcholinesterase in CSF of patients with Alzheimer’s disease. J Alzheimers Dis. 2012;28(2):443–458. | ||
Lane RM, He Y. Butyrylcholinesterase genotype and gender influence Alzheimer’s disease phenotype. Alzheimers Dement. 2013;9(2):e1–e73. | ||
Podoly E, Shalev DE, Shenhar-Tsarfaty S, et al. The butyrylcholinesterase K variant confers structurally derived risks for Alzheimer pathology. J Biol Chem. 2009;284(25):17170–17179. | ||
Michaelson DM. APOE ε4: the most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement. 2014;10(6):861–868. | ||
Ward A, Crean S, Mercaldi CJ, et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology. 2012;38(1):1–17. | ||
Liu M, Bian C, Zhang J, Wen F. Apolipoprotein E gene polymorphism and Alzheimer’s disease in Chinese population: a meta-analysis. Sci Rep. 2014;4:4383. | ||
Farlow M, Lane R, Kudaravalli S, He Y. Differential qualitative responses to rivastigmine in APOE epsilon 4 carriers and noncarriers. Pharmacogenomics J. 2004;4(5):332–335. | ||
Blesa R, Aguilar M, Casanova JP, et al. Relationship between the efficacy of rivastigmine and apolipoprotein E (epsilon 4) in patients with mild to moderately severe Alzheimer disease. Alzheimer Dis Assoc Disord. 2006;20(4):248–254. | ||
Erkinjuntti T, Román G, Gauthier S, Feldman H, Rockwood K. Emerging therapies for vascular dementia and vascular cognitive impairment. Stroke. 2004;35(4):1010–1017. | ||
Tomimoto H. Subcortical vascular dementia. Neurosci Res. 2011;71(3):193–199. | ||
Kalaria RN, Erkinjuntti T. Small vessel disease and subcortical vascular dementia. J Clin Neurol. 2006;2(1):1–11. | ||
Sadowsky CH, Micca JL, Grossberg GT, Velting DM. Rivastigmine from capsules to patch: therapeutic advances in the management of Alzheimer’s disease and Parkinson’s disease dementia. Prim Care Companion CNS Disord. 2014;16(5). | ||
Román GC. Rivastigmine for subcortical vascular dementia. Expert Rev Neurother. 2005;5(3):309–313. | ||
Moretti R, Torre P, Antonello RM, Cazzato G. Rivastigmine in subcortical vascular dementia: a comparison trial on efficacy and tolerability for 12 months follow-up. Eur J Neurol. 2001;8(4):361–362. | ||
Moretti R, Torre P, Antonello RM, Cazzato G, Bava A. Rivastigmine in subcortical vascular dementia: an open 22-month study. J Neurol Sci. 2002;203–204:141–146. | ||
Moretti R, Torre P, Antonello RM, Cazzato G, Bava A. Rivastigmine in subcortical vascular dementia: a randomized, controlled, open 12-month study in 208 patients. Am J Alzheimers Dis Other Demen. 2003;18(5):265–272. | ||
Moretti R, Torre P, Antonello RM, Cazzato G, Griggio S, Bava A. An open-label pilot study comparing rivastigmine and low-dose aspirin for the treatment of symptoms specific to patients with subcortical vascular dementia. Curr Ther Res. 2002;63(7):443–458. | ||
Moretti R, Torre P, Antonello RM, et al. Rivastigmine superior to aspirin plus nimodipine in subcortical vascular dementia: an open, 16-month, comparative study. Int J Clin Pract. 2004;58(4):346–353. | ||
Moretti R, Torre P, Antonello RM, Cazzato G, Pizzolato G. Different responses to rivastigmine in subcortical vascular dementia and multi-infarct dementia. Am J Alzheimers Dis Other Demen. 2008;23(2):167–176. | ||
Mok V, Wong A, Ho S, Leung T, Lam WW, Wong KS. Rivastigmine in Chinese patients with subcortical vascular dementia. Neuropsychiatr Dis Treat. 2007;3(6):943–948. | ||
Giladi N, Shabtai H, Gurevich T, Benbunan B, Anca M, Korczyn AD. Rivastigmine (Exelon) for dementia in patients with Parkinson’s disease. Acta Neurol Scand. 2003;108(5):368–373. | ||
Wesnes KA, McKeith I, Edgar C, Emre M, Lane R. Benefits of rivastigmine on attention in dementia associated with Parkinson disease. Neurology. 2005;65(10):1654–1656. | ||
Li Z, Yu Z, Zhang J, et al. Impact of rivastigmine on cognitive dysfunction and falling in Parkinson’s disease patients. Eur Neurol. 2015;74(1–2):86–91. | ||
Weintraub D, Somogyi M, Meng X. Rivastigmine in Alzheimer’s disease and Parkinson’s disease dementia: an ADAS-cog factor analysis. Am J Alzheimers Dis Other Demen. 2011;26(6):443–449. | ||
Schmitt FA, Farlow MR, Meng X, Tekin S, Olin JT. Efficacy of rivastigmine on executive function in patients with Parkinson’s disease dementia. CNS Neurosci Ther. 2010;16(6):330–336. | ||
Tekin S, Cummings JL. Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002;53(2):647–654. | ||
Bonelli RM, Cummings JL. Frontal-subcortical circuitry and behavior. Dialogues Clin Neurosci. 2007;9(2):141–151. | ||
Burn D, Emre M, McKeith I, et al. Effects of rivastigmine in patients with and without visual hallucinations in dementia associated with Parkinson’s disease. Mov Disord. 2006;21(11):1899–1907. | ||
Poewe W, Wolters E, Emre M, et al; EXPRESS Investigators. Long-term benefits of rivastigmine in dementia associated with Parkinson’s disease: an active treatment extension study. Mov Disord. 2006;(4):456–461. | ||
Oh YS, Kim JS, Lee PH. Effect of rivastigmine on behavioral and psychiatric symptoms of Parkinson’s disease dementia. J Mov Disord. 2015;8(2):98–102. | ||
Emre M, Aarsland D, Albanese A, et al. Rivastigmine for dementia associated with Parkinson’s disease. N Engl J Med. 2004;351(24):2509–2518. | ||
Schmitt FA, Aarsland D, Brønnick KS, Meng X, Tekin S, Olin JT. Evaluating rivastigmine in mild-to-moderate Parkinson’s disease dementia using ADAS-cog items. Am J Alzheimers Dis Other Demen. 2010;25(5):407–413. | ||
Emre M, Poewe W, De Deyn PP, et al. Long-term safety of rivastigmine in parkinson disease dementia: an open-label, randomized study. Clin Neuropharmacol. 2014;37(1):9–16. | ||
Oertel W, Poewe W, Wolters E, et al. Effects of rivastigmine on tremor and other motor symptoms in patients with Parkinson’s disease dementia: a retrospective analysis of a double-blind trial and an open-label extension. Drug Saf. 2008;31(1):79–94. | ||
Olin JT, Aarsland D, Meng X. Rivastigmine in the treatment of dementia associated with Parkinson’s disease: effects on activities of daily living. Dement Geriatr Cogn Disord. 2010;29(6):510–515. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.