")
Back to Journals » International Journal of Nanomedicine » Volume 17
Rhodojaponin III-Loaded Chitosan Derivatives-Modified Solid Lipid Nanoparticles for Multimodal Antinociceptive Effects in vivo
Authors Yang Q, Yang J, Sun S, Zhao J, Liang S, Feng Y, Liu M, Zhang J
Received 14 February 2022
Accepted for publication 4 August 2022
Published 16 August 2022 Volume 2022:17 Pages 3633—3653
DOI https://doi.org/10.2147/IJN.S362443
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yan Shen
Qingyun Yang,* Jian Yang,* Shuigen Sun, Jingyi Zhao, Shuang Liang, Yi Feng, Minchen Liu, Jiquan Zhang
Engineering Research Center of Modern Preparation Technology of TCM of Ministry of Education, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yi Feng; Jiquan Zhang, Engineering Research Center of Modern Preparation Technology of TCM of Ministry of Education, Shanghai University of Traditional Chinese Medicine, 1200 Cailun Road, Shanghai, 201203, People’s Republic of China, Tel +86-21-51322431 ; +86 13816062506, Email [email protected]; [email protected]
Background: Rhodojaponin III (RJ-III) is a bioactive diterpenoid, which is mainly found in Rhododendron molle G. Don (Ericaceae), a potent analgesia in traditional Chinese medicine with several years of clinical applications in the country. However, its clinical use is limited by its acute toxicity and poor pharmacokinetic profiles. To reduce such limitations, the current study incorporated RJ-III into the colloidal drug delivery system of hydroxypropyl trimethyl ammonium chloride chitosan (HACC)-modified solid lipid nanoparticles (SLNs) to improve its sustained release and antinociceptive effects in vivo for oral delivery.
Results: The optimized RJ-III@HACC-SLNs were close to spherical, approximately 134 nm in size, and with a positive zeta potential. In vitro experiments showed that RJ-III@HACC-SLNs were stable in the simulated gastric fluid and had a prolonged release in PBS (pH = 6.8). Pharmacokinetic results showed that after intragastric administration in mice, the relative bioavailability of RJ-III@HACC-SLNs was 87.9%. Further, it was evident that the peak time, half-time, and mean retention time of RJ-III@HACC-SLNs were improved than RJ-III after the administration. In addition, pharmacodynamic studies revealed that RJ-III@HACC-SLNs markedly reduced the acetic acid, hot, and formalin-induced nociceptive responses in mice (P < 0.001), and notably increased the analgesic time (P < 0.01). Moreover, RJ-III@HACC-SLNs not only showed good biocompatibility with Caco-2 cells in vitro but its LD50 value was also increased by 1.8-fold as compared with that of RJ-III in vivo.
Conclusion: These results demonstrated that RJ-III@HACC-SLNs improved the pharmacokinetic characteristics of the RJ-III, thereby exhibiting toxicity-attenuating potential and antinociceptive enhancing properties. Consequently, HACC-SLNs loaded with RJ-III could become a promising oral formulation for pain management that deserves further investigation in the future.
Keywords: rhodojaponin III, multimodal antinociceptive, oral administration, solid lipid nanoparticles, hydroxypropyl trimethyl ammonium chloride chitosan, pharmacokinetic
Graphical Abstract:

Introduction
Pain, an unpleasant emotional and sensory experience,1,2 is the most common symptom for seeking medical care.3,4 With regards to disability, prevalence, and economic burden, unrelieved pain is a major human health concern.5,6 It is estimated that the age-standardized prevalence of chronic pain conditions 12–25% in developed countries and 34–41% in developing countries.7,8 The available pain management strategies heavily rely on agents with analgesic properties, such as opioids, non-steroidal anti-inflammatory drugs, antidepressants, and anticonvulsant agents.4 These analgesic drugs are extensively used in clinical; however, only about one in four of individuals with pain achieves satisfactory relief.9 Moreover, these drugs are associated with harmful side effects, poor tolerability, long-term safety concerns, and the potential for abuse, as well as the inconvenience of use.9 Therefore, it is necessary and important for new non-opioid analgesic agents that are of high efficacy.
For thousands of years, Rhododendron molle G. Don has been clinically used to treat pain in China.10 Rhodojaponin III (RJ-III) is a grayanane-type diterpenoid isolated from the herb that shows extensive biological activities11–13 such as antinociceptive effects,14 anti-rheumatoid arthritis,15 lowering the blood pressure,16 slowing the heart rate,16 and inhibiting inflammatory response.17 Notably, RJ-III exhibits more efficacy as compared with morphine in both acute and inflammatory pain models.14 In diabetic neuropathic pain models, its potency was 100-fold that of the first-line drug gabapentin.14 In addition, naloxone, a morphine antagonist, does not exert significant antagonism on the analgesic effects of RJ-III in an acetic acid-mediated writhing test.14 Accordingly, based on the previously conducted studies, it is evident that RJ-III is a potential non-opioid analgesic for pain management. However, when it was orally administered, RJ-III showed severe acute toxicity with LD50 of 7.609 mg/kg in mice.18 We studied the pharmacokinetic properties of RJ-III in mice, and the results showed that the Tmax was 0.08 h, t1/2 was 0.76 h, and oral absolute bioavailability was 73.6%, which showed RJ-III possessed characteristics with rapid absorption, rapid elimination, and good oral bioavailability.19 Further, the results suggested that an oral drug delivery system with sustained-release capability could provide better antinociceptive effects and reduced acute toxicity of RJ-III by prolonging the action time and reducing the fluctuation of plasma concentration.
Currently, colloidal drug delivery systems have emerged as prominent solutions to enhance the oral pharmacokinetic characteristics of therapeutic agents and their bioavailability.20,21 Solid lipid nanoparticles (SLNs), a colloidal drug delivery system, have received more attention than other nano-delivery systems in oral administration owing to prolonging drug release by the hindrance of solid lipid shells, enhancing absorption through lymphatic delivery through microfold cells, and extensive drug payload characteristics.20–22 Although conventional SLNs are associated with various advantages, its oral delivery application is limited by its burst release behavior under acidic conditions in stomach and enzymatic degradation in the gastrointestinal tract.23–26 Moreover, in some cases, electrostatic repulsion between the negatively charged surface of SLNs and those of mucus membranes hinders, which might limit drug absorption resulting in decreased cellular uptake, and oral bioavailability.24–26 Therefore, it is crucial to develop effective and feasible surface-modified SLNs to prevent the burst release and maintain appropriate oral availability of RJ-III.
Chitosan is an abundant natural polymer of great interest in biomedical applications. It has been used in a wide range of applications of drug delivery such as nanoparticle, microspheres, and membranes, because it is biodegradable, non-toxic, and biocompatible.27–30 Hydroxypropyl trimethyl ammonium chloride chitosan (HACC) is a quaternary ammonium chitosan derivative,31 which is more beneficial for oral delivery than conventional chitosan only dissolves in acidic solutions.32 It displays aqueous solubility behavior pH-independent, is electropositive, biocompatible and mucoadhesive.31–35 Furthermore, HACC acts as a stabilizer at acid pH and an absorption enhancer in oral delivery.32 A previous study revealed that HACC-modified SLNs enhance gastrointestinal stability of nanoparticles and oral bioavailability of docetaxel as compared with chitosan-modified SLNs.32 Nowadays, HACC can potentially be used in various fields as its quaternized cationic nature allows strong electrostatic interactions with negative charges.31–35 However, the application of HACC-modified SLN both in loading RJ-III and in analgesia has not been reported.
In this study, a drug delivery system of RJ-III@HACC-SLNs was for the first time developed to improve the pharmacokinetics, antinociceptive effect, and safety of RJ-III. Meanwhile, the carrier materials were further optimized to ensure stable delivery of RJ-III in the gastrointestinal environment. Pharmacokinetic experiments were also conducted to investigate whether RJ-III@HACC-SLNs has sustained-release effects. In addition, the anti-nociceptive effects of RJ-III@HACC-SLNs were examined through multimodal experiments on using acetic acid-mediated writhing and the hot-plate tests to simulate acute pain models, whereas the formalin test was used to simulate both and persistent inflammatory pain. The safety of RJ-III@HACC-SLNs was also investigated through cytotoxicity test of Caco-2 cells in vitro and acute toxicity test on mice in vivo. Obtained results of the present study will help evaluate whether the protonation, and sustained-release effect of HACC-SLNs can prolong the antinociceptive time of RJ-III in vivo, as well as provide a new perspective towards an effective oral drug delivery system and analgesic treatment.
Materials and Methods
Materials
Standard rhodojaponin III (>98% purity) was acquired from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China); Rhodojaponin III was obtained from the Catch Bio-Science & Technology Co., Ltd. (Jiangsu, China) with purity of more than 92%. PC-98T egg yolk lecithin (AL15018, purity = 98%) was procured from A.V.T. Pharmaceutical Co., Ltd (Shanghai, China). Glycerol monostearate (GM), glyceryl tristearate (GT), stearic acid (SA), and Tween® 80 were bought from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Precirol ATO 5 (ATO 5) and Compritol 888 ATO (888 ATO) were gifts from GATTEFOSSé (Saint-Priest, France). HACC (Lushen Bioengineering Co, Ltd. (Nantong, China)), Dulbecco’s Modified Eagle’s Medium (DMEM), penicillin/streptomycin, fetal bovine serum (FBS), Hank’s Balanced Salt Solution (HBSS), and 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) were the products of Thermo Fisher (Massachusetts, USA).
Cell Culture
Caco-2 Cell lines were purchased from American Type Culture Collection (Virginia, USA) and cultured in 10% (v/v) FBS-supplemented DMEM with 1% penicillin–streptomycin as well as 1% (v/v) non-essential amino-acids in a 5% CO2, 90% relative humidity atmosphere at 37°C. Medium change was carried out every other day, while cell passaging was performed every 4–6 days through dissociation utilizing trypsin (0.25%) – EDTA (0.02%) solution.
Animals
The ICR female and male mice (Grade II, 18–22 g) were obtained from Zhejiang Wei-Tong-Li-Hua laboratory animal technology Co. LTD (Zhejiang, China), which has a production license number of SCXK (Zhejiang) 2019-0001. Mice were kept in air-conditioned rooms at 22–24°C, 12 h light/dark cycle and provided with food and water ad libitum. Prior to experiments, mice were fasted overnight. Animal experiments were conducted according to the Animal Welfare Act, and the experimental procedures were approved by the Institutional Animal Care and Use Committee of Shanghai University of Traditional Chinese Medicine (Approval No. PZSHUTCM211101032).
Preparation of RJ-III@HACC-SLNs
Rhodojaponin III-loaded solid lipid nanoparticles (RJ-III@SLNs) were prepared through an emulsification-diffusion approach with minor modification.36 Before the optimization, the main preparation process was as follows: 20 mg egg yolk lecithin and 3 mg RJ-III were completely dissolved in absolute alcohol and thereafter mixed with 30 mg glycerol monostearate (GM) to form an oil phase. Vacuum rotary evaporation was performed to remove the organic solvent to obtain a lipid film layer. Under sustained ultrasound, 10 mL water phase supplemented with 0.2% (w/v) Tween-80 was added to the lipid film in 30 min using a needle. The RJ-III@SLNs were obtained after intermittent sonication using a probe sonicator (Xinzhi, Ningbo, China) at 400 w for 4 min (2s/3s). RJ-III@HACC-SLNs was obtained by binding HACC to the surface of RJ-III@SLNs through electrostatic adsorption.37 Briefly, 4 mL RJ-III@SLNs was added to 8 mL HACC solution (0.1%, w/v), followed by further stirring for 1 h.
Optimization of RJ-III@HACC-SLNs
The single-factor experiment and central composite design/response surface method were conducted to develop efficient RJ-III@HACC-SLNs. First, the parameters in the modification process of RJ-III@HACC-SLNs, including volume ratio of HACC to RJ-III@SLNs, stirring time, were optimized by single-factor investigation. The particle size, zeta potential, and encapsulation efficiency (EE) were used as the evaluation index. The core lipids were then selected by comparing the EE, zeta potential, and particle size of RJ-III@HACC-SLNs based on the five common and different molecular weights used in solid lipids (GM, GT, SA, ATO 5, and 888 ATO). Table 1 shows the formulations of the SLNs. Importantly, the amount of lipid-to-drug ratio, the mass ratio of co-surfactant (CoS) to surfactant (S), and the volume ratio of HACC to RJ-III@SLNs were optimized using central composite design/response surface method (Table 2).
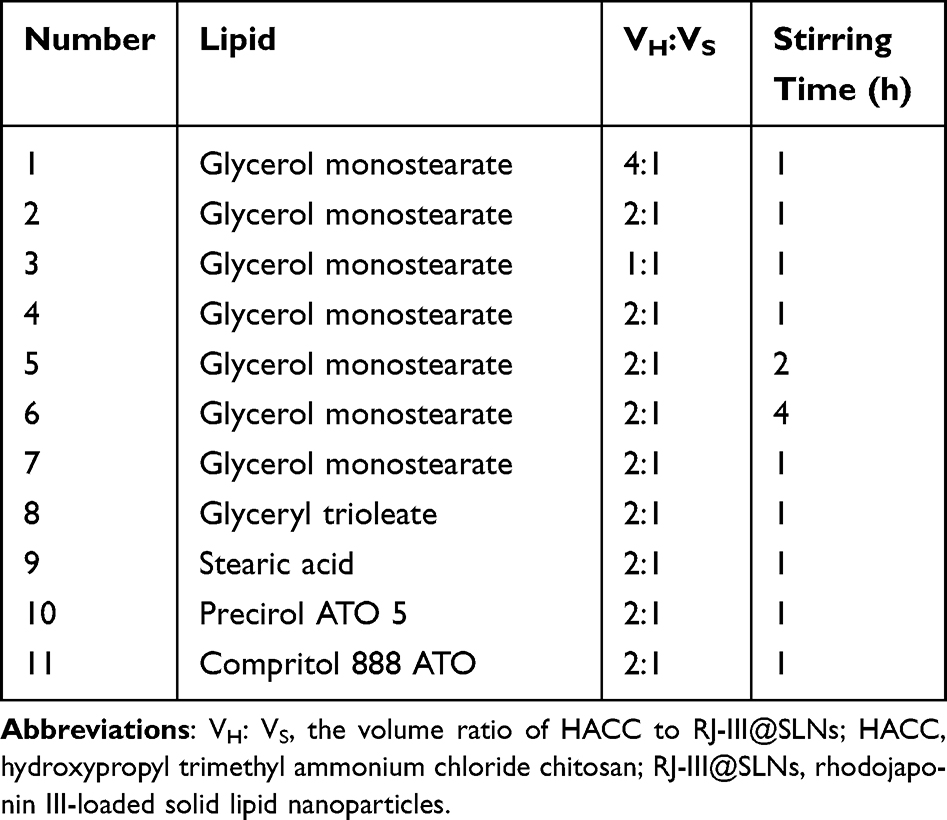 |
Table 1 Formulations of the Various Solid Lipid Nanoparticles |
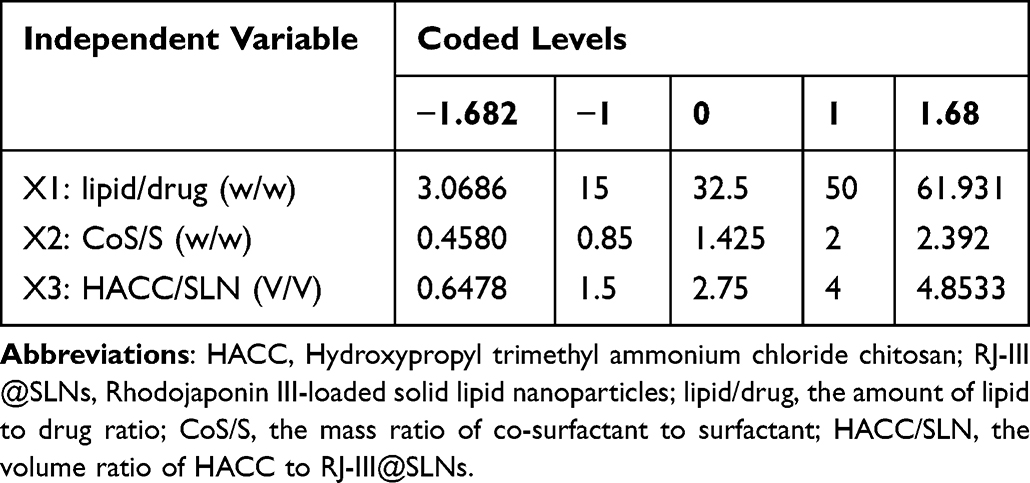 |
Table 2 Independent Variables and Their Levels of Central Composite Design-Response Surface Method |
Characterization of RJ-III@HACC-SLNs
The morphology of RJ-III@HACC-SLNs was assessed through transmission electron microscopy (FEI Talos, Thermo Fisher Scientific, USA). In addition, the zeta potentials and particle sizes were measured using a Zeta Potential/Particle Sizer (Nicomp 380 ZLS, PSS⋅NICOMP, USA). The changes in solid-state forms of RJ-III in SLNs were evaluated by X-ray diffractograms (Rikagu, D/Max-3C, Japan). FT-IR spectrophotometer (IRAffinity-1S, SHIMADZU, Japan) was used to evaluate the surface chemical structure of RJ-III, SLNs, RJ-III@SLNs, RJ-III@HACC-SLNs, to assess the surface characterization of RJ-III@HACC-SLNs. The EE of RJ-III@HACC-SLNs was assessed by high-performance liquid chromatography (HPLC) and ultrafiltration centrifugation. Briefly, the unencapsulated RJ-III was isolated from RJ-III@HACC-SLNs through a 20 kDa ultrafiltration centrifuge tube (Millipore, USA) at 10,000 rpm for 30 min. The total amount of drug in drug-loaded SLNs was evaluated by dissolving SLNs in methanol to release encapsulated RJ-III, and assayed by HPLC. The conditions for the HPLC system (Agilent-1260B, Agilent, USA) equipped with an evaporative light scattering detector (Agilent-G460B, Agilent, USA) were as follows: CAPCELL PAK C18 column (4.6 × 150 mm, 5 μm); mobile phase, acetonitrile–water (30:70, v/v, 1.0 mL/min); injection temperature, 25°C; evaporation temperature, 60°C; atomization temperature, 30°C; sample volume, 20 μL. The equation for calculating EE is
EE% = m2/m1 × 100%
where m1 represents the amount of total drug in RJ-III@HACC-SLNs suspension and m2 represents the amount of drug loaded in HACC-SLNs.
In vitro Stability and Release of RJ-III@HACC-SLNs
The stability in vitro of RJ-III@SLNs and RJ-III@HACC-SLNs was studied according to previous reports.37 Incubation of nanoparticles was done at 37°C in simulated gastric fluid (SGF). Samples were obtained at 0, 1, 2, and 3 h, and assessed for changes in EE. SGF was constituted using 2 g sodium chloride, 36.5% 7 mL hydrochloric acid, and pepsin (3.2 g) in 1000 mL of water. The in vitro release investigation of RJ-III@HACC-SLNs, RJ-III@SLNs, and RJ-III was performed in PBS of pH 6.8 by the dialysis bag (molecular weight cutoff 8–14 kDa) diffusion technique. The test samples were stored inside the bag (equivalent to 3 mg RJ-III), dipped into 200 mL medium at 37 ± 0.5°C in a conical flask with a stirring speed of 100 rpm. Two-milliliter samples were taken and instantly replaced with an equal volume of fresh release medium at pre-set time 0.25, 0.5, 1, 2, 4, 8, 12, 24 h. Amounts of RJ-III in the medium were also determined using the HPLC method.
Pharmacokinetic Studies
The RJ-III@HACC-SLNs’ oral absorption was evaluated by pharmacokinetic parameters, and the pharmacokinetic study was performed in mice. The mice were randomized into several groups (6 mice per group) and intragastrically administered with RJ-III or RJ-III@HACC-SLNs containing RJ-III 0.2 mg/kg.19 With regards to pharmacokinetic assays, mice have anesthetized using diethyl ether at 0.033, 0.083, 0.25, 0.5, 1, 2, 3, 4, 6, and 8 h time points, after administration, venous blood samples were obtained from saphenous veins of the thighs into tubes containing EDTA-K2. Plasma samples were obtained by centrifugation (6000 rpm, 8 min, 4°C), and were then quantified according to the LC-MS/MS method previously studied by the research group.19 Chromatographic separation was performed using an ACQUITY UPLC HSS T3 (1.8 µm, 2.1×50 mm) reverse-phase column (Waters technology (Shanghai) Co., LTD (Shanghai, China)). The column was equipped with an AF0-8497 guard column (Phenomenex, CA, USA) and maintained at room temperature. The flow rate and sample injection volume were 0.5 mL/min and 10 μL, respectively.19 A non-compartmental analysis was conducted using the WinNonlin®8.2.0 software (Pharsight, CA, USA) to obtain pharmacokinetic parameters.
Multimodal Analgesia Studies of RJ-III@HACC-SLNs
The multimodal analgesic effects of RJ-III@HACC-SLNs were determined using the acetic acid writhing and hot plate, and formalin tests. The acetic acid writhing test was separated into two parts. In the first part, 40 mice were assigned into 4 groups (n = 10) and pretreated with RJ-III@HACC-SLNs (0.10 mg/kg of RJ-III, i.g), RJ-III (0.10 mg/kg, i.g), aspirin (200 mg/kg, i.g) or normal saline (0.9% NaCl, i.g). After 15 min, mice were administered with 0.8% acetic acid (10 mL/kg, i.p) and the nociception intensity evaluated by counting the number of abdominal contortions, such as abdominal muscle contractions and extensions of hind paws for 30 min. In the second part of the experiment as before, but the time of acetic acid intervention was 45 min after administration. The pharmacodynamics of every preparation was evaluated by comparing the number of writhing observed in mice.
In the hot plate test, first, each mouse was placed thrice on the heated plate 53 ± 0.5°C at a 15 min interval to obtain a basal pain threshold which is the reaction time about paw lick time or jump. Mice with reaction <5 s or >30 s longer were omitted. Then, the animals (n = 10) received RJ-III@HACC-SLNs (contain 0.20 mg/kg RJ-III, i.g), RJ-III (0.20 mg/mg, i.g), aspirin (200 mg/kg, i.g) or normal saline (0.9% NaCl, i.g). Reaction times were evaluated at 15 and 45 min after administration, with 30 s as the cutoff time to avoid injury to the paw.
The formalin test was divided into two parts similar to acetic acid writhing test. In the first part, the mice (n = 10) were treated with RJ-III@HACC-SLNs (contain 0.10 mg/kg RJ-III, i.g), RJ-III (0.10 mg/kg, i.g), aspirin (200 mg/kg, i.g) or normal saline (0.9% NaCl, i.g) 15 min before 25 μL of 2.5% formalin subcutaneous injection. The licking as well as biting time about the injected left hind paw, indicating pain, was recorded from 0 to 5 min (phase I, neurogenic phase) and from 15 to 30 min (Phase II, inflammatory phases). Findings were presented as licking time, in seconds (s). The second part of the experiment was carried out as described earlier, but the time of formalin intervention was 45 min after administration. Finally, the antinociceptive effect of each preparation was determined by comparing the time of licking and biting for the different sets of experiments.
In vitro Cytotoxicity
The biocompatibility of RJ-III@HACC-SLNs was assessed by MTT on Caco-2 cells.38 In the cytotoxicity study, the Caco-2 cells were cultured at 1.5×105 cells/well in 96-well plates and incubated for form cell layers. Test solutions were assigned into five groups of Blank SLNs (SLNs), Blank SLNs modified by HACC (HACC-SLNs), RJ-III, RJ-III@SLNs, and RJ-III@HACC-SLNs. The cell layers were obtained and rinsed 3 times using HBSS. Then, cells were incubated with 200 μL of various concentrations of blank nanocarriers (1, 2, 5, 10, 50, 100, and 500 µg/mL), and nano-formulations with RJ-III at different doses (total amount of RJ-III in SLNs of 0.1, 0.2, 0.5, 1, 2, 5, and 10 µg/mL) for 24 h. Cell layers treated with blank culture medium (200 μL) were the controls of 100% viability. After incubation, the addition of MTT solution (20 μL; 5 mg/mL) was followed by further incubation for 4 h. Subsequently, the MTT dye was removed from wells and 200 µL of dimethyl sulfoxide was added to each well to solubilize the formazan crystals. The results were quantified using a microplate reader at 570 nm.
Cell viability % = OD2/OD1 × 100%
where OD1 represents the absorbance intensity of the untreated cells, and OD2 represents the absorbance intensity of the treated cells.
In vitro Fluorescent Live/Dead Assay
Calcein-AM/PI double staining kit (KeyGen, China) was used for live/dead staining of cells to qualitatively investigate the effect of RJ-III, RJ-III@SLNs and RJ-III@HACC-SLNs on the cell viability of Caco-2 cells. Briefly, cells were treated with various concentrations of samples in 12-well plates. After 4 h of treatment, cells were stained with a mixture of Calcein-AM and PI staining for live and dead cells. The staining solution was replaced by PBS after 15 min. Live cells appeared green, whereas dead cells appeared red. The stained specimens were observed and imaged under a fluorescent microscope.
In vivo Acute Toxicity Test
To evaluate the safety of RJ-III@HACC-SLNs in vivo, acute lethal characteristics were assayed as previously reported, with minor changes.14 Briefly, after 3 days of adaptation, 80 mice were randomized into 10 groups (n = 8). Ever group was administered RJ-III (1.711, 2.312, 3.125, 4.223, and 5.707 mg/kg, obtained from our previous studies) or RJ-III@HACC-SLNs (total amount of RJ-III in SLNs of 3.423, 4.625, 6.250, 8.446, and 11.413 mg/kg, based on a preliminary experiment) by single intragastric administration, and cumulative mortality within 7 days was recorded to calculate median lethal dose (LD50) by the Bliss method.39
Statistical Analyses
Results were expressed as mean ± standard error (n = 3). The one-way or two-way ANOVA was performed using Origin 2021b software. P < 0.05, P < 0.01 and P < 0.001 were the set significance thresholds.
Results
Prepared and Optimization of RJ-III@HACC-SLNs
RJ-III@SLNs were successfully prepared by emulsification-diffusion.36 Then, the RJ-III@HACC-SLNs were obtained as recognized as a clear solution by electrostatic adsorption between RJ-III@SLNs and HACC solution, as shown in Figure 1. In a preliminary study, variables such as the volume ratio of VH to VS, stirring time, and solid lipid that influences the particle size, zeta potential, and the EE of RJ-III@HACC-SLNs were investigated by the single-factor experiment. Previous studies have shown that the nanoparticles considered as members of the nanotoxicological classification system class I with particle size of above 100 nm and made of biodegradable materials,40 zeta potential ± 15 mV or higher ensures adequate repulsion of the nearby nanoparticles in the suspension.36,41 Therefore, the optimization principle of RJ-III@HACC-SLNs was to select the option with the best EE when the particle size was above 100 nm while the zeta potential was higher than 15 mV.
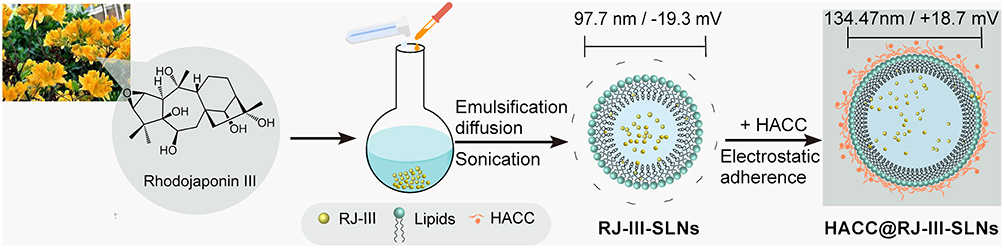 |
Figure 1 The preparation procedure of RJ-III@HACC-SLNs. Abbreviations: RJ-III, rhodojaponin III; HACC, hydroxypropyl trimethyl ammonium chloride chitosan; RJ-III@SLNs, rhodojaponin III-loaded solid lipid nanoparticles; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles. |
As shown in Figure 2A-C, in the modification process, the EE, zeta potential and particle size of RJ-III@HACC-SLNs were affected by the different volume ratios of VH to VS In particle size, it can be concluded that particle size decreases with the decrease of volume ratios of VH to VS Further analysis revealed that the 2:1 group was significantly better than the 4:1 group and the 1:1 group in EE and zeta potential. Besides, as shown in Figure 2D-F, in stirring time, it was inferred that increasing the time decreased the lower EE and higher zeta potential of nanoparticles. In terms of particle size, there was a significant difference between the 1 h group and the 2 h group (P < 0.01). Meanwhile, the zeta potential of RJ-III@HACC-SLNs was above 15 mV within 1–4 h, but the zeta potential of 2 h and 4 h groups was markedly higher relative to that of 1 h group (P < 0.001, P < 0.001). However, in terms of EE, the 1 h group was markedly higher than those of the 2 h and 4 h groups (P < 0.001). Therefore, 1 h was chosen as the optimal stirring time.
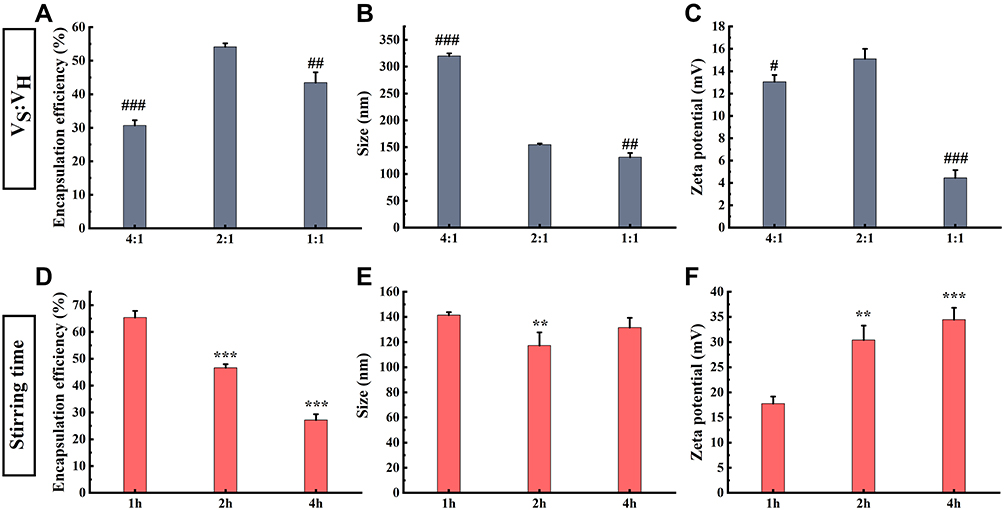 |
Figure 2 The optimized modification process of RJ-III@HACC-SLNs (Mean ± SD, n = 3). Influences of various volume ratios of HACC to RJ-III@HACC-SLNs on encapsulation efficiency (A), particle size (B), and zeta potential (C) of RJ-III@HACC-SLNs. The effects of different stirring time encapsulation efficiency (D), particle sizes (E), and zeta potentials (F) of RJ-III@HACC-SLNs. #P < 0.05, ##P < 0.01, ###P < 0.001 compared with the 2:1 group; **P < 0.01, ***P < 0.001 compared with the 1 h group. Abbreviations: HACC, hydroxypropyl trimethyl ammonium chloride chitosan; RJ-III@SLNs, rhodojaponin III-loaded solid lipid nanoparticles; VH: VS, the volume ratio of HACC to RJ-III@SLNs; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles. |
One solid lipid was selected as a lipid matrix in five commonly used oral delivery and biodegradable solid lipids of GM, GT, SA, ATO 5, and 888 ATO (Figure 3).22,26 Before modification, particle sizes of RJ-III@SLNs increased with molecular weights of the different lipids, and all the zeta potentials of RJ-III@SLNs were negative potential, and the EE of RJ-III@SLNs varied due to the properties of the different lipids. The SA was not subjected to further investigation because of its low EE and absolute value of potential. However, during the modification process, flocculation occurred in RJ-III@SLNs based on 888 ATO, which had the highest EE. Therefore, 888 ATO was excluded. After modification, particle size of HACC-modified SLNs was larger than that of unmodified SLNs, zeta potential of SLNs changed from negative potential to positive potential, implying that HACC was adsorbed onto drug-loaded SLNs through non-covalent bonds. Furthermore, the RJ-III@HACC-SLNs based on GM possessed a smaller particle size, higher zeta potential, and EE than the others. Furthermore, many original publications had shown that SLNs based on GM could enhance bioavailability and prolonged circulation time.26 Therefore, GM was selected as the core lipid.
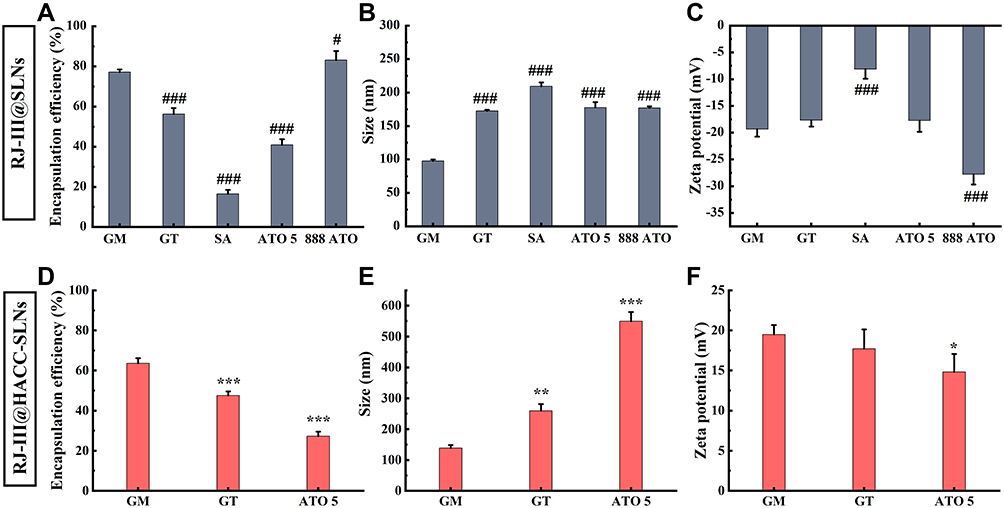 |
Figure 3 The screening of the core lipids of nanoparticles of RJ-III@HACC-SLNs (Mean ± SD, n = 3). The effect of different lipids on particle size (A), zeta potential (B), and encapsulation efficiency (C) of RJ-III@SLNs. ###P < 0.001 compared with the GM group. The influences of different lipids on particle size (D), zeta potential (E), and encapsulation efficiency (F) of RJ-III@HACC-SLNs. *P < 0.05, **P < 0.01, ***P < 0.001 compared with the GM group. Abbreviations: RJ-III@SLNs, rhodojaponin III-loaded solid lipid nanoparticles; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles; GM, glycerol monostearate; GT, glyceryl trioleate; SA, stearic acid; ATO 5, Precirol ATO 5; 888 ATO, Compritol 888 ATO. |
In a subsequent study, single-factor experiments were carried out to investigate the appropriate ranges of three critical variables, including the lipid-to-drug ratio (X1) (Figure S1), cosurfactant-to-surfactant ratio (X2) (Figure S2), and the volume ratio of HACC to SLNs (X3) (Figure 2A and B) that affected the particle size and EE of RJ-III@HACC-SLNs. Meawhile, the volume of the aqueous phase was determined to be 10ml (Figure S3).Then, the formulation of RJ-III@HACC-SLNs was optimized by central composite design/response surface method. The observed particle size and EE values from the 19 experiments are as shown in Table 3. The results of statistical parameters calculation indicated that quadratic polynomial model was the best fitted for the experimental data for all responses. The R2 values were 0.9998 and 0.9982 for Y1 (particle size) and Y2 (EE) models, respectively. This means that the relationship between the variables and responses was well depicted by second-order model. In addition, the P-values were <0.001, which indicates that the equation model was significant and had better correlation. Regression analysis was performed for determining the optimal region for response studies (Figure 4). The model equations are as follows:
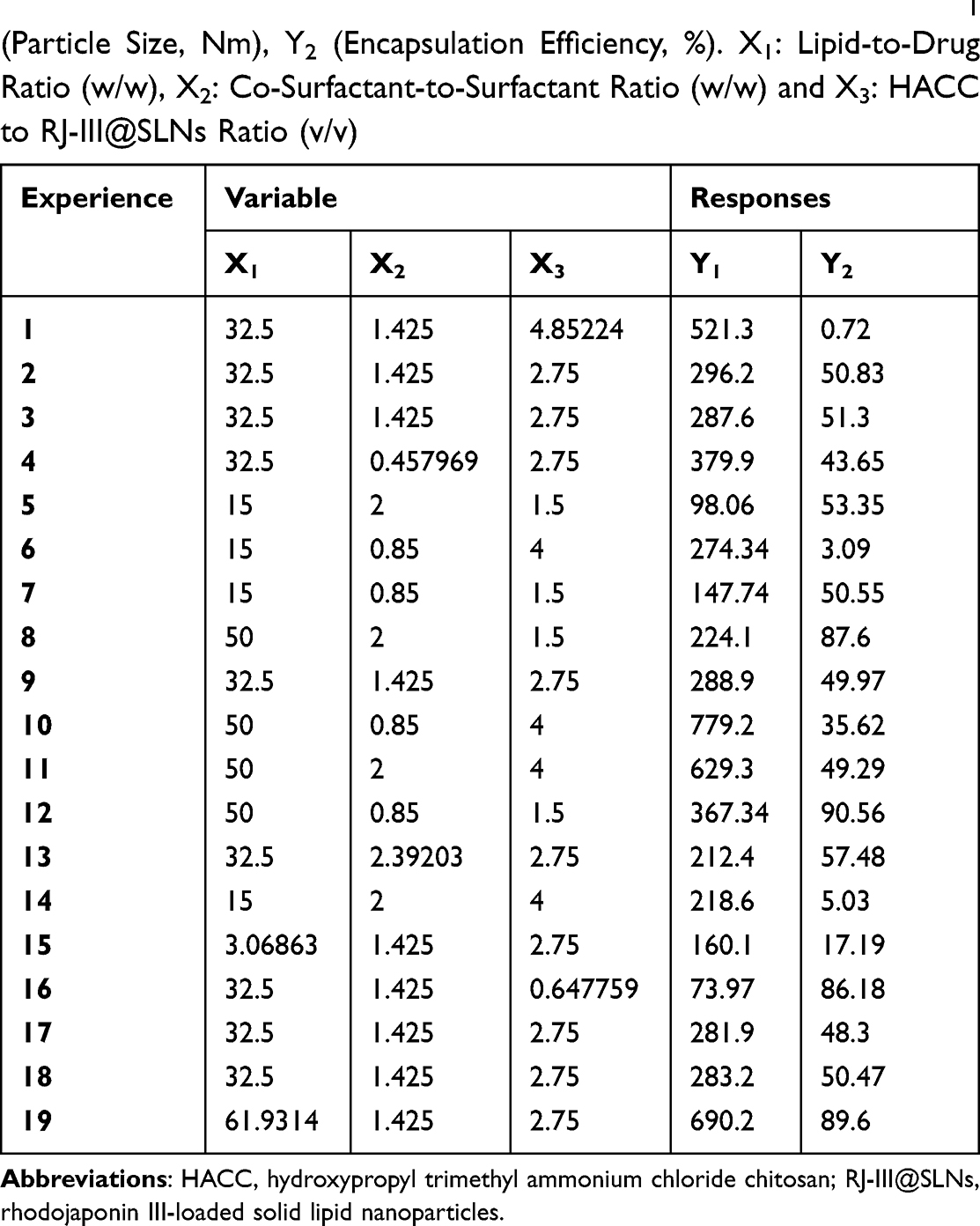 |
Table 3 The Central Composite Design and Resulting Values of Y1 (Particle Size, Nm), Y2 (Encapsulation Efficiency, %). X1: Lipid-to-Drug Ratio (w/w), X2: Co-Surfactant-to-Surfactant Ratio (w/w) and X3: HACC to RJ-III@SLNs Ratio (v/v) |
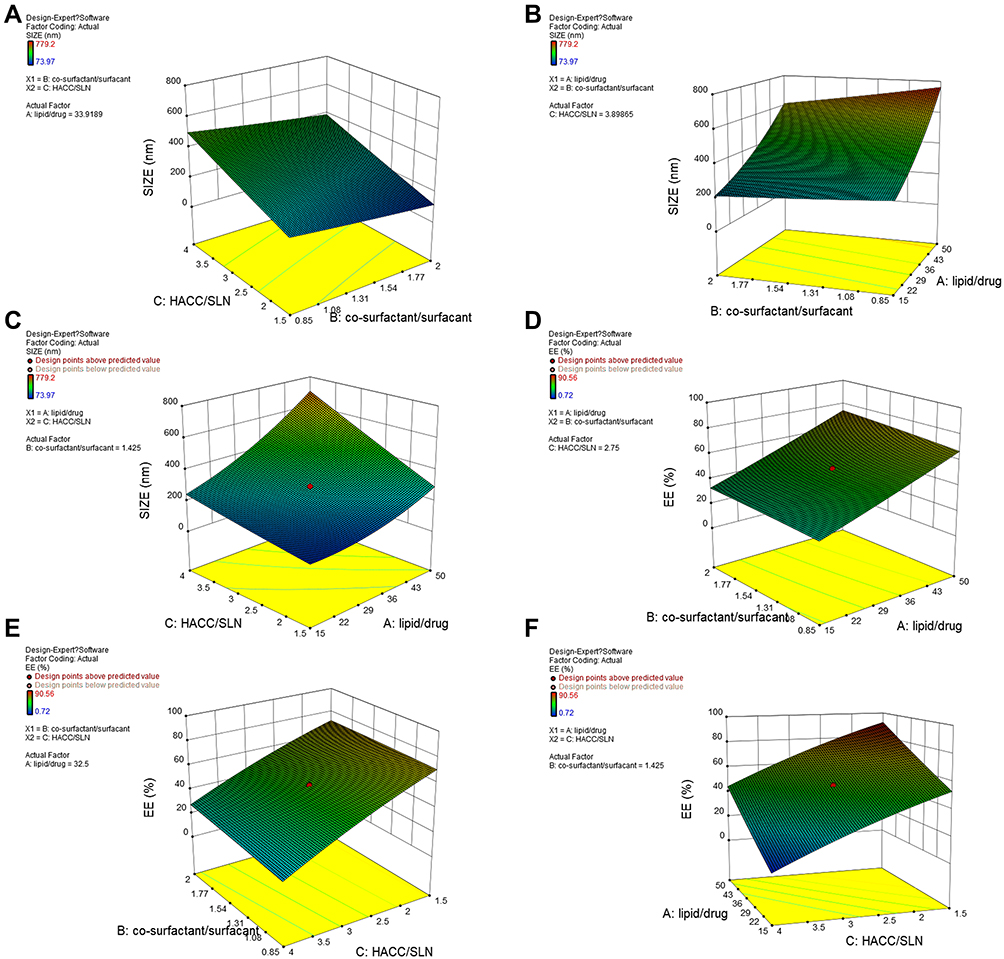 |
Figure 4 The response surface and contour plots of the fitting equations of the particle size and encapsulation efficiency. (A–C) showing the effects of variables on size of RJ-III@HACC-SLNs, (A) the co-surfactant/surfactant and the HACC/SLN, (B) the lipid/drug and the co-surfactant/surfactant, and (C) the lipid/drug and the HACC/SLN. (D–F) showing the effects of variables on EE of RJ-III@HACC-SLNs, (D) the lipid/drug and the co-surfactant/surfactant, (E) the HACC/SLN and the co-surfactant/surfactant, and (F) the HACC/SLN and the lipid/drug. Abbreviations: RJ-III, rhodojaponin III; HACC, hydroxypropyl trimethyl ammonium chloride chitosan; RJ-III@SLNs, rhodojaponin III-loaded solid lipid nanoparticles; lipid/drug, the amount of lipid-to-drug ratio; co-surfactant/surfactant, the mass ratio of co-surfactant to surfactant; HACC/SLN, the volume ratio of HACC to RJ-III@SLNs; EE, encapsulation efficiency. |
Y1 = 287.58+ 157.63X1 – 49.81X2 +133.01X3 – 23.47X1X2 + 71.24X1X3 − 1.59X2X3 + 48.55X12 + 2.94X22 + 3.46X32 (R2 = 0.9998, P < 0.0001)
Y2 = 50.26+ 19.18X1 +2.831X2 – 24.37X3 + 0.75X1X2 + 0.32X1X3 +1.97X2X3 +0.67X12 −0.33X22 −2.48X32 (R2 = 0.9982, P < 0.0001)
The optimal prescription of RJ-III@HACC-SLNs was as follows: the amount of lipid-to-drug ratio was 28.66, the mass ratio of cosurfactant to surfactant was 1.38, and the volume ratio of HACC to SLNs was 1.5. The obtained RJ-III@HACC-SLNs exhibited a light blue opalescence (Figure 5A), particle size of (134.47 ± 2.73) nm, EE of (73.80 ± 8.86)%, and zeta potential of (18.72 ± 1.15) mV (Figure 5B). It was subjected to further physicochemical characterization, pharmacokinetics, antinociceptive, and safety studies.
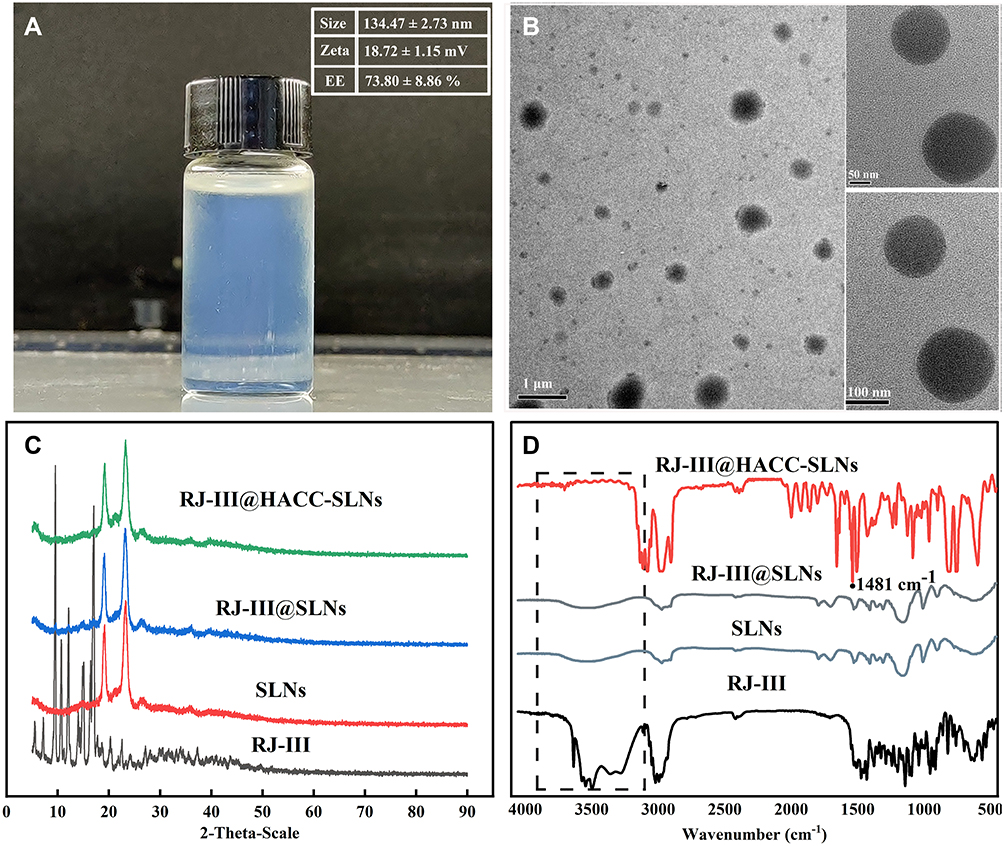 |
Figure 5 Structural characterization of RJ-III@HACC-SLNs. (A) The photograph of RJ-III@HACC-SLNs; (B) Transmission electron microscopy of RJ-III@HACC-SLNs; (C) X-ray diffractograms of RJ-III, SLNs, RJ-III@SLNs, and RJ-III@HACC-SLNs; (D) FT-IR spectra of RJ-III, SLNs, RJ-III@SLNs, and RJ-III@HACC-SLNs. Abbreviations: RJ-III, rhodojaponin III; HACC, hydroxypropyl trimethyl ammonium chloride chitosan; RJ-III@SLNs, rhodojaponin III-loaded solid lipid nanoparticles; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles; SLNs, solid lipid nanoparticles without RJ-III; EE, encapsulation efficiency. |
Structural Characterization of RJ-III@HACC-SLNs
The data shown in Figure 5B demonstrate that RJ-III@HACC-SLNs nearly acquired a spherical shape. The XRD is important for evaluating changes in RJ-III crystallinity or its precipitation abilities in amorphous forms and presented in Figure 5C. Further, the XRD pattern of RJ-III reveals the crystalline characteristic of the drug. Sharp crystalline peaks at 2θ scattered angles between 5° and 17° disappeared in RJ-III@HACC-SLNs and RJ-III@SLNs but instead showed two mountain peaks at 19° and 23°, suggesting that the disordered crystalline state of RJ-III entrapped by the core of SLNs. The surface characterization of prepared SLNs was further investigated using FT-IR. In Figure 5D, the FT-IR spectra revealed the disappearance of the bimodal peak near 3407–3638 cm−1 which belongs to hydroxyl group [−OH] of RJ-III, and the peaks in 800–1500 cm−1 of RJ-III significantly decreased in RJ-III@SLNs. After coating the HACC, the peak appearing at 1481 cm−1 was due to [−CH3] of quaternary ammonium group skeleton stretching vibration, which supported the HACC coating on the surface of RJ-III@SLNs.34
In vitro Stability and Release of RJ-III@HACC-SLNs
The oral therapeutic delivery strategy developed improved the pharmacological effects and decreased acute toxicity of RJ-III by prolonging the action time and reducing fluctuation of plasma concentration. Therefore, in vitro stability and release of nanoparticles are essential in investigating the dosage form performance. Figure 6A shows a strong stability of RJ-III@HACC-SLNs as compared with that of RJ-III@SLN. The EE of RJ-III@HACC-SLNs did not significantly decrease (P > 0.05), that RJ-III@SLNs had an extremely significant decrease (P < 0.001), during 3 h incubated in SGF. The above changes in nanoparticle properties suggest the better stability of HACC modification agents in artificial gastric fluids, possibly because HACC prevents the amino, and hydroxy groups of chitosan from generating hydrogen bonds, improving water solubility and positive activity for protonation under acidic conditions, and protecting nanoparticle stability.31,33
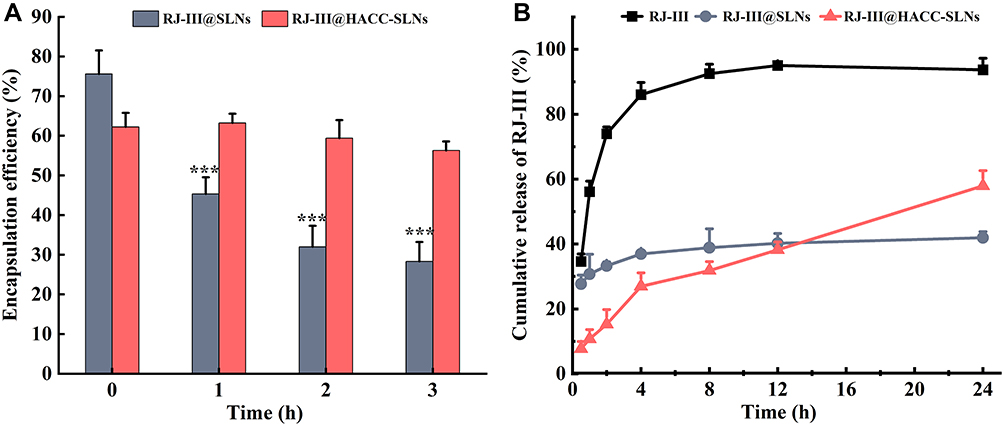 |
Figure 6 In vitro stability and release profile of RJ-III@HACC-SLNs (Mean ± SD, n = 3). (A) The stability of RJ-III@SLNs and RJ-III@HACC-SLNs in a simulated gastric fluid. ***p < 0.001 compared with the encapsulation efficiency of RJ-III@SLNs at 0 h. (B) The release profile of RJ-III, RJ-III@SLNs, and RJ-III@HACC-SLNs into PBS at pH 6.8. Abbreviations: RJ-III, rhodojaponin III; HACC, hydroxypropyl trimethyl ammonium chloride chitosan; RJ-III@SLNs, rhodojaponin III-loaded solid lipid nanoparticles; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles. |
The in vitro release of RJ-III@HACC-SLNs was investigated at 37°C under pH 6.8 PBS solutions. As shown in Figure 6B, the RJ-III solution was 95% within 24 h, indicating rapid diffusion of RJ-III, whereas the release rate of RJ-III encapsulated in SLNs or HACC-SLNs was relatively gentle indicating that the nanocarriers showed some resistance to the release of RJ-III. Further, the RJ-III@SLNs exhibited a higher RJ-III release rate than RJ-III@HACC-SLNs, while the cumulative RJ-III release of RJ-III@HACC-SLNs was higher than RJ-III@SLNs. In the first 2 h, over 33% of the drug had been released from RJ-III@SLN, while 15% of the drug had been released from RJ-III@HACC-SLNs. Then, the release was sustained for up to 24 h, with in a total release of 41% or 56%, respectively. The distinct release profiles suggested that the HACC coated on the surfaces of the RJ-III@SLNs could sustain release by effectively suppressing drug release rate through adsorption interactions between HACC and SLNs. Meanwhile, the free drug is important to exert immediate antinociceptive when treated with RJ-III@HACC-SLNs, which indicated RJ-III@HACC-SLNs may exert rapid and lasting analgesic effects in vivo.
Pharmacokinetic Study
The pharmacokinetic characteristics were performed using various formulations: RJ-III solution and RJ-III@HACC-SLNs were tested using 0.2 mg/kg administered in mice. RJ-III@SLNs was not selected for the experiments because of their instability and fast release. Figure 7 respectively showed the plasma concentration–time curves of RJ-III and mean pharmacokinetic parameters. Wide variabilities in PK parameters were noted between the various RJ-III formulations. The RJ-III solution was the control in this assay.
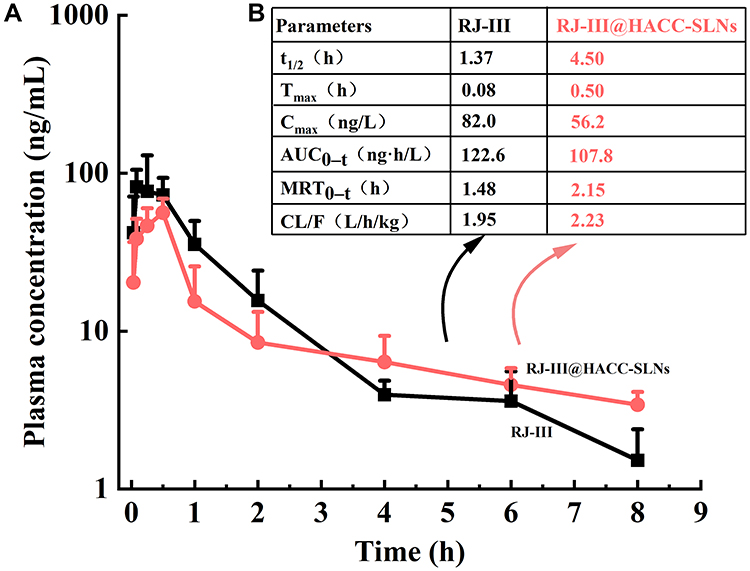 |
Figure 7 (A) Plasma concentration–time curves of RJ-III after RJ-III and RJ-III@HACC-SLNs (equivalent to 0.2 mg/kg as RJ-III) were intragastrically administered to mice. (Mean ± SD, n = 6). (B) Pharmacokinetic parameters of RJ-III, HACC@RJ-III-SLNs after oral administration (0.2 mg/kg) in mice (Mean, n = 6). Abbreviations: RJ-III, rhodojaponin III; HACC, hydroxypropyl trimethyl ammonium chloride chitosan; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles; AUC, area under the curve; Cmax, peak concentration; MRT, mean retention time; CL/F, clearance rate; t1/2, half-life; Tmax, peak time. |
The RJ-III solution resulted in Tmax at 0.083 h and t1/2 at 1.37 h, which illustrated oral bioavailability problems with rapid absorptions and RJ-III elimination. In contrast, the RJ-III@HACC-SLNs yielded a Cmax of 56.22 ± 12.72 ng/mL at 0.5 hours post-administration, and a longer t1/2 as well as MRT, implying that in this form, RJ-III has a long drug residence and active time. Furthermore, relative bioavailability of RJ-III@HACC-SLNs was calculated at 87.9%, showing that RJ-III@HACC-SLNs maintain good oral absorption while prolonging RJ-III release in vivo. The results hence lay the foundation for further in vivo multimodal analgesic pharmacodynamic studies.
Multimodal Antinociceptive Effect of RJ-III@HACC-SLNs
The in vivo antinociceptive effect of RJ-III@HACC-SLNs was investigated in the present study. Different stimuli, hot-plate, acetic acid, and formalin-induced nociception were selected as pain models. According to preliminary results, 0.1 mg/kg RJ-III was selected as the optimal dose in the acetic acid-induced writhing and formalin tests, 0.2 mg/kg RJ-III was chosen as the dose in the hot-plate test, to inhibit the occurrence of toxic effects, 300 mg/kg aspirin was selected as the positive drug group.
In the first series of experiments, the acetic acid-induced writhing test, a sensitive, predictive acute pain animal model42,43 was used to assess the antinociceptive effects of RJ-III@HACC-SLNs. As illustrated in Figure 8A, relative to the normal saline group at administration for 15 min and 45 min, there were marked differences in the number of writhes among the mice in RJ-III group (P < 0.001, P < 0.001), the RJ-III@HACC-SLNs group (P < 0.001, P < 0.001), and the aspirin group (P < 0.001, P < 0.01), indicating that group RJ-III, RJ-III@HACC-SLNs, and aspirin, exerted analgesic effects within 45 min. Furthermore, the RJ-III@HACC-SLNs group exhibited a more lasting antinociceptive effect as compared with groups RJ-III and aspirin, which RJ-III@HACC-SLNs group showed no marked differences in the number of writhes between 15 and 45 min (P > 0.05), while groups RJ-III and aspirin showed statistically increases in number of writhes (P < 0.001, P < 0.01). Remarkably, the RJ-III@HACC-SLNs group showed a greater analgesic efficacy as compared with groups RJ-III and aspirin after administration for 45 min, which there is a significant difference in writhing number of RJ-III@HACC-SLNs contained 0.10 mg/kg RJ-III compared with the 0.10 mg/kg RJ-III group and the 300mg/kg aspirin group (P < 0.05, P < 0.01).
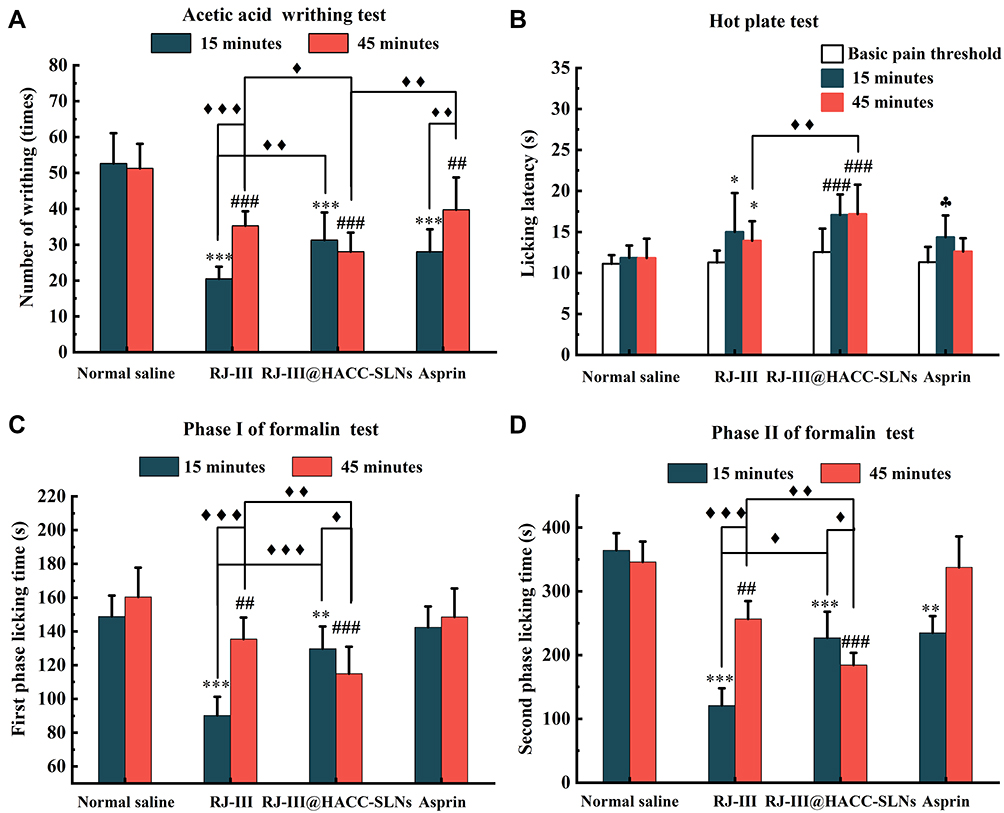 |
Figure 8 The multimodal antinociceptive effects of RJ-III@HACC-SLNs (Mean ± SD, n = 10). (A) Antinociceptive effects of RJ-III@HACC-SLNs in acetic acid-induced writhing test (♦P <0.05; ♦♦P <0.01; ♦♦♦P <0.001). Mice were administered with normal saline (0.9% NaCl, i.g), RJ-III (0.1 mg/kg, i.g), RJ-III@HACC-SLNs (0.10 mg/kg of RJ-III, i.g), and aspirin (200 mg/kg, i.g). ***P < 0.001 vs normal saline group for administering 15 min; ###P < 0.001 and ##P < 0.01 vs normal saline group after administeration for 45 min. (B) Influence of RJ-III (0.2 mg/kg), RJ-III@HACC-SLNs (0.20 mg/kg of RJ-III), and aspirin in the hot-plate test (♦♦P < 0.01). *P < 0.05 vs the basic pain threshold of before administration of RJ-III; ###P < 0.001 vs the basic pain threshold of before administration of RJ-III@HACC-SLNs; ♣P < 0.05 vs the basic pain threshold of before administration of aspirin. (C) and (D) The effects of RJ-III (0.1 mg/kg) and RJ-III@HACC-SLNs (0.10 mg/kg of RJ-III) on phases I and II of the formalin test (♦P <0.05; ♦♦P < 0.01; ♦♦♦P < 0.001). **P < 0.01 and ***P < 0.001 vs normal saline group for administering 15 min; ###P < 0.001 and ##P < 0.01 vs normal saline group after administeration for 45 min. Abbreviations: RJ-III, rhodojaponin III; HACC, hydroxypropyl trimethyl ammonium chloride chitosan; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles. |
Central antinociceptive effects of RJ-III@HACC-SLNs were assessed in the hot-plate test, using classic acute pain models that record responses to thermal stimuli.44,45 As shown in Figure 8B, after administration of 15 min, 300 mg/kg aspirin and 0.2 mg/kg RJ-III increased the latency time compared to pretreatment (P < 0.05, P < 0.05). In contrast, the RJ-III@HACC-SLNs showed superior potency at doses approximately 1500-fold lower than aspirin (P < 0.001). After administration for 45 min, RJ-III, RJ-III@HACC-SLNs maintained its antinociception effects relative to the pretreatment group (P < 0.05, P < 0.001). Moreover, antinociceptive was more effective of RJ-III@HACC-SLNs compared with RJ-III after administration for 45 min (P < 0.01). Notably, the aspirin group showed no significant analgesic effect after administration for 45 minutes (P > 0.05).
To investigate antinociceptive characteristics, RJ-III and RJ-III@HACC-SLNs were investigated in the formalin test.43,46 Responses in Phase I correlated with acute neurogenic pain, which was largely due to direct stimulation of nociceptors.43,46 Contrastingly, the mechanisms involved in Phase II responses were highly complex, involving inflammatory processes accompanied by spontaneous primary afferent neuronal activities as well as central alterations of pain processing.43,46 Figure 8C and D show that within 15 min of administration, the non-steroidal anti-inflammatory drug aspirin suppressed behavioral responses in phase II (the inflammatory phase) (P < 0.01). However, this had no effects on phase I responses (P > 0.05) as shown in the previous studies.47 In comparison, it was evident that RJ-III and RJ-III@HACC-SLNs were active in both phases I and II (P < 0.01, P < 0.001) within administration 15 min. At this point, the analgesic effect of RJ-III in phase I and phase II was stronger than that of RJ-III@HACC-SLNs (P < 0.01, P < 0.001). RJ-III and RJ-III@HACC-SLNs still showed an analgesic effect in phases I and II after administration for 45 min, but RJ-III and RJ-III@HACC-SLNs showed a completely different antinociceptive trend, significantly enhanced in RJ-III@HACC-SLNs (P < 0.05), and significantly decreased in RJ-III (P < 0.001) as compared with the respective administration for 15 min. Remarkably, 45 min post-treatment, the antinociceptive effect of RJ-III@HACC-SLNs in phase I and phase II was stronger than that of RJ-III (P < 0.01, P < 0.01).
These data showed that RJ-III@HACC-SLNs is durable and effective at suppressing responses in physical or chemically induced acute and inflammatory pain as compared with RJ-III and aspirin.
In vitro Cytotoxicity Studies
The study of cytotoxicity was done to elucidate on the biocompatibility of oral absorptions of HACC-RJ-III@SLNs. The cytotoxic effect of the SLNs formulations, with and without RJ-III, against Caco-2 cells is shown in Figure 9. A cell viability of >80% was considered non-toxic to cells.37 The SLNs and HACC-SLNs without RJ-III, were noncytotoxic to the cell, even at doses of up to 500 μg/mL under the experimental conditions (Figure 9A). This suggested that SLNs and HACC-SLNs may be good carriers for RJ-III delivery. The effect of different concentrations of RJ-III on cytotoxicity caused by RJ-III@SLNs and RJ-III@HACC-SLNs were also explored in the present study. The cytotoxicity studies (Figure 9B) showed cell viability was over 90% from 0 to 20 μg/mL RJ-III concentration for 24 h. This indicates that RJ-III@HACC-SLNs did not cause significant toxicity to Caco-2 cells across tested concentrations, which matches previous reports,37 provided an experimental basis for the subsequent oral absorption mechanism research of RJ-III@HACC-SLNs.
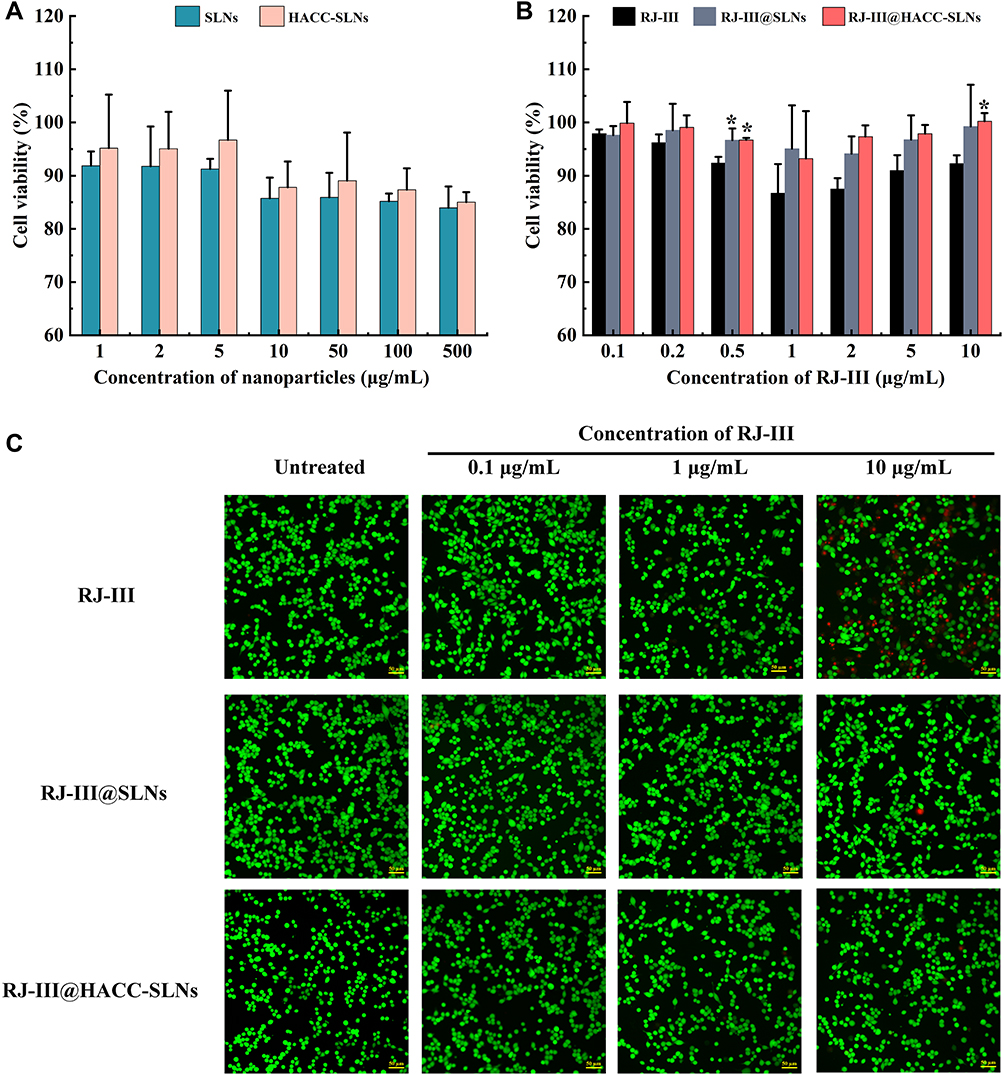 |
Figure 9 The safety of HACC-RJ-III@SLNs. (A) Cytotoxicity of blank SLNs (B-SLNs) and HACC-modified SLNs without RJ-III (HACC-SLNs) in Caco-2 cells after cultured with different SLNs for 24 h (Mean ± SD, n = 3). (B) Cytotoxicity of RJ-III, RJ-III@SLNs, and RJ-III@HACC-SLNs in Caco-2 cells after being cultured with different SLNs for 24 h (Mean ± SD, n = 3), *P < 0.05 compared with RJ-III group. (C) Live/Dead double staining, cells on RJ-III, RJ-III@SLNs, and RJ-III@HACC-SLNs 4h after seeding. Red staining represents dead cells while green fluorescence indicates live cells. Scale bars: 50 μm. Abbreviations: RJ-III, rhodojaponin III; HACC, hydroxypropyl trimethyl ammonium chloride chitosan; SLNs, solid lipid nanoparticles; HACC-SLNs, HACC-modified solid lipid nanoparticles; RJ-III@SLNs, rhodojaponin III-loaded solid lipid nanoparticles; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles. |
In vitro Fluorescent Live/Dead Assay
To confirm the results obtained using MTT assays, a fluorescent live/dead assay-based Calcein-AM/PI double staining kit was performed for Caco-2 cells. This allows direct counting of the numbers of live and dead cells. Calcein-AM can specifically bind to living cells and produce green fluorescence, whereas PI dye can specifically bind to dead cells and produce red fluorescence. As shown Figure 9C, after 4 h of treatment, this assay yielded comparable cell viability as that of the MTT assay in the three preparations. Moreover, when cells were treated with the samples containing 10 μg/mL RJ-III content, the red fluorescence was weakest in RJ-III@HACC-SLNs, indicating that the RJ-III@HACC-SLNs group had the best safety for Caco-2 cells as compared with the RJ-III and RJ-III@SLNs groups.
Acute Toxicity
To further evaluate the safety of RJ-III@HACC-SLNs, the acute lethal test, RJ-III, and RJ-III@HACC-SLNs were administered through single oral administration at increasing dosage, and mortality was observed over the 7 days. Figure 10 shows that the LD50 of RJ-III and RJ-III@HACC-SLNs was 3.62 (2.96–4.29, at a 95% confidence limit) and 6.42 mg/kg (5.18–7.66, at a 95% confidence limit), respectively, indicating that the acute toxicity of RJ-III@HACC-SLNs was approximately 1/2 of that of RJ-III, suggested HACC-modified SLNs reducing the acute toxicity of RJ-III, and indicated that RJ-III@HACC-SLNs had sufficient safety when playing analgesic effect.
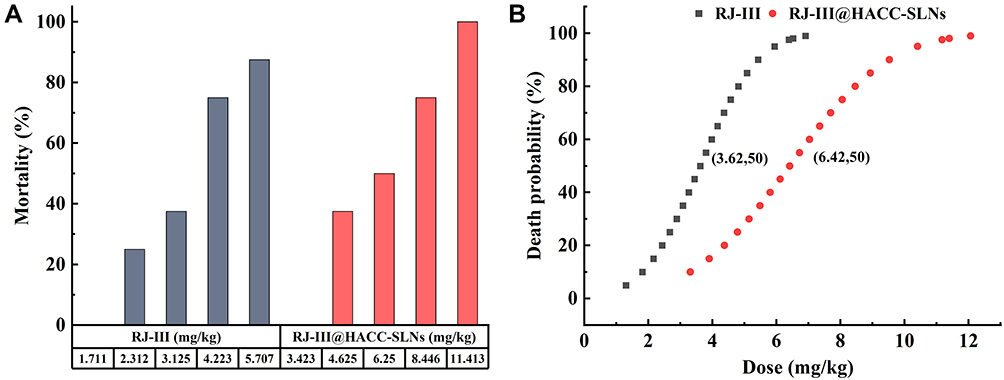 |
Figure 10 (A) Acute lethal effects of RJ-III and RJ-III@HACC-SLNs after single intragastric administration in ICR mice (n = 8). (B) The dosage–mortality curve graph of RJ-III and RJ-III@HACC-SLNs as determined with the Bliss method (n = 8). Abbreviations: RJ-III, rhodojaponin III; HACC, hydroxypropyl trimethyl ammonium chloride chitosan; RJ-III@HACC-SLNs, rhodojaponin III-loaded HACC-modified solid lipid nanoparticles. |
Discussion
With increased awareness, chronic pain has been defined as a disease in medicine and a global public health priority in the field of public health.7 Pain impairs the quality of life and is associated with enormous societal economic losses.5,6,48 Annually, it is estimated that pain costs more than $100 billion in costs of direct healthcare and lost work time in the US.5,6,48 Further, it has been found that management of pain is a vast clinical challenge. Currently, lack of effective and non-opioids analgesics has contributed to the recent opioid drug abuse tragedy experience in some parts of the world.9 Therefore, the urgency of the situation demands a major effort to identify new drugs for the treatment of pain.
With thousands of years of clinical use and the reported efficacy of relieve pain, traditional Chinese medicine may provide an opportunity for a rapid search for new analgesic agents. In China, 426 analgesic traditional drugs are recorded in the Chinese Pharmacopoeia alone, while also identifying several monomeric compounds to serve as new analgesic compounds, including alkaloids, flavonoids, terpenoids, coumarins, etc.49
Rhodojaponin III (RJ-III) is one among the identified compounds with a lot of potential for development of analgesic agents. However, development of an analgesia agent from RJ-III is hindered by its high acute toxicity18 and rapid in vivo elimination.19 Therefore, it is key to further research on how to reduce toxicity and enhance the efficacy of such small-molecule drugs. The current study provides a feasible dosage form for further analgesic development of RJ-III. In addition, a new perspective is provided for the incorporation of HACC-SLNs in development of analgesics. In the present study, it was evident that the sustained release of HACC-SLNs suppressed fluctuations in the plasma levels of RJ-III, which in turn improved the analgesic efficacy of RJ-III and reduced its acute toxicity.
The choice of lipid is crucial in the ingredient design during the preparation of HACC-SLNs. The lipids must be selected based on their ability to solubilize the encapsulated drug. In the preliminary study, core lipids were selected by comparing the EE and particle size of RJ-III@HACC-SLNs. Ultimately, it was decided that GM should be used as the core lipid in the present study. The lipid has a single fatty acid chain bonded to a glycerol backbone, possesses amphiphilic nature, can self-assemble in water or oil into various mesophases, and has been extensively used in food as well as personal care products.50,51 Furthermore, it was found that the amount of lipid-to-drug ratio, the mass ratio of CoS to S, and the volume ratio of HACC to SLNs also had significant effects on the EE as well as the particle size of RJ-III@HACC-SLNs by previous single-factor experiments (Supplementary Information). To obtain a suitable particle size and high EE for satisfying the further studies, the formulation was optimized by using the central composite design/response surface method, a broadly used method for complex prescription optimization in pharmacy. In the present study, it was found that RJ-III@HACC-SLNs based on the optimal formulation had better physical characteristics. It had a particle size of around 139 nm, suggesting that it may be absorbed by intestinal epithelial cells and reach the circulatory system with an intact particle.52–54 In addition, it had a zeta potential of about 19 mV which indicated potential stability. This may be important to drug absorption due to electrostatic adsorption between the cell membranes and positively charged HACC-SLNs.52–54
In vitro stability and release testing are essential analytical tools to investigate the dosage form performance and release mechanism for complex nanocarriers.55 To select the suitable nano-formulation for in vivo study, RJ-III@SLNs and RJ-III@HACC-SLNs have discussed its stability of EE in SGF and observed for drug release studies in PBS (pH = 6.8). Previous studies have shown that HACC-modified SLNs significantly affect the stability of nanoparticles and release of drugs, which might be due to the deprotonation of HACC under acidic conditions.31,33,37 Therefore, RJ-III@HACC-SLNs was selected as the optimal formulation, and subsequent studies conducted on its pharmacokinetic, antinociceptive, and safety. RJ-III@SLNs were not further investigated because of their instability and fast release.
To evaluate the sustained-release capacity of RJ-III@HACC-SLNs, it is important to elucidate its pharmacokinetic properties. In this study, RJ-III generally presented the characteristics of high oral absorption rate and rapidly metabolized as had been shown in our previous results.19 After oral RJ-III administration in the form of HACC-SLNs, Tmax and t1/2 were delayed via endocytosis, reaching peak time at about 0.5 h and half-life time at approximately 4.50 h. It was found that there was a lower Cmax and longer MRT as compared with that in the RJ-III group. This indicated that RJ-III@HACC-SLNs could prevent strong fluctuations in plasma levels and achieve sustained release of RJ-III in vivo.
The analgesic effects of RJ-III@HACC-SLNs in mice were determined using the acetic acid writhing and hot plate tests as the acute pain models.42–44 Further, the formalin test was used as a model with both acute and persistent inflammatory pain.42–44 Aspirin, a non-steroidal anti-inflammatory drug, was used as the positive drug in the present study because it is an effective and versatile medication for mild-to-moderate pain.56 It was evident that intragastric administration of RJ-III@HACC-SLNs showed a fast and durable effective analgesic effect in physical or chemically induced acute pain and chemically induced inflammatory pain in mice. The effective analgesic effect could be attributed to the prolonged release of nanoparticles in vivo. The pro-inflammatory cytokines, including IL-1b, TNF-a, and IL-6 play an active role in nociception.57–59 Furthermore, relevant studies have shown that RJ-III exhibits a strong anti-inflammatory potential by suppressing the levels of pro-inflammatory cytokine (IL-1β, IL-6, and TNF-α).15 Therefore, the anti-inflammatory effect of RJ-III could be the motive for its anti-nociceptive but subsequent experiments should be carried out on antinociceptive mechanisms of RJ-III@HACC-SLNs.
Safety has always been a concern regardless of a strong bioactive RJ-III or SLNs with widely used due to the unique physicochemical characteristics. Therefore, evaluation of the safety of RJ-III@HACC-SLNs is one of the most important parts of effectiveness studies. Consequently, the current study also carried out both in vitro cytotoxicity and in vivo acute toxicity tests. Cytotoxicity tests were conducted through MTT and a fluorescent live/dead analysis in Caco-2 cells which is a widely used intestinal cell barrier model.38 Results of the present study showed that SLNs and HACC@SLNs were biocompatible, which was consistent with the results of a previous study.37 Further, the safety of RJ-III@HACC-SLNs was found to have a wide range of safe concentrations. In addition, it was found that the cellular activity of RJ-III@HACC-SLNs remains unaffected when the RJ-III concentration reaches 10 μg/mL and this provided the basis for requirement of cellular uptake and cellular transport studies. The LD50 is often used to gauge the toxicity of drugs and chemicals. Results of the present study found that the LD50 value of RJ-III@HACC-SLNs was 1.8 times higher than that of RJ-III, suggesting that HACC-modified SLNs reduced the acute toxicity of RJ-III and exerted antinociceptive effects.
Conclusion
The present study prepared RJ-III@HACC-SLNs and investigated its multimodal in vivo antinociceptive properties. The study prepared RJ-III@HACC-SLNs that offered particle sizes in a nanometer range, good stability, and a prolonged-release profile. Results of the in vivo experiments demonstrated that RJ-III@HACC-SLNs reduced the fluctuation plasma concentration, prolonged active time, and enhanced antinociceptive effects in mice. Further, RJ-III@HACC-SLNs showed a good biocompatibility with Caco-2 cells and better safety whose LD50 value was 1.8 times that of RJ-III. The results of the current study form the basis for research and development of RJ-III for application against nociception. We investigate the long-term analgesic effects of RJ-III@HACC-SLNs in chronic pain models and elucidate the antinociceptive mechanisms in which RJ-III@HACC-SLNs are involved.
Acknowledgments
This work was financially supported by the science and technology support project of the Shanghai Science and Technology Commission (14401901400).
Disclosure
The authors declare that they have no competing interests.
References
1. Loeser JD, Melzack R. Pain: an overview. Lancet. 1999;353(9164):1607–1609. doi:10.1016/S0140-6736(99)01311-2
2. Mogil JS. Qualitative sex differences in pain processing: emerging evidence of a biased literature. Nat Rev Neurosci. 2020;21:353–365.
3. Melnikova I. Pain market. Nat Rev Drug Discov. 2010;9:589–590.
4. Shaheed CA, Machado GC, Underwood M. Drugs for chronic pain. Br J Gen Pract. 2020;70:576–577.
5. Kuehn B. Chronic pain prevalence. JAMA. 2018;320:1632.
6. Arenas OM, Lumpkin EA. Touching base with mechanical pain. Cell. 2020;180:824–826.
7. Goldberg DS, McGee SJ. Pain as a global public health priority. BMC Public Health. 2011;11:770.
8. Jackson T, Thomas S, Stabile V, et al. A systematic review and meta-analysis of the global burden of chronic pain without clear etiology in low- and middle-income countries: trends in heterogeneous data and a proposal for new assessment methods. Anesth Analg. 2016;123:739–748.
9. Woodcock J, Difficult balance–pain A. Management, drug safety, and the FDA. N Engl J Med. 2009;361:2105–2107.
10. Cai Y-Q, Hu J-H, Qin J, et al. Rhododendron Molle (Ericaceae): phytochemistry, Pharmacology, and Toxicology. Chin J Nat Med. 2018;16(6):401–410. doi:10.1016/S1875-5364(18)30073-6
11. Cai YQ, Hu JH, Qin J, et al. Rhododendron molle (Ericaceae): phytochemistry, pharmacology, and toxicology. Chin J Nat Med. 2018;16:401–410.
12. Zhi X, Xiao L, Liang S, et al. Chemical constituents of Rhododendron molle. Chemistry Natural Compounds. 2013;49:454–456.
13. Zou HY, Luo J, Xu DR, et al. Tandem solid-phase extraction followed by HPLC-ESI/QTOF/MS/MS for rapid screening and structural identification of trace diterpenoids in flowers of Rhododendron molle. Phytochemical Analysis. 2014;25:255–265.
14. Li Y, Liu YB, Zhang JJ, et al. Antinociceptive grayanoids from the roots of Rhododendron molle. J Nat Prod. 2015;78:2887–2895.
15. He YC, Yao YM, Xue QW, et al. Anti-rheumatoid arthritis potential of diterpenoid fraction derived from Rhododendron molle fruits. Chin J Nat Med. 2021;19:181–187.
16. Mao HY, Li CY, Cui JJ, et al. Rhomotoxin pharmacologic action in lowering blood pressure and slowing heart rate. Chin Med J. 1982;95:311–318.
17. Zhou JF, Liu TT, Zhang HQ, et al. Anti-inflammatory grayanane diterpenoids from the leaves of Rhododendron molle. J Nat Prod. 2018;81:151–161.
18. Huizhen C, Boping D, Nianbao Z, et al. Extraction of Rhomotoxin and Its LD50. China Pharmaceuticals. 2010;19:10–11.
19. Zhang JQ, Zhao CC, Yang QY, et al. Pharmacokinetics, bioavailability and tissue distribution studies of Rhodojaponin III in mice using Qtrap LC–MS/MS. Biomedical Chromatography. 2019;1:548.
20. Nunes S, Madureira AR, Campos D, et al. Solid lipid nanoparticles as oral delivery systems of phenolic compounds: overcoming pharmacokinetic limitations for nutraceutical applications. Crit Rev Food Sci Nutr. 2017;57:1863–1873.
21. Mirchandani Y, Patravale VB, Solid Lipid SB. Nanoparticles for hydrophilic drugs. J Control Release. 2021;335:457–464.
22. Mehnert W, Mäder K. Solid lipid nanoparticles: production, characterization and applications. Adv Drug Deliv Rev. 2001;47:165–196.
23. Wong CY, Al-Salami H, Dass CR. Potential of insulin nanoparticle formulations for oral delivery and diabetes treatment. J Control Release. 2017;264:247–275.
24. Du Y, Ling L, Ismail M, et al. Redox sensitive lipid-camptothecin conjugate encapsulated solid lipid nanoparticles for oral delivery. Int J Pharm. 2018;549:352–362.
25. Ganesan P, Ramalingam P, Karthivashan G, et al. Recent developments in solid lipid nanoparticle and surface-modified solid lipid nanoparticle delivery systems for oral delivery of phyto-bioactive compounds in various chronic diseases. Int J Nanomedicine. 2018;13:1569–1583.
26. Salah E, Abouelfetouh MM, Pan Y, et al. Solid lipid nanoparticles for enhanced oral absorption: a review. Colloids Surf B Biointerfaces. 2020;196:111305.
27. Gao Y, Ma Q, Cao J, et al. Recent advances in microfluidic-aided chitosan-based multifunctional materials for biomedical applications. Int J Pharm. 2021;600:120465.
28. Chen C, Liu Y, Wang H, et al. Multifunctional chitosan inverse opal particles for wound healing. ACS Nano. 2018;12:10493–10500.
29. Yang XL, Ju XJ, Mu XT, et al. Core-shell chitosan microcapsules for programmed sequential drug release. ACS Appl Mater Interfaces. 2016;8:10524–10534.
30. Chen J, Zhan Y, Wang Y, et al. Chitosan/silk fibroin modified nanofibrous patches with mesenchymal stem cells prevent heart remodeling post-myocardial infarction in rats. Acta Biomater. 2018;80:154–168.
31. Cho J, Grant J, Piquette-Miller M, et al. Synthesis and physicochemical and dynamic mechanical properties of a water-soluble chitosan derivative as a biomaterial. Biomacromolecules. 2006;7:2845–2855.
32. Shi LL, Lu J, Cao Y, et al. Gastrointestinal stability, physicochemical characterization and oral bioavailability of chitosan or its derivative-modified solid lipid nanoparticles loading docetaxel. Drug Dev Ind Pharm. 2017;43:839–846.
33. Shi C, Zhu P, Chen N, et al. Preparation and sustainable release of modified konjac glucomannan/chitosan nanospheres. Int J Biol Macromol. 2016;91:609–614.
34. Xu X, Li Y, Wang F, et al. Synthesis, in vitro and in vivo evaluation of new norcantharidin-conjugated hydroxypropyltrimethyl ammonium chloride chitosan derivatives as polymer therapeutics. Int J Pharm. 2013;453:610–619.
35. Xiao B, Ma P, Ma L, et al. Effects of tripolyphosphate on cellular uptake and RNA interference efficiency of chitosan-based nanoparticles in raw 264.7 macrophages. J Colloid Interface Sci. 2017;490:520–528.
36. Zhang L, Zhu K, Zeng H, et al. Resveratrol solid lipid nanoparticles to trigger credible inhibition of doxorubicin cardiotoxicity. Int J Nanomedicine. 2019;14:6061–6071.
37. Shi LL, Xie H, Lu J, et al. Positively charged surface-modified solid lipid nanoparticles promote the intestinal transport of docetaxel through multifunctional mechanisms in rats. Mol Pharm. 2016;13:2667–2676.
38. Yu Z, Fan W, Wang L, et al. Effect of surface charges on oral absorption of intact solid lipid nanoparticles. Mol Pharm. 2019;16:5013–5024.
39. Bai K, Hong B, He J, et al. Preparation and antioxidant properties of selenium nanoparticles-loaded chitosan microspheres. Int J Nanomedicine. 2017;12:4527–4539.
40. Keck CM, Müller RH. Nanotoxicological Classification System (NCS) - a guide for the risk-benefit assessment of nanoparticulate drug delivery systems. Eur J Pharm Biopharm. 2013;84:445–448.
41. Omwoyo WN, Melariri P, Gathirwa JW, et al. Development, characterization and antimalarial efficacy of dihydroartemisinin loaded solid lipid nanoparticles. Nanomedicine. 2016;12:801–809.
42. Chien TY, Huang SK, Lee CJ, et al. Antinociceptive and anti-inflammatory effects of zerumbone against mono-iodoacetate-induced arthritis. Int J Mol Sci. 2016;17:249.
43. Hernandez-Leon A, Gonzalez-Trujano ME, Narvaez-Gonzalez F, et al. Role of beta-caryophyllene in the antinociceptive and anti-inflammatory effects of Tagetes lucida Cav. Essential Oil Molecules. 2020;1:25.
44. Gomes Junior AL, Islam MT, Nicolau LAD, et al. Anti-inflammatory, antinociceptive, and antioxidant properties of anacardic acid in experimental models. ACS Omega. 2020;5:19506–19515.
45. Costa LEC, Brito TV, Damasceno ROS, et al. chemical structure, anti-inflammatory and antinociceptive activities of a sulfated polysaccharide from Gracilaria intermedia algae. Int J Biol Macromol. 2020;159:966–975.
46. Montiel-Ruiz RM, Cordova-de la Cruz M, Gonzalez-Cortazar M, et al. Antinociceptive effect of hinokinin and kaurenoic acid isolated from Aristolochia odoratissima L. Molecules. 2020;2:25.
47. Wang D, Yang H, Liang Y, et al. Antinociceptive effect of spirocyclopiperazinium salt compound DXL-A-24 and the underlying mechanism. Neurochem Res. 2019;44:2786–2795.
48. Dahlhamer J, Lucas J, Zelaya C, et al. Prevalence of chronic pain and high-impact chronic pain among adults - United States, 2016. MMWR Morb Mortal Wkly Rep. 2018;67:1001–1006.
49. Wang R, Han L, Gao Q, et al. Progress on active analgesic components and mechanisms of commonly used traditional Chinese medicines: a comprehensive review. J Pharm Pharm Sci. 2018;21:437–480.
50. Wang FC, Marangoni AG. Internal and external factors affecting the stability of glycerol monostearate structured emulsions. RSC Adv. 2015;5:93108–93116.
51. Talele P, Sahu S, Mishra AK. Physicochemical characterization of solid lipid nanoparticles comprised of glycerol monostearate and bile salts. Colloids Surf B Biointerfaces. 2018;172:517–525.
52. Hu X, Yang G, Chen S, et al. Biomimetic and bioinspired strategies for oral drug delivery. Biomater Sci. 2020;8:1020–1044.
53. Liu L, Yao W, Rao Y, et al. pH-responsive carriers for oral drug delivery: challenges and opportunities of current platforms. Drug Deliv. 2017;24:569–581.
54. Banerjee A, Qi J, Gogoi R, et al. Role of nanoparticle size, shape and surface chemistry in oral drug delivery. J Control Release. 2016;238:176–185.
55. Iqbal A, Zaman M, Wahab Amjad M, et al. Solid lipid nanoparticles of mycophenolate mofetil: an attempt to control the release of an immunosuppressant. Int J Nanomedicine. 2020;15:5603–5612.
56. Peck J, Urits I, Zeien J, et al. A comprehensive review of over-the-counter treatment for chronic migraine headaches. Curr Pain Headache Rep. 2020;24:19.
57. Sabat R, Jemec GBE, Matusiak L, et al. Hidradenitis suppurativa. Nat Rev Dis Primers. 2020;6:18.
58. Chen G, Zhang YQ, Qadri YJ, et al. Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron. 2018;100:1292–1311.
59. Risbud MV, Shapiro IM. Role of cytokines in intervertebral disc degeneration: pain and disc content. Nat Rev Rheumatol. 2014;10:44–56.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.