")
Back to Journals » International Journal of Women's Health » Volume 14
Rhabdomyosarcoma in Adults: Case Series and Literature Review
Authors Chen J, Liu X, Lan J, Li T, She C, Zhang Q, Yang W
Received 7 December 2021
Accepted for publication 3 March 2022
Published 28 March 2022 Volume 2022:14 Pages 405—414
DOI https://doi.org/10.2147/IJWH.S352143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Elie Al-Chaer
Jian Chen,1 Xiaoyun Liu,1 Jian Lan,1 Tingchao Li,2 Chaokun She,1 Qingyun Zhang,1 Wei Yang3
1Department of Gynecology and Obstetrics, The Third Affiliated Hospital of Zunyi Medical University (The First People’s Hospital of Zunyi), Zunyi City, Guizhou Province, People’s Republic of China; 2Pathology Department, The Third Affiliated Hospital of Zunyi Medical University (The First People’s Hospital of Zunyi), Zunyi City, Guizhou Province, People’s Republic of China; 3Imaging Department, The Third Affiliated Hospital of Zunyi Medical University (The First People’s Hospital of Zunyi), Zunyi City, Guizhou Province, People’s Republic of China
Correspondence: Xiaoyun Liu; Jian Lan, Department of Gynecology and Obstetrics, The Third Affiliated Hospital of Zunyi Medical University (The First People’s Hospital of Zunyi), No. 98, Fenghuang Road, Zunyi, Guizhou, 563000, People’s Republic of China, Email [email protected]; [email protected]
Abstract: Rhabdomyosarcoma, a common soft tissue malignant tumor in children and adolescents, is exceedingly rare in adults. Nevertheless, The outcome in adults is very poor, especially when compared to outcomes in children in whom significant improvements in treatment has been achieved. The first case was of a 24-year-old pregnant Chinese woman with a rare primary site of rhabdomyosarcoma in the perineal body. She presented with a perineal mass and was diagnosed during the second trimester of pregnancy, which is a very rare occurrence. The second case was a 70-year-old Chinese woman who suffered from right lower abdominal pain for 1 month and was misdiagnosed with an epithelial ovarian carcinoma. Mesenteric pleomorphic rhabdomyosarcoma was later confirmed by postoperative pathology. Both cases had undergone preoperative examination with chest and abdominal computed tomography (CT) and pelvic magnetic resonance imaging (MRI) examinations, as well as examination of complete blood count, liver panel, renal panel, and serum tumor markers. Diagnosis was based on histopathology and immunohistochemistry. The patient in the first case received chemotherapy after which the mass decreased in size; however, the patient was lost to follow-up. The second case underwent tumor resection and received chemotherapy and radiotherapy.
Keywords: rhabdomyosarcoma, adult, pregnancy, diagnosis, heterogeneity, prognosis
Introduction
Rhabdomyosarcoma is a primitive mesenchymal malignant tumor with a tendency for striated muscle tissue differentiation. It can arise in virtually any anatomic site of the body; however, the extremities are the most common site in adults, followed by the trunk, genitourinary tract, head, and neck.1
According to the clinical characteristics, microscopic morphology, and molecular genetic characteristics of this disease, rhabdomyosarcoma is classified into one of the following three variants: embryonal, alveolar, and pleomorphic (as the second case). Embryonal rhabdomyosarcoma can be further classified into three categories: conventional (as the first case), spindle-cell, and botryoid. The prognosis shows remarkable differences in patients with different histological types. Pleomorphic rhabdomyosarcoma has the worst prognosis, and embryonic rhabdomyosarcoma has a better prognosis than alveolar rhabdomyosarcoma. Likewise, in the further classification of embryonal rhabdomyosarcoma, botryoid and spindle-cell categories are associated with a more favorable prognosis than conventional rhabdomyosarcoma.
According to the proportion of rhabdomyoblasts, embryonic rhabdomyosarcoma can be divided into three types:2 primitive type (rhabdomyoblast < 10%, as the first case), intermediate type (rhabdomyoblast, 10–50%), and differentiated type (rhabdomyoblast > 50%). The primitive type is mainly composed of small round or spindle primitive mesenchymal cells, and well-differentiated rhabdomyoblasts are often difficult to see or are absent. In the intermediate type, in addition to poorly differentiated small round or spindle primitive mesenchymal cells, the rhabdomyoblasts, which are larger, round-to-oval cells with eosinophilic cytoplasm, can also be seen. Differentiated embryonic rhabdomyosarcoma composed entirely of well-differentiated round, spindle or polygonal rhabdomyoblasts is rare. However, it may be observed in relapsing or metastatic tumors after treatment.
Due to the remarkable clinical and biological heterogeneity of rhabdomyosarcoma, there remains some challenges in correctly diagnosing rhabdomyosarcoma, and limited information about this rare disease suggests that multiple adverse clinical manifestations are more common in adult patients. Moreover, there are no definitive, optimal regimens for the management of rhabdomyosarcoma in adults. This article presents the cases of two female adults with rhabdomyosarcoma. The first case was a 24-year-old young Chinese woman who was admitted to our Hospital at 18 weeks’ gestation with the progressive enlargement of a perineal mass of 15 days duration. A biopsy of the mass revealed embryonal rhabdomyosarcoma. The second case was a 70-year-old Chinese woman who suffered from right lower abdominal pain for one month. Mesenteric pleomorphic rhabdomyosarcoma was later confirmed by postoperative pathology. By presenting these two cases, we aimed to raise awareness of rhabdomyosarcoma in adults.
Case Presentation
Case #1
A 24-year-old pregnant young woman was admitted to our hospital because of a perineal mass that progressively enlarged for 15 days. She was gravida 3 para 0 (G3 P0), and clinical examination showed that the mass was approximately 6.0×5.0 cm on the left side of the perineal body. Microscopical examination of the biopsy specimen from the perineal mass showed that the tumor cells were predominantly undifferentiated, small round cells with hyperchromatic nuclei and scanty cytoplasm, similar to undifferentiated primitive mesenchymal cells. In some focal areas rhabdomyoblastic differentiation was manifested by strap-shaped cells with eosinophilic cytoplasm and cross-striations. In addition, large tumor cells with scattered or clustered hyperchromatic nuclei, also known as anaplastic rhabdomyosarcoma, were observed (Figure 1A). The histomorphology of the tumor was similar to that of embryonic striated muscle, which is composed of aggregated tumor cells and loose myxoid mesoderm tissue. The variable cellularity, hypocellular areas with myxoid stroma and densely cellular zones were alternately distributed (Figure 1B). A limited immunohistochemical panel showed strong and diffuse staining of desmin (Figure 1C) and actin in tumor cells. This immunophenotype supports the morphological diagnosis of embryonic rhabdomyosarcoma. CT and MRI scans revealed that the perineal body lesion (6.5 × 6.3 cm) invaded the internal and external sphincter of the anal canal and was suspected to involve the lower segment of the vagina (Figure 2A). Lymph node metastasis was found in the left pelvic wall, groin, and from the obturator fossa level to the level of the fifth lumbar vertebrae (Figure 2B). Chest CT showed no evidence of metastatic disease (Stage IV, Group 4, according to the Intergroup Rhabdomyosarcoma Study Group staging and grouping system).3
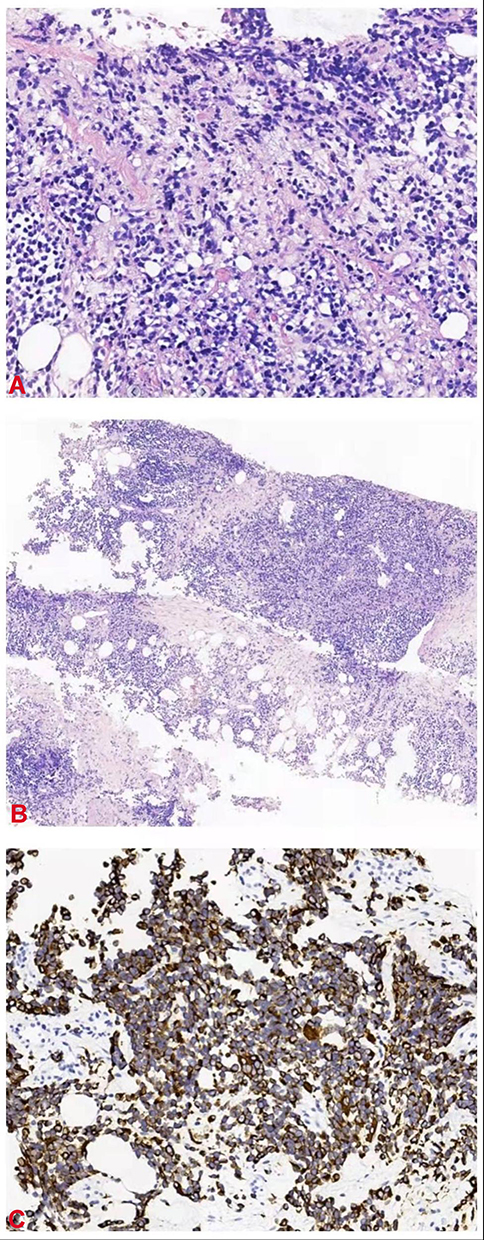 |
Figure 1 (A) Hematoxylin and eosin stain (HE), original magnification 200×. (B) HE stain 100×. (C) Desmin 400×. |
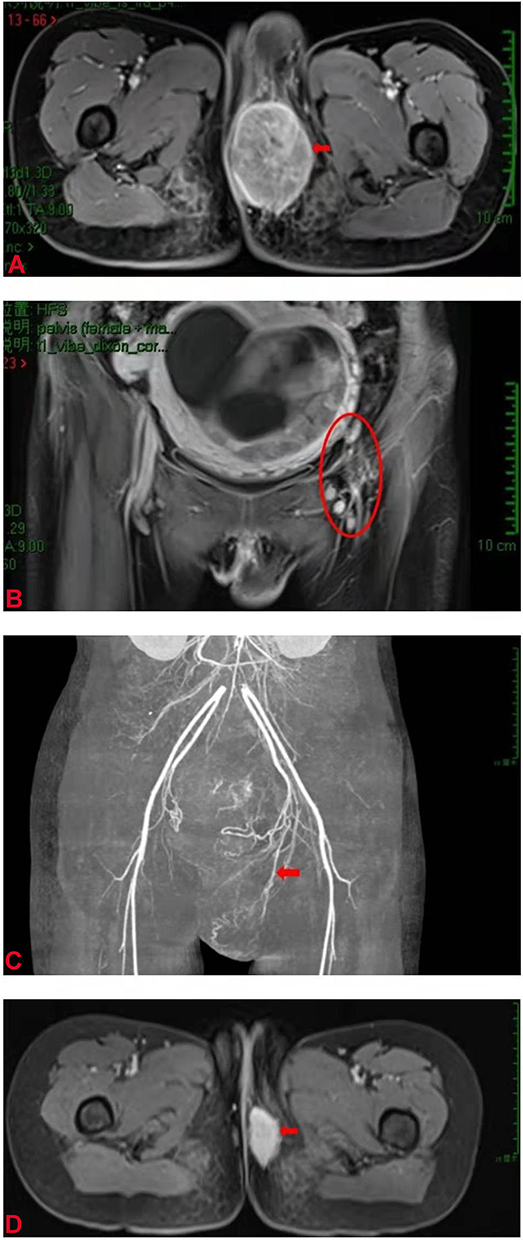 |
Figure 2 MRI and CT scans showed the focus of the perineal body. (A) MRI scan revealed a tumor in the perineum (arrow). (B) MRI scan showed enlarged lymph nodes in the left pelvic wall (circle). (C) Angiography showed that the blood supply of the lesion originated from the internal pudendal artery (arrow). (D) MRI scan showed that the focus was significantly reduced. |
Since a soft tissue sarcoma (STS) of the perineum was present, an artificial abortion was performed with the consent of the patient. The appearance of the dead infant was normal. Furthermore, angiography revealed that the blood supply of the focus was abundant and originated from the internal pudendal artery (Figure 2C). Therefore, local infusion chemotherapy (cisplatin and pirarubicin) was administered and permanent embolization of the supplying vessel was performed. The patient received ifosfamide intravenous infusion chemotherapy again after an interval of eight days. Later, the patient continued to receive intravenous infusion chemotherapy of ifosfamide combined with cisplatin for three cycles, and after a total of four cycles of chemotherapy, pelvic MRI showed that the focus had markedly decreased in size (2.1 × 3.4 cm) (Figure 2D), and the previously enlarged lymph nodes were unclear. However, the patient was referred to another hospital and lost to follow-up.
Case #2
A 70-year-old female patient G4 P3, presented to our hospital with right lower abdominal pain of one month duration. Physical examination and palpation revealed a mass in the right lower abdomen about 15.0×10.0 cm in size. Abdominal MRI and CT scanning (Figure 3A) revealed a cystic solid mass in the right lower abdomen about 16.6×10.1 × 9.9 cm in size. The mass may have originated from the ovary and metastasized to the peritoneum, omentum, and pelvic lymph nodes, but we also suspect that the mass originated from intestinal tract or mesentery with abdominal and pelvic effusion. Further cytological examination of the ascites revealed suspicious cancer cells, cancer antigen-125 (527.90 U/mL), and carcinoembryonic antigen (3.5 ng/mL). Electronic colonoscopy was normal. Thus, a diagnosis of epithelial ovarian carcinoma was established (stage III C, according to International Federation of Gynecology and Obstetrics, 2014). In the meantime, since the patient had hypoalbuminemia, typical chemotherapy with a combination of albumin paclitaxel and cisplatin was administered. The ascites significantly improved after two cycles of chemotherapy. However, abdominal CT showed that the tumor was larger than before (24.3 × 20.5×8.8 cm) (Figure 3B). Thus, interval cytoreductive surgery was performed after a multidisciplinary team decision. Intraoperatively, the uterus, ovaries, and fallopian tubes were unremarkable, and the polycystic tumor was located in the pelvic and abdominal cavity with a size of 20.0×18.0 cm; it adhered to the omentum majus and the mesentery of the small intestine. Complete tumor resection and omentectomy was performed. Postoperative pathology confirmed mesenteric pleomorphic rhabdomyosarcoma with omentum majus metastasis (Stage IV and Group 4).
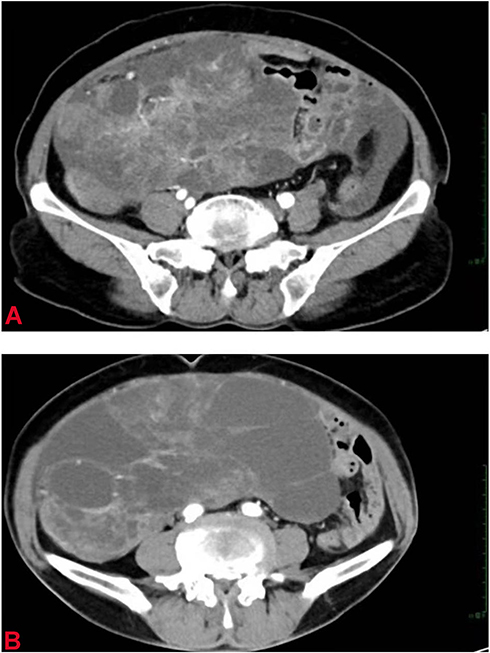 |
Figure 3 (A) CT shows a cystic-solid tumor in the abdomen. (B) CT shows a larger abdominal tumor (after two cycles of chemotherapy). |
Histopathologically, microscopic examination of the postoperative specimen showed obvious atypical large, round or polygonal tumor cells, and tadpole, strap-shaped, and racquet cells. The cytoplasm was deeply eosinophilic, similar to the rhabdomyoblasts of embryonic rhabdomyosarcoma, but the cell-size was larger with an irregular shape, and it was difficult to find cross-striations in the cytoplasm. Additionally, the cells were large, deformed, with deeply eosinophilic cytoplasm, which is the most helpful morphological feature for the diagnosis of this histological type. Some tumor cells had a rhabdoid shape, that is, the nucleus is large, round or ovoid, often tilted to one side, with vesicular chromatin and eye-catching nuclei and abundant cytoplasm. Furthermore, multinucleated giant cells were seen in some foci, resembling pleomorphic undifferentiated sarcoma (Figure 4A).
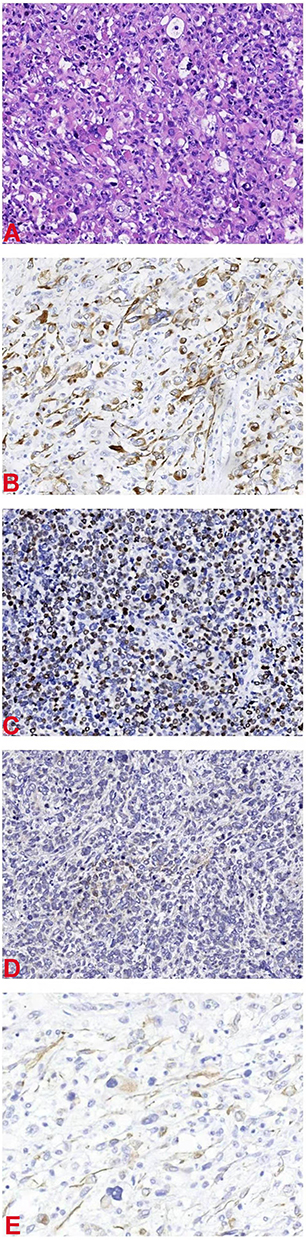 |
Figure 4 (A) Hematoxylin and eosin stain 200×. (B) Desmin 200×. (C) MyoD1 200×. (D) Myogenin 200×. (E) Actin 200×. |
Immunohistochemical examination showed that desmin, MyoD1, myogenin and actin staining (Figure 4B–E) were positive in tumor cells. After surgery, chemotherapy and/or radiotherapy will be continued for this patient.
Discussion
Rhabdomyosarcoma is a common STS in children and adolescents but is less common in adults.4 It is typically a rapidly progressive tumor in patients who otherwise seem healthy. Rhabdomyosarcoma is difficult to diagnose due to its relatively rare occurrence, its significant clinical and biological heterogeneity (eg, numerous primary anatomic sites, population of different ages, multiple histological types, and varied extent of disease at the time of diagnosis). Thus, in 1972, the Intergroup Rhabdomyosarcoma Study Group (IRSG) was established by three pediatric cooperative cancer study groups to combine their patients and investigative resources for further research. To date, the IRSG has completed several continuous trials of rhabdomyosarcoma treatment in thousands of patients. These trials, along with those conducted by other international organizations, have improved patient outcomes, identified prognostic variables, and developed risk-based treatments.5,6
Given the relative rarity of this disease, there is limited data available on the management of rhabdomyosarcoma in adults, and the National Comprehensive Cancer Network guidelines for STSs strongly encouraged that all patients consult a specialist with expertise in the treatment of rhabdomyosarcoma.7 A multidisciplinary team of pediatric, medical, surgical, and radiation oncologists is highly recommended for patient evaluation. All patients require multimodal treatment planning (surgery, radiotherapy, and chemotherapy) and risk stratification.
Combination chemotherapy, which is used to reduce the size of the primary tumor and to eradicate gross or microscopic metastatic foci, is the mainstay treatment in children. Generally, surgical resection is performed if it will not impair function or cosmesis. Complete resection cannot be performed in most patients (as in our first case) due to the inconvenient location of the primary site of most rhabdomyosarcomas. For some patients with the best prognostic factors, resection is not necessary because these tumors are highly curable with a combination of chemotherapy and radiotherapy.8 Local control of the primary tumor is a prerequisite condition for long-term management of this disease, usually accomplished by surgery, radiotherapy, or both.
As early as the 1960s, a combination regimen including vincristine, actinomycin and cyclophosphamide was proved to significantly improve the response rate.9 This approach became the standard chemotherapy regimen for pediatric non-metastatic rhabdomyosarcoma (intermediate or high-risk).10 This regimen is well tolerated, with acute toxic effects that are mild and late toxic effects that are minimal.9 The IRSG study showed that the 5-year failure-free survival rates of newly diagnosed low-risk rhabdomyosarcoma patients treated with vincristine and actinomycin were similar to those treated with vincristine, actinomycin and cyclophosphamide (89% and 85%, respectively), suggesting that vincristine and dactinomycin may be suitable candidates for patients with newly diagnosed, low-risk rhabdomyosarcoma.11 Moreover, omitting cyclophosphamide, an alkylating agent, helps reduce the risk of secondary hematological diseases and infertility.
Chemotherapy regimens used in adults with rhabdomyosarcoma are usually derived from the pediatric clinical trials on rhabdomyosarcoma conducted by the IRSG. In the MD Anderson Cancer Center study, the 10-year overall, disease-free, and metastasis-free survival rates for adults with rhabdomyosarcoma treated with chemotherapy regimens containing vincristine and cyclophosphamide with dactinomycin or doxorubicin were 47%, 45%, and 59%, respectively.12 At present, agents such as carboplatin, irinotecan, topotecan, and vinorelbine have also shown significant efficacy in the treatment of pediatric patients with metastatic, relapsing, or refractory rhabdomyosarcoma.13,14
Surgery is the primary treatment for patients with pleomorphic rhabdomyosarcoma (74% vs 34% for non-pleomorphic histology), and the event-free survival rate was 37% for patients who underwent complete resection compared to 0% in patients with unresectable tumors.4 The incidence of this type increases with age, and one of the historic dilemmas of this disease has been its true relationship with other types of rhabdomyosarcoma. For example, molecular inheritance studies have shown that compared with other types of rhabdomyosarcoma, the imbalance region of pleomorphic rhabdomyosarcoma is more similar to that of malignant fibrous histiocytoma, which is also more common in adults.15
In terms of radiotherapy, this treatment is beneficial to patients with advanced disease. It is typically used to control residual bulky or microscopical tumor, especially when the tumor is located in an unresectable site. At the same time, with the continuous improvement of diagnostic technology, advanced radiotherapy techniques such as conformal radiotherapy, intensity-modulated radiotherapy and proton therapy reduce radiation toxic complications and provide a good local control effect in the treatment of rhabdomyosarcoma in children.6,16
Historically, all patients with this rare tumor were believed to have a dismal prognosis. However, for early pediatric patients with localized disease, favorable sites and histological types, with aggressive multimodality therapy, long-term survival rates of nearly 90% can be achieved.17 This manifestation has not been shown in adults, who continue to have a very poor prognosis. It is paramount to recognize the disease as early as possible, but there are still some challenges in making a correct diagnosis. Rhabdomyosarcoma has significant clinical and biological heterogeneity, so it is easily misdiagnosed as a more common disease. For example, the patient who primary site in the genital tract was misdiagnosed as benign polyps initially, and relapsed within a short period of time after treatment. Later, the initial biopsy specimens were reinterpreted as embryonal rhabdomyosarcoma.17 However, benign polyps lack specific expression of rhabdomyosarcoma such as mitotic activity, cambium layer, and rhabdomyoblasts that would help differentiate between them. Additionally the differential diagnosis of botryoid rhabdomyosarcoma is Mullerian adenosarcoma because both are characterized by the condensation of atypical spindle sarcoma cells under the epithelium (the so-called cambium layer).18 The absence of phyllodes-like areas and intraluminal papillae excludes adenosarcoma. Additionally, as in the second case, the tumor was initially misdiagnosed as epithelial ovarian cancer based on the clinical suspicion of common and frequently-occurring diseases, but this mistake may be avoided by carefully examining the blood supply of the lesions in three-dimensional angiography.
Rhabdomyosarcoma’s spread is hematogenous with common sites of metastases seen in the lung parenchyma, bones, locoregional lymph nodes, liver, and brain. Staging procedures in new patients include CT or MRI scanning of the primary site, CT scanning of the chest, bone scanning, examination of bone marrow aspirates, and lumbar puncture (when the primary site is located in a parameningeal site). Positron emission tomography scanning may contribute to initial staging because of the possibility of lymph node metastases and the appearance of unusual sites of initial metastatic disease in adult patients.19
Immunohistochemically, tumor cells express desmin, MyoD1, myoglobin, myogenin, and muscle-specific actin. Occasionally, tumor cells can also express WT-1, which has a significant value in the differential diagnosis from other small round cell tumors. Additionally, the expression of related immune antibodies also depends on the degree of tumor differentiation. In both reported cases, a limited immunohistochemical panel showed strong and diffuse staining of the tumor cells for desmin and actin, both of which are sensitive but not highly specific markers of rhabdomyoblastic differentiation. When the morphologic and immunohistochemical findings of a tumor are equivocal, specific findings on electron microscopy or the detection of characteristic chromosomal translocations are helpful for diagnosis (eg, more than 80% of patients with alveolar rhabdomyosarcoma have one of two types of chromosome translocations).20 Moreover, the genetic characteristics of the tumor cells may provide more important information in the future.21
The prognostic risk factors of pediatric patients include the site of the primary tumor, the extent of the initial surgical resection, the age at diagnosis, the histologic type, the tumor-node-metastasis stage, and most importantly, the response to treatment. In the MD Anderson Cancer Center study, the 10-year metastasis-free survival was 72% for patients with rhabdomyosarcoma who responded to chemotherapy compared to 19% for patients who failed to respond.12 Meanwhile, Sultan et al reported that the primary site, histological subtype, and disease stage were significantly correlated with the prognosis of rhabdomyosarcoma in adults.1 Hawkins et al also reported that postoperative margin status predicted disease-specific survival in adults (105 months in patients who underwent complete resection and 9 months in patients with positive margins).22
The limited information regarding this disease suggests that multiple adverse clinical factors seem to converge in adults with rhabdomyosarcoma, although major discrepancies in biology have not been described. Related studies has shown that a more pronounced expression of multidrug-resistance proteins in adult patients, and the tolerance of adults to intensive treatment is lower. These factors may be the reason for the poor prognosis of patients with rhabdomyosarcoma.23 In a SEER database analysis, the 5-year, 10-year, and 15-year survival rates including all patients were estimated to be 46%, 42% and 41%, respectively. The results of children were significantly better than adults. The corresponding child survival rates were 61%, 58%, and 57%, and the adult survival rates were 27%, 21%, and 18%, respectively. The most outstanding discrepancies were that in patients with early stages and localized diseases, the 5, 10, and 15-year survival rates were much lower in adults (estimated at 47%, 36%, and 30%, respectively) than in children (82%, 80%, and 79%, respectively).1 These disparities might reflect differences in pathogenesis, and what is controversial is whether chemotherapy, the main therapy of pediatric rhabdomyosarcoma treatment, should be used in all adults with rhabdomyosarcoma. In a retrospective study, adult patients were more likely to show similar outcomes to children when receiving a treatment strategy more similar to adult STS, which is based on standard surgery-based treatment, usually supplemented with radiotherapy, but rarely with chemotherapy.4
In conclusion, rhabdomyosarcoma is a rare soft tissue malignant tumor in adults, and it has significant clinical and biological heterogeneity. Thus, there are still some challenges to make a correct diagnosis. When the morphologic and immunohistochemical findings of a tumor are equivocal, specific findings on electron microscopy, or the detection of characteristic chromosomal translocations are helpful for diagnosis. The prognosis of adults is significantly worse than that of children, and the reasons for this continued deterioration in outcome remain largely unknown. The expression of multidrug resistance protein is more obvious in adult patients, and the lower tolerance of adults to intensive treatment may be the reason of poor prognosis in adults with rhabdomyosarcoma. Meanwhile, due to the limited information available about this rare disease, there are no definitive, optimal regimens for the treatment of rhabdomyosarcoma in adults. Additional studies on the management plan and the pathogenesis of rhabdomyosarcoma in adults are needed.
Abbreviations
CT, computed tomography; MRI, magnetic resonance imaging; STS, soft tissue sarcoma; IRSG, Intergroup Rhabdomyosarcoma Study Group.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the first author on reasonable request.
Ethics Approval and Informed Consent
The writing and publication of the two case reports were approved by the Third Affiliated Hospital of Zunyi Medical University (The First People’s Hospital of Zunyi).
Consent for Publication
Written informed consent was obtained from both patients for the publication of these case reports and associated images. Copies of written consent are available upon request.
Acknowledgment
The authors would like to thank our patients and her family for their support.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, agreed to the submitted journal, and agreed to be accountable for all aspects of the work.
Funding
This study was supported by the Key Project of Zunyi Science and Technology Plan [Zunshi Science and Technology Co-operative Society (2017) No. 28].
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2600 patients. J Clin Oncol. 2009;27:3391–3397. doi:10.1200/JCO.2008.19.7483
2. Schmidt D, Reimann O, Treuner J, Harms D. Cellular differentiation and prognosis in embryonal rhabdomyosarcoma. A report from the Cooperative Soft Tissue Sarcoma Study 1981 (CWS 81). Virchows Arch a Pathol Anat Histopathol. 1986;409:183–194. doi:10.1007/BF00708327
3. Crist WM, Anderson JR, Meza JL, et al. Intergroup rhabdomyosarcoma study-IV: results for patients with nonmetastatic disease. J Clin Oncol. 2001;19:3091–3102. doi:10.1200/JCO.2001.19.12.3091
4. Ferrari A, Dileo P, Casanova M, et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer. 2003;98:571–580. doi:10.1002/cncr.11550
5. Borinstein SC, Steppan D, Hayashi M, et al. Consensus and controversies regarding the treatment of rhabdomyosarcoma. Pediatr Blood Cancer. 2018;65:e26809. doi:10.1002/pbc.26809
6. Terezakis SA, Wharam MD. Radiotherapy for rhabdomyosarcoma: indications and outcome. Clin Oncol. 2013;25(1):27–35. doi:10.1016/j.clon.2012.07.009
7. von Mehren M, Randall RL, Benjamin RS, et al. Soft tissue sarcoma, Version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2021;21:86–88.
8. van Ewijk R, Vaarwerk B, Breunis WB, et al. The value of early tumor size response to chemotherapy in pediatric rhabdomyosarcoma. Cancers. 2021;13:510. doi:10.3390/cancers13030510
9. Friedmann AM, Tarbell NJ, Schaefer PW, Hoch BL. Case records of the Massachusetts general hospital. Weekly clinicopathological exercises. Case 4-2004. A nine-month-old boy with an orbital rhabdomyosarcoma. N Engl J Med. 2004;350:494–502. doi:10.1056/NEJMcpc030036
10. Bisogno G, De Salvo GL, Bergeron C, et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): a multicentre, open-label, randomised, Phase 3 trial. Lancet Oncol. 2019;20:1566–1575. doi:10.1016/S1470-2045(19)30617-5
11. Raney RB, Walterhouse DO, Meza JL, et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J Clin Oncol. 2011;29:1312–1318. doi:10.1200/JCO.2010.30.4469
12. Liu YT, Wang CW, Hong RL, Kuo SH. Prognostic factors and treatment outcomes of adult patients with rhabdomyosarcoma after multimodality treatment. Anticancer Res. 2019;39:1355–1364. doi:10.21873/anticanres.13249
13. Setty BA, Stanek JR, Mascarenhas L, et al. VIncristine, irinotecan, and temozolomide in children and adolescents with relapsed rhabdomyosarcoma. Pediatr Blood Cancer. 2018;65:e26728. doi:10.1002/pbc.26728
14. Defachelles AS, Bogart E, Casanova M, et al. Randomized Phase II trial of vincristine-irinotecan with or without temozolomide, in children and adults with relapsed or refractory rhabdomyosarcoma: a European paediatric soft tissue sarcoma study group and innovative therapies for children with cancer trial. J Clin Oncol. 2021;39:2979–2990. doi:10.1200/JCO.21.00124
15. Feroce F, Cantile M, Aquino G, et al. Molecular characterization of a bladder pleomorphic rhabdomyosarcoma in an adult patient. Pathol Res Pract. 2020;216:153033. doi:10.1016/j.prp.2020.153033
16. Das M. Intelligence outcomes after proton versus photon therapy. Lancet Oncol. 2020;21:e16. doi:10.1016/S1470-2045(19)30802-2
17. Garrett LA, Harmon DC, Schorge JO. Embryonal rhabdomyosarcoma of the uterine corpus. J Clin Oncol. 2013;31:e48–50. doi:10.1200/JCO.2012.43.1841
18. Park HM, Park MH, Kim YJ, et al. Mullerian adenosarcoma with sarcomatous overgrowth of the cervix presenting as cervical polyp: a case report and review of the literature. Int J Gynecol Cancer. 2004;14:1024–1029. doi:10.1136/ijgc-00009577-200409000-00042
19. Tateishi U, Hosono A, Makimoto A, et al. Comparative study of FDG PET/CT and conventional imaging in the staging of rhabdomyosarcoma. Ann Nucl Med. 2009;23:155–161. doi:10.1007/s12149-008-0219-z
20. Song W, Platteel I, Suurmeijer AJH, van Kempen LC. Diagnostic yield of NanoString nCounter FusionPlex profiling in soft tissue tumors. Genes Chromosomes Cancer. 2020;59:318–324. doi:10.1002/gcc.22834
21. Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: a report from the children’s oncology group. J Clin Oncol. 2002;20:2672–2679. doi:10.1200/JCO.2002.03.137
22. Hawkins WG, Hoos A, Antonescu CR, et al. Clinicopathologic analysis of patients with adult rhabdomyosarcoma. Cancer. 2001;91:794–803. doi:10.1002/1097-0142(20010215)91:4<794::AID-CNCR1066>3.0.CO;2-Q
23. Komdeur R, Klunder J, van der Graaf WT, et al. Multidrug resistance proteins in rhabdomyosarcomas: comparison between children and adults. Cancer. 2003;97:1999–2005. doi:10.1002/cncr.11259
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.