")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Risk of epilepsy in patients with systemic lupus erythematosus – a retrospective cohort study
Authors Tsai J, Lin C, Lin C, Sung F , Lue K
Received 18 March 2014
Accepted for publication 12 June 2014
Published 2 September 2014 Volume 2014:10 Pages 1635—1643
DOI https://doi.org/10.2147/NDT.S64323
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Jeng-Dau Tsai,1,2 Cheng-Li Lin,3,4 Cheng-Chieh Lin,5–7 Fung-Chang Sung,3,4,7,* Ko-Huang Lue1,2,*
1Institute of Medicine, Chung Shan Medical University Hospital, 2Department of Pediatrics, Chung Shan Medical University and Hospital, 3Institute of Clinical and Medical Science, China Medical University, 4Management Office for Health Data, China Medical University Hospital, 5Department of Family Medicine, China Medical University School of Medicine, 6Division of Family Medicine, China Medical University Hospital, 7Department of Healthcare Administration, College of Health Science, Asia University, Taichung, Taiwan
*These authors contributed equally to this work
Background: Systemic lupus erythematosus (SLE) affects central and peripheral nervous systems, manifesting neuropsychiatric disorders that vary from subtle signs to life-threatening complications. This study compared the risk of epilepsy between a general population and patients with SLE.
Methods: From the national insurance claims data of the Taiwan National Health Research Institutes, we identified 32,301 patients with newly diagnosed SLE from 1997–2010 and, for comparison, 129,204 randomly selected people without SLE; the frequencies of both groups were matched by sex, age, and diagnosis date. The incidence of epilepsy was estimated for both cohorts by the end of 2010.
Results: The incidence of epilepsy was 2.86-fold higher in the SLE cohort than in the non-SLE cohort (9.10 per 10,000 person-years versus 3.18 per 10,000 person-years), with a Cox method estimated adjusted hazard ratio (aHR) of 2.33 (95% confidence interval [CI] =1.89–2.88) for the SLE cohort. The incidence increased with age in the non-SLE cohort, while it decreased with the increase of age in the SLE cohort. Compared with the non-SLE cohort, the age-specific aHR of epilepsy for the SLE cohort decreased from 8.05 (95% CI =4.30–15.0) for those aged ≤20 years to 0.90 (95% CI =0.57–1.42) for those aged 60 years and above (P=0.01). Comorbidities that had a significant association with epilepsy included infarction (aHR =7.62), intracerebral hemorrhage (aHR =5.75), aseptic meningoencephalitis (aHR =5.35), and psychiatric disorder (aHR =3.31).
Conclusion: Patients with SLE are at higher risk of epilepsy than the general population, especially younger SLE patients. Neurologic comorbidities and psychiatric disorders increase the epilepsy risk further.
Keywords: epilepsy, incidence, lupus, health insurance data, cohort study
Introduction
Systemic lupus erythematosus (SLE) is a well-known inflammatory autoimmune disease, causing dysfunction and damage to systemic organ and nervous systems.1–3 Neuropsychiatric manifestations, common in SLE patients, involve active systemic diseases, psychiatric and neurologic disorders, and they lead to other complications and increased mortality.4,5 The American College of Rheumatology (ACR) has defined 19 neuropsychiatric manifestations of SLE (NPSLE) that include several neurological disorders.6 The ACR formulated a nomenclature system to define the 19 forms of NPSLE, including central and peripheral neuropathy.7,8 The Systemic Lupus International Collaborating Clinics criteria published in 2012 also include some of these SLE syndromes.9
Among these conditions, seizures are recognized as either a direct result of the primary complication or as the secondary proinflammatory process of SLE.10,11 Cross-sectional and follow-up studies have reported that 6.7%–14.4% of patients with SLE suffer from seizures.5,8,12 In a 7.8-year follow-up study, Appenzeller et al8 identified 60 (11.6%) epileptic seizures in 519 SLE patients, with 19 episodes appearing at the onset of SLE symptoms. In an Italian 10-year multicenter study, Govoni et al13 followed a cohort of 959 SLE patients and found 14.4% with epileptic seizures. These studies show that epilepsy is the unfortunate consequence of SLE, occurring before or at the onset of SLE, or after diagnosis of the disease. However, most studies observed only patients with SLE, without using non-SLE subjects as a reference group for the risk comparison. It is possible that SLE and seizures occur because of a coincidental coexistence, common to a primary disease that causes the epilepsy.14,15
The relative risk of epilepsy occurring after the SLE diagnosis is, in fact, not clear. To the best of our knowledge, no cohort study has measured the risk of epilepsy for SLE patients compared to that for the general population. In this study, we thus investigate the age-specific mean prevalence and incidence of epilepsy in SLE patients and the general population by using the health insurance database in Taiwan. We further performed a retrospective cohort study to evaluate the risk of epilepsy between patients with and without SLE.
Materials and methods
Data sources
The Department of Health in Taiwan incorporated 13 insurance programs into a universal National Health Insurance program in 1995. Since 1998, this program has provided health care coverage to 99% of the entire 23.74 million people in Taiwan.16 With governmental authorization, the Taiwan National Health Research Institutes (NHRI) has been responsible for managing the insurance data. We obtained longitudinal claims datasets from the NHRI for the population insured in the period from 1997–2010. The NHRI had scrambled patient identifiers for privacy protection before the dataset was released to users. This study was exempted from full ethical review.
Study subjects
All diagnoses of diseases in the claims data were coded using the International Classification of Diseases, 9th Revision, Clinical Modification (ICD-9-CM). From the claims data, we identified 32,301 patients with SLE (ICD-9-CM code 710.0), newly diagnosed as catastrophic illness patients in 1997–2010, without a history of epilepsy, as the SLE cohort. Diagnosis of SLE followed the 1997 ACR classification criteria for SLE;6 at least four criteria had to be met. The date of SLE diagnosis was used as the index date. Patients with missing information on age or sex at baseline were excluded. For each SLE case identified, four subjects without a history of SLE and epilepsy were randomly selected into the non-SLE cohort from the same insurance claims dataset, and were frequency-matched by age, sex, and the diagnosis month and year of SLE.
Criteria and definition
The follow-up time was estimated for each subject from the index date until the diagnosis of epilepsy, or until the subject was censored because of loss to follow-up, death, withdrawal from the insurance system, or to the end of 2010. The comorbidities with a potential association with epilepsy were identified at baseline from the inpatient claims data, including psychiatric disorder (ICD-9-CM codes 295–296, 300.0, 301, 307.40–307.49, and 309), aseptic meningoencephalitis (ICD-9-CM codes 322.2, 322.9, and 323.8–323.9), intracerebral hemorrhage (ICD-9-CM code 431), infarction (ICD-9-CM codes 433–434), headache (ICD-9-CM code 784.0), and peripheral neuropathy (ICD-9-CM codes 337, 356.4, 356.8, 356.9, and 357.4).
Statistical analysis
The data analysis first calculated the age-specific (<20 years, 20–29 years, 30–39 years, 40–49 years, 50–59 years, 60–69 years, and ≥70 years) mean incidence and prevalence rates for both SLE patients and the general population from 1997–2010. For the retrospective cohort study, we compared the distributions of sex, age (<20 years, 20–39 years, 40–59 years, and ≥60 years), and comorbidities between the SLE and non-SLE cohorts, and we examined distributions using the chi-square test for categorical variables and the Student’s t-test for continuous variables. The sex- and age-specific incidence rates of epilepsy for both cohorts were calculated. Follow-up time was used to estimate the incidence density rate per 10,000 person-years by demographic status and comorbidity. The SLE cohort-to-non-SLE cohort incidence rate ratio (IRR) with a 95% confidence interval (CI) for each demographic variable and comorbidity was calculated using Poisson regression analysis. A multivariable Cox proportion hazards regression model was used to assess the hazard ratio (HR) of epilepsy for the SLE cohort compared with the non-SLE cohort, with demographic characteristics and comorbid conditions included in the model. The Kaplan–Meier method was used to assess the age-specific cumulative proportional incidence of epilepsy for the two cohorts by age, and the differences were tested with a log–rank test. SAS (version 9.1 for Windows; SAS Institute Inc., Cary, NC, USA) was used for all statistical analyses. All significance levels were set at a P-value of 0.05.
Results
Figure 1 shows the mean age-specific average incidence and prevalence of epilepsy in the general population and among patients with SLE over the 14-year period. Both the incidence and prevalence rates in the general population were the lowest in the 20–30-year age group (0.14 per 100 and 0.50 per 100, respectively), and they increased with age in a U-shaped trend to their highest levels in the 70 years and older group (3.5 per 100 and 7.0 per 100, respectively). On the other hand, the incidence and prevalence of epilepsy in SLE patients were the highest in the youngest group (1.29 per 100 and 1.94 per 100, respectively), which declined slightly and fell somewhat flat with increasing age. The incidence of epilepsy was 4.5-fold higher in SLE patients than in the general population (1.29 per 100 versus 0.29 per 100, respectively) for subjects aged <20 years. The incidence became 9.7-fold higher in the general population when compared to the SLE patients (3.50 per 100 versus 0.36 per 100). A cross-over in the incidence rates between SLE patients and the general population occurred in the 30–39-year-olds. Similar trends also appeared for the prevalence rates between SLE patients and the general population, with the cross-over occurring in the 30–39-year-old groups.
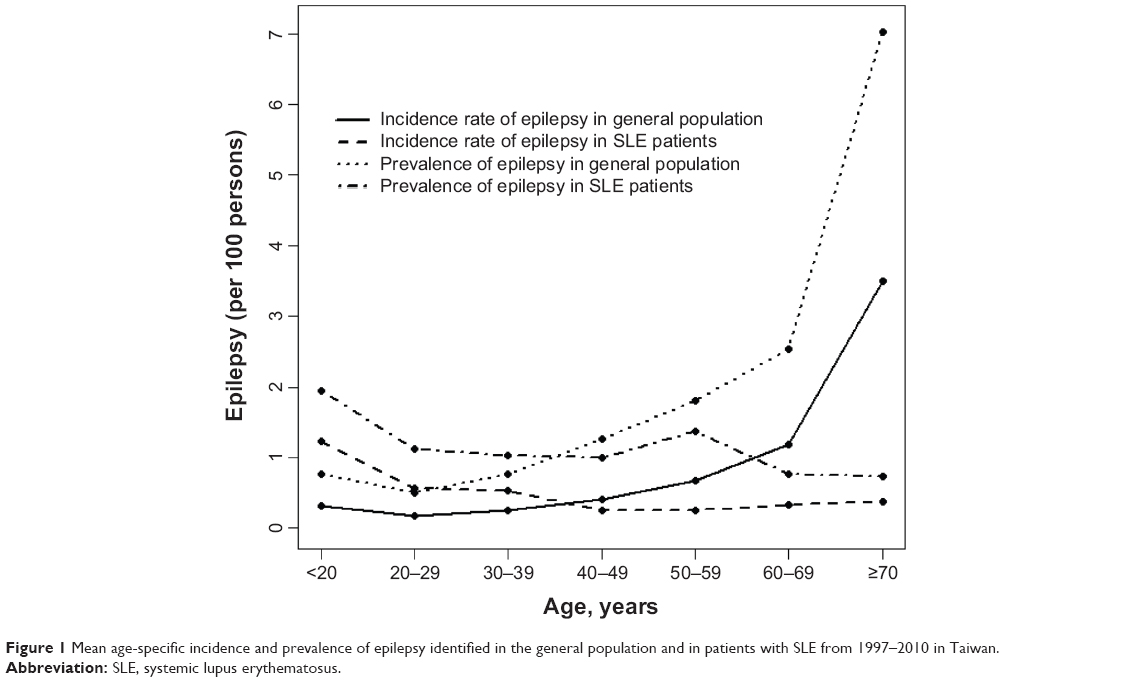 | Figure 1 Mean age-specific incidence and prevalence of epilepsy identified in the general population and in patients with SLE from 1997–2010 in Taiwan. |
For the retrospective cohort study, we established a cohort of 32,301 SLE patients and a non-SLE cohort of 129,204 subjects. Age and sex distributions were similar in both the SLE and non-SLE cohorts with a mean age of 44.3 years; the majority were women (86.1%) (Table 1). The SLE cohort, when compared to the non-SLE cohort, had a higher prevalence rate of comorbidities that included psychiatric disorders, aseptic meningoencephalitis, intracerebral hemorrhage, infarction, headache, and peripheral neuropathy (P<0.0001).
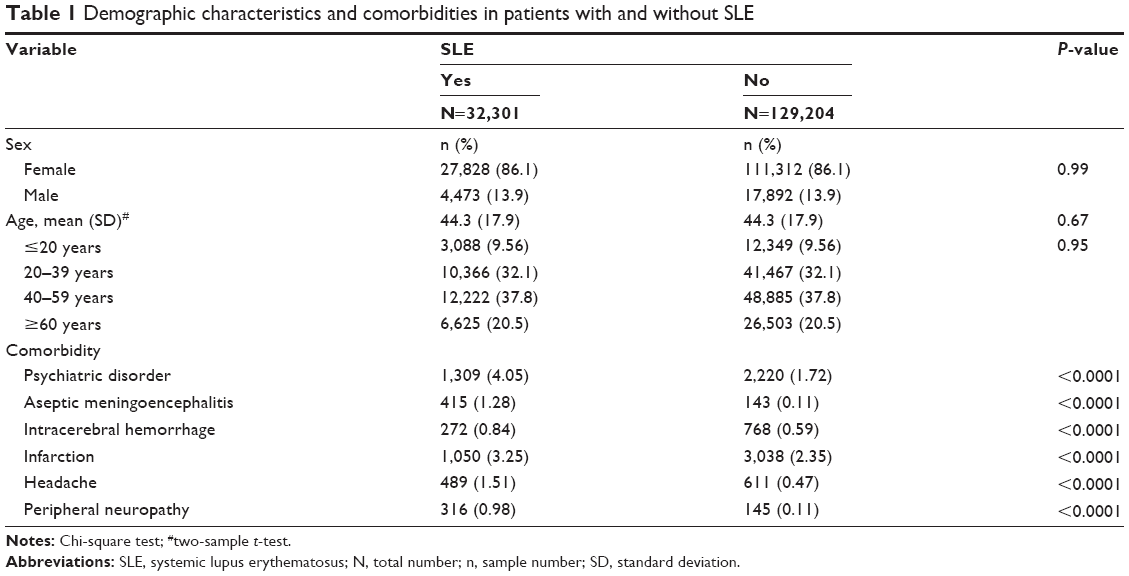 | Table 1 Demographic characteristics and comorbidities in patients with and without SLE |
In the longitudinal cohort analysis, the overall incidence of epilepsy was 2.86-fold higher in the SLE cohort than in the non-SLE cohort (9.10 per 10,000 person-years versus 3.18 per 10,000 person-years) (Table 2), with a multivariate Cox model measured adjusted HR (aHR) of 2.33 (95% CI =1.89–2.88). The SLE cohort-to-non-SLE cohort IRR of epilepsy was higher for women than men (IRR =3.24; 95% CI =3.12–3.37 versus IRR =1.88; 95% CI =1.70–2.08). However, the aHR was significantly higher for men when compared with women, with an aHR of 1.49 (95% CI =1.18–1.90) (data not shown).
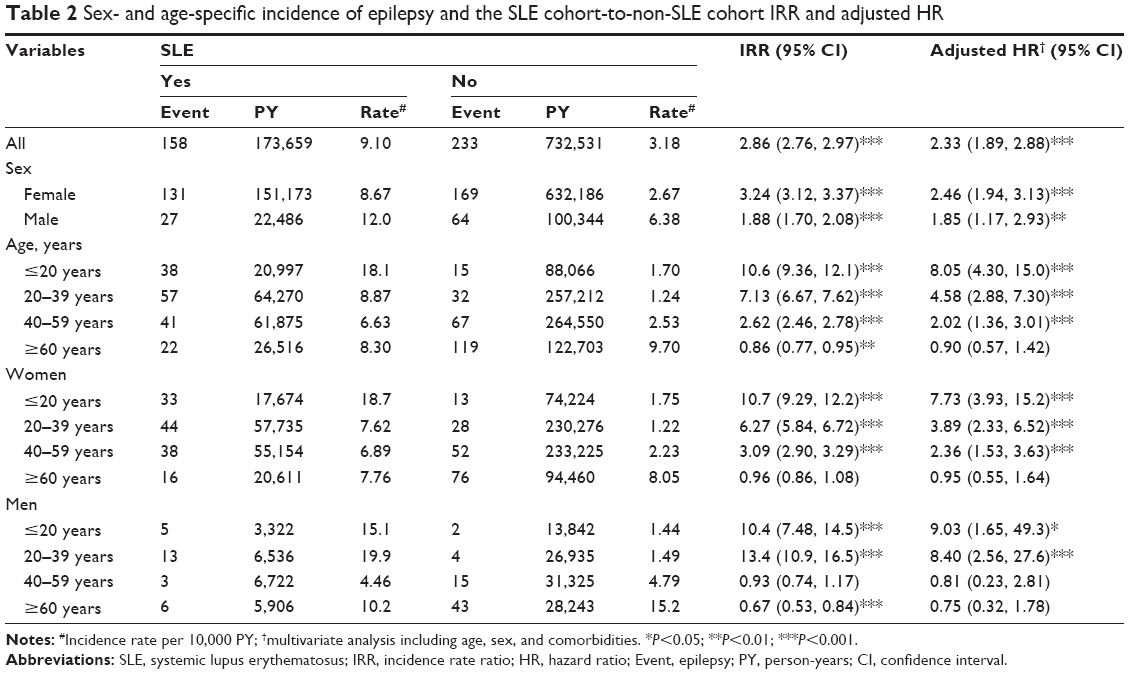 | Table 2 Sex- and age-specific incidence of epilepsy and the SLE cohort-to-non-SLE cohort IRR and adjusted HR |
We conducted a further data analysis to measure the epilepsy incidence by the type of epilepsy (generalized, partial, and unclassified seizures) (Table 3). Most cases were unclassified, with an SLE cohort-to-non-SLE cohort IRR of 2.74. The IRR was the highest for partial epilepsy. Table 4 shows that comorbidity increased the incidence of epilepsy. SLE patients with a comorbidity of intracerebral hemorrhage had the highest incidence of epilepsy (97.8 per 10,000 person-years). However, SLE patients with infarction had the highest aHR of 7.62; 95% CI of 5.87–2.58, followed by a comorbidity of intracerebral hemorrhage (aHR =5.75; 95% CI =4.03–8.19), aseptic meningoencephalitis (aHR =5.35; 95% CI =3.46–8.27), and psychiatric disorder (aHR =3.31; 95% CI =2.40–4.58).
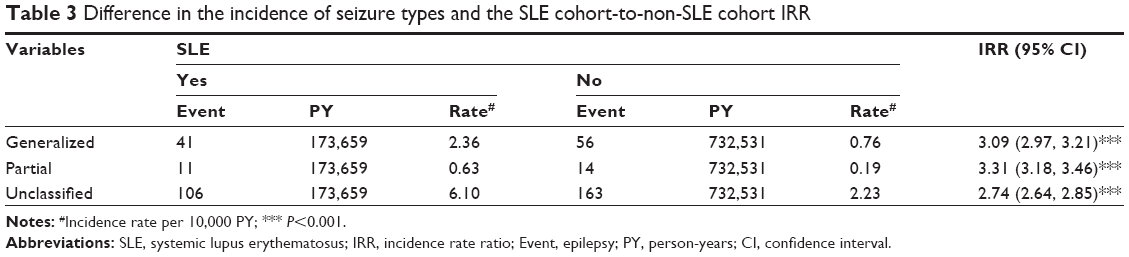 | Table 3 Difference in the incidence of seizure types and the SLE cohort-to-non-SLE cohort IRR |
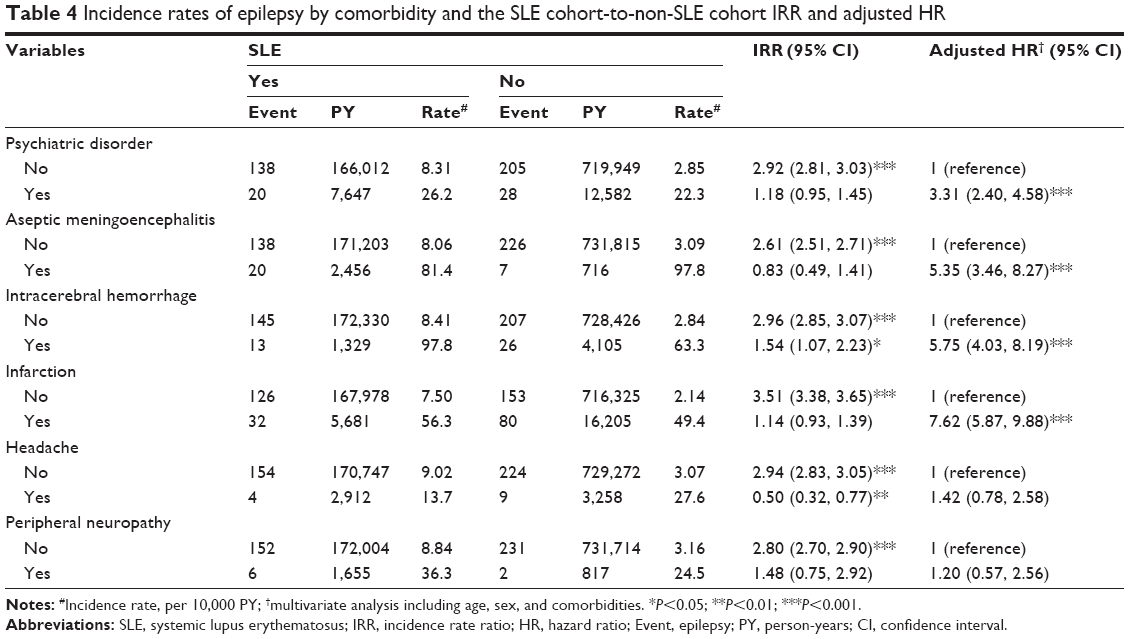 | Table 4 Incidence rates of epilepsy by comorbidity and the SLE cohort-to-non-SLE cohort IRR and adjusted HR |
The mean follow-up periods were 5.38 years in the SLE cohort and 5.67 years in the non-SLE cohort. Figures 2A–E show the age-specific cumulative proportional incidence of epilepsy by the end of follow-up, compared between the two cohorts. The SLE cohort had a significantly higher epilepsy incidence than that of the non-SLE cohort, which was measured for all subjects and for younger age groups (P<0.0001), except for the elderly. The difference was the greatest for the subcohort of <20 years of age, as it was 1.55% greater for the SLE subjects than for the non-SLE subjects (1.75% versus 0.20%, respectively; P<0.0001). One-third of epilepsy cases in the young SLE patients occurred within 2 years after the diagnosis of SLE.
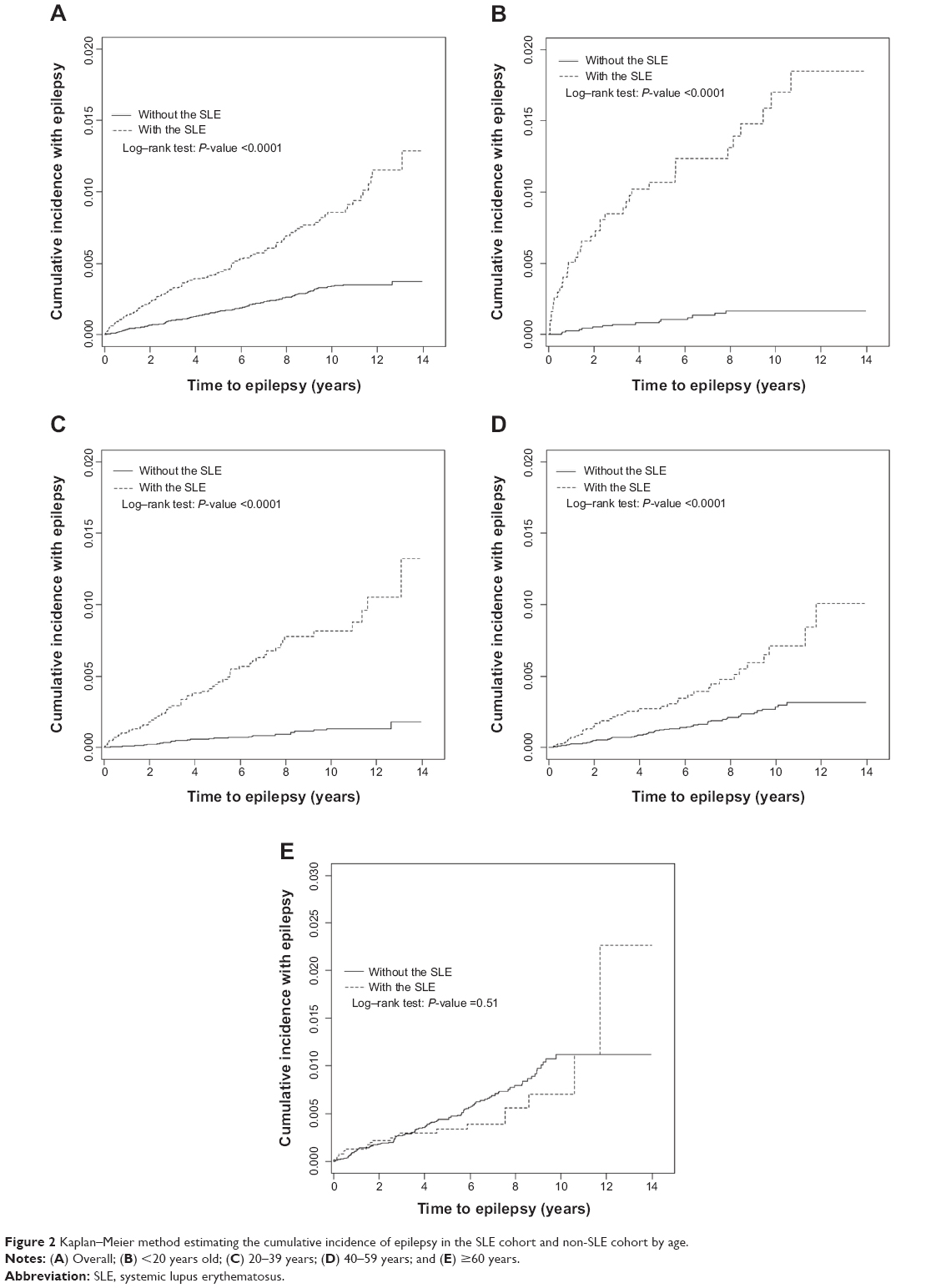 | Figure 2 Kaplan–Meier method estimating the cumulative incidence of epilepsy in the SLE cohort and non-SLE cohort by age. |
Discussion
The strength of the present study lies in using population data in order to represent the national population and to allow us to estimate the age-specific occurrences of epilepsy in both the general population and in patients with SLE. The rheumatologists and clinical physicians in Taiwan follow the 1997 ACR classification criteria for SLE and NPSLE, which standardizes the database.17 The retrospective follow-up design compared the epilepsy incidences between patients with SLE and the general population. Using the extended availability of claims data (with its high validity) also enables us to eliminate potential coding biases.18
Patients with lupus may suffer from various neurological symptoms, which contribute to a number of complications and mortality.6,19,20 Among the 19 ACR-defined neuropsychiatric symptoms, ranging from subtle signs of headache and mood disorders to severe life-threatening conditions of cerebrovascular accidents, myelopathy, and acute confusion, the risk of epilepsy needs adequate measurement.6–8 The primary central nervous system involvement for SLE patients is a challenge for diagnosis, as manifestations can be focal, diffuse, or a combination thereof.8,15 As a result, studies have focused on the neurological manifestations in order to reduce complications and mortality.19 The diagnosis of NPSLE concurrent with SLE is one of the most difficult challenges for clinicians because of the diversity of complications.7
Results of the retrospective cohort analysis in this study demonstrated an overall incidence of epilepsy that was nearly threefold higher in the SLE cohort than in the non-SLE cohort, though the severities of the disorders are not available from the claims database. Findings from this study confirm our hypothesis that, with a reference cohort, the epilepsy manifestation may occur at a high risk after SLE is diagnosed.21 Younger SLE patients are at the highest risk, and females are more vulnerable than males for the risk of SLE.
The pathophysiologic mechanisms of neuronal damage from seizures and psychotic episodes for SLE patients have been associated with inflammatory cytokines, autoantibodies, and specific and nonspecific immune complexes.14,22 SLE patients are thus at an elevated risk of vasculopathic, cytotoxic, and autoantibody-mediated neuronal injury. The consequent microvasculopathy is common in SLE patients, with microscopic brain findings mainly relating to complement activation and antiphospholipid antibodies (APS).23 The exact cause of APS has not been well clarified, but the activation of the coagulation system is evident.24 For this reason, secondary APS may cause blood to flow improperly and provoke blood clots in vessels leading to subsequent disorders in the central and peripheral nervous systems.25,26 Serum antineuronal antibodies are prevalent in patients with SLE suffering from neurological manifestations subsequent to encephalopathies and seizures.27
This study also shows that SLE patients are therefore more likely to suffer from strokes than those in the general population without lupus. The APS antibody may trigger the formation of blood clots and may attack cell membranes inside blood vessels, which travel to the brain and can cause embolic strokes.28 Inflammation inside blood vessel walls that causes vasculitis is particularly dangerous in the brain, and can lead to a stroke episode.29 However, therapeutic strategies recommended for NPSLE manifestations remain limited. Biologic agents targeting specific pathways have been proposed as new tools for SLE treatment. Among them, B-cell-targeted therapeutic experiments are considered to be at the forefront in the treatment of SLE. The open-label and randomized clinical trials have shown treatment efficacy for SLE, and treatments may also be potentially effective for NPSLE.30
It is important to note in the present study that the mean annual age-specific occurrences of epilepsy are different between SLE patients and the non-SLE general population. The incidence and prevalence rates of epilepsy slightly decrease with age in patients with SLE, while the incidence and prevalence rates of the disease sharply increase with age, beginning at 40–50 years of age in the general population. The U-shaped age-specific prevalence of epilepsy in the general population is consistent with findings from a previous study.31 A Finland study32 also found that epilepsy prevalence was 0.39 per 100 for 6–15-year-old children in the general population, which is comparable to the prevalence of 0.51 per 100 in 6–12-year-old children in Norway.33 Both prevalence rates are lower than our finding of 0.75 per 100 for children. An Italian study also found a U-shaped prevalence of epilepsy as a function of age that was much higher in children aged 5–14 years (5.05 per 1,000), which was approximately 1.7-fold higher than the average prevalence rate in the whole population.34 The age-specific analyses in our study are characterized by the cross-over in incidence and prevalence rates between SLE patients and the general population. In the retrospective cohort analysis, we found that children with SLE are at the greatest risk of developing epilepsy.
It is likely that the etiology of epilepsy is different between the general population and patients with SLE.35,36 In non-SLE subjects, the occurrence of epilepsy could mainly be due to other disorders and aging, especially starting at 40 years of age. Reproductive events, higher APS, and other psychiatric and neurodevelopmental conditions may have a stronger association with developing epilepsy events in the general population.36–38 Our study found that comorbidities increase the epilepsy risk dramatically, particularly among those with cerebrovascular disorders and aseptic meningoencephalitis. Intracerebral hemorrhage and infarction that increase with age may have greater contributions to the epilepsy risk in the older general population. The lower incidence and prevalence of epilepsy in older SLE patients may be due to higher mortality from SLE.
This study used a population dataset to conduct a natural history study evaluating the epilepsy risk for SLE patients. However, certain limitations are still present in this study. Although SLE is considered to be a severe catastrophic illness in Taiwan, and although patients are registered with the physician’s approval and evaluation by the National Bureau of Insurance, some patients are reluctant to apply for catastrophic care, and these patients may thus be included in the non-SLE cohort.39 However, the size of this population is likely to be insignificant. Furthermore, given that information on clinical conditions, laboratory data, images, and the severity of SLE may not all be available, we were unable to account for these factors in our evaluation of epilepsy risk. Another limitation is that information on SLE-related disease activity and damage was also unavailable from the insurance database. Given that we were unable to make a clear distinction between acute symptomatic and unprovoked epilepsy in this nationwide population-based study, residual confounding variables cannot be completely ruled out based on the available data. Although the ACR has defined 19 forms of NPSLE, the numbers of epilepsy cases identified in this study are not large enough to differentiate between how other manifestations are associated with the occurrence of epilepsy.
Despite the limitations, this study indicates that the age-specific incidence and prevalence of epilepsy tend toward opposite directions between SLE patients and the general population, which may reflect etiological variation. While the causes of occurrences of epilepsy are often difficult to determine, SLE is a known cause; the current study clearly showed that seizures tend to appear early in the course of SLE. Due to the often subtle and variable courses of NPSLE, clinical physicians are aware of the need to carefully search for the potential etiologies of undetermined neurological diseases. An epilepsy prevention program for young SLE patients deserves greater attention.
Acknowledgments
This study was supported by the Taiwan Department of Health (grant number DOH102-TD-B-111-004 and DOH102-TD-C-111-005), the National Science Council (grant numbers NSC 100-2621-M-039-001), and the China Medical University Hospital (grant number 1MS1).
Disclosure
The authors report no conflicts of interest in this work.
References
D’Cruz DP, Khamashta MA, Hughes GR. Systemic lupus erythematosus. Lancet. 2007;369(9561):587–596. | ||
Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med. 2008;358(9):929–939. | ||
Popescu A, Kao AH. Neuropsychiatric systemic lupus erythematosus. Curr Neuropharmacol. 2011;9(3):449–457. | ||
Benedict RH, Shucard JL, Zivadinov R, Shucard DW. Neuropsychological impairment in systemic lupus erythematosus: a comparison with multiple sclerosis. Neuropsychol Rev. 2008;18(2):149–166. | ||
Andrade RM, Alarcón GS, González LA, et al. LUMINA Study Group. Seizures in patients with systemic lupus erythematosus: data from LUMINA, a multiethnic cohort (LUMINA LIV). Ann Rheum Dis. 2008; 67(6):829–834. | ||
The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999; 42(4):599–608. | ||
Buća A, Perković D, Martinović-Kaliterna D, Vlastelica M, Titlić M. Neuropsychiatric systemic lupus erythematosus: diagnostic and clinical features according to revised ACR criteria. Coll Antropol. 2009;33(1):281–288. | ||
Appenzeller S, Cendes F, Costallat LT. Epileptic seizures in systemic lupus erythematosus. Neurology. 2004;63(10):1808–1812. | ||
Petri M, Orbai AM, Alarcón GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum. 2012;64(8):2677–2686. | ||
Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. | ||
Hanly JG, Urowitz MB, Su L, et al. Seizure disorders in systemic lupus erythematosus results from an international, prospective, inception cohort study. Ann Rheum Dis. 2012;71(9):1502–1509. | ||
Honczarenko K, Budzianowska A, Ostanek L. Neurological syndromes in systemic lupus erythematosus and their association with antiphospholipid syndrome. Neurol Neurochir Pol. 2008;42(6):513–517. | ||
Govoni M, Bombardieri S, Bortoluzzi A, et al; Italian Society of Rheumatology. Factors and comorbidities associated with first neuropsychiatric event in systemic lupus erythematosus: does a risk profile exist? A large multicentre retrospective cross-sectional study on 959 Italian patients. Rheumatology (Oxford). 2012;51(1):157–168. | ||
Shorvon SD. The etiologic classification of epilepsy. Epilepsia. 2011; 52(6):1052–1057. | ||
West SG. Lupus and the central nervous system. Curr Opin Rheumatol. 1996;8(5):408–414. | ||
Cheng T-M. Taiwan’s National health insurance system: high value for the dollar. In: Okma KGH, Crivelli L, eds. Six countries, six reform models: the health reform experience of Israel, The Netherlands, New Zealand, Singapore, Switzerland and Taiwan. Hackensack, NJ: World Scientific, 2009:171–204. | ||
Yu HH, Lee JH, Wang LC, Yang YH, Chiang BL. Neuropsychiatric manifestations in pediatric systemic lupus erythematosus: a 20-year study. Lupus. 2006;15(10):651–657. | ||
Fang K, Shia BC, Ma S. Health insurance coverage, medical expenditure and coping strategy: evidence from Taiwan. BMC Health Serv Res. 2012;12:442. | ||
Souza DC, Santo AH, Sato EI. Mortality profile related to systemic lupus erythematosus: a multiple cause-of-death analysis. J Rheumatol. 2012;39(3):496–503. | ||
Bernatsky S, Boivin JF, Joseph L, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006;54(8):2550–2557. | ||
Rai D, Kerr MP, McManus S, Jordanova V, Lewis G, Brugha TS. Epilepsy and psychiatric comorbidity: a nationally representative population-based study. Epilepsia. 2012;53(6):1095–1103. | ||
Vincent A, Crino PB. Systemic and neurologic autoimmune disorders associated with seizures or epilepsy. Epilepsia. 2011;52(Suppl 3):12–17. | ||
Sciascia S, Murru V, Sanna G, Roccatello D, Khamashta MA, Bertolaccini ML. Clinical accuracy for diagnosis of antiphospholipid syndrome in systemic lupus erythematosus: evaluation of 23 possible combinations of antiphospholipid antibody specificities. J Thromb Haemost. 2012;10(12):2512–2518. | ||
Sokol DK, O’Brien RS, Wagenknecht DR, Rao T, McIntyre JA. Antiphospholipid antibodies in blood and cerebrospinal fluids of patients with psychosis. J Neuroimmunol. 2007;190 (1–2):151–156. | ||
Borowoy AM, Pope JE, Silverman E, et al. Neuropsychiatric lupus: the prevalence and autoantibody associations depend on the definition: results from the 1,000 faces of lupus cohort. Semin Arthritis Rheum. 2012;42(2):179–185. | ||
Rand JH. Antiphospholipid antibody syndrome: new insights on thrombogenic mechanisms. Am J Med Sci. 1998;316(2):142–151. | ||
Cojocaru IM, Cojocaru M, Botnaru L, Miu G, Sapira V, Tănăsescu R. Detection of serum of IgG anti-neuronal antibodies in systemic lupus erythematosus patients with central nervous system manifestations. Rom J Intern Med. 2010;48(3):267–269. | ||
Belmont HM, Abramson SB, Lie JT. Pathology and pathogenesis of vascular injury in systemic lupus erythematosus. Interactions of inflammatory cells and activated endothelium. Arthritis Rheum. 1996; 39(1):9–22. | ||
Kampylafka EI, Alexopoulos H, Kosmidis ML, et al. Incidence and prevalence of major central nervous system involvement in systemic lupus erythematosus: a 3-year prospective study of 370 patients. PLoS One. 2013;8(2):e55843. | ||
Murdaca G, Colombo BM, Puppo F. Emerging biological drugs: a new therapeutic approach for systemic lupus erythematosus. An update upon efficacy and adverse events. Autoimmun Rev. 2011;11(1):56–60. | ||
Chen CC, Chen LS, Yen MF, Chen HH, Liou HH. Geographic variation in the age- and gender-specific prevalence and incidence of epilepsy: analysis of Taiwanese National Health Insurance-based data. Epilepsia. 2012;53(2):283–290. | ||
Eriksson KJ, Koivikko MJ. Prevalence, classification, and severity of epilepsy and epileptic syndromes in children. Epilepsia. 1997; 38(12):1275–1282. | ||
Waaler PE, Blom BH, Skeidsvoll H, Mykletun A. Prevalence, classification, and severity of epilepsy in children in western Norway. Epilepsia. 2000;41(7):802–810. | ||
Gallitto G, Serra S, La Spina P, et al. Prevalence and characteristics of epilepsy in the Aeolian islands. Epilepsia. 2005;46(11):1828–1835. | ||
González-Duarte A, Cantú-Brito CG, Ruano-Calderón L, García-Ramos G. Clinical description of seizures in patients with systemic lupus erythematosus. Eur Neurol. 2008;59(6):320–323. | ||
Adelöw C, Andersson T, Ahlbom A, Tomson T. Unprovoked seizures in multiple sclerosis and systemic lupus erythematosus: a population-based case-control study. Epilepsy Res. 2012;101(3):284–287. | ||
Nicoletti A, Reggio A, Bartoloni A, et al. Prevalence of epilepsy in rural Bolivia: a door-to-door survey. Neurology. 1999;53(9):2064–2069. | ||
Shoenfeld Y, Lev S, Blatt I, et al. Features associated with epilepsy in the antiphospholipid syndrome. J Rheumatol. 2004;31(7):1344–1348. | ||
Lai NS, Tsai TY, Koo M, Huang KY, Tung CH, Lu MC. Patterns of ambulatory medical care utilization and rheumatologist consultation predating the diagnosis of systemic lupus erythematosus: a national population-based study. PLoS One. 2014;9(7):e101485. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.