")
Back to Journals » Adolescent Health, Medicine and Therapeutics » Volume 5
Rhabdomyosarcoma in adolescent and young adult patients: current perspectives
Authors Egas-Bejar D, Huh W
Received 22 January 2014
Accepted for publication 11 April 2014
Published 17 June 2014 Volume 2014:5 Pages 115—125
DOI https://doi.org/10.2147/AHMT.S44582
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Daniela Egas-Bejar, Winston W Huh
Division of Pediatrics, The Children’s Cancer Hospital, The University of Texas MD Anderson Cancer Center, Houston, TX, USA
Abstract: Rhabdomyosarcoma (RMS), a malignant tumor of mesenchymal origin, is the third most common extracranial malignant solid tumor in children and adolescents. However, in adults, RMS represents <1% of all solid tumor malignancies. The embryonal and alveolar histologic variants are more commonly seen in pediatric patients, while the pleomorphic variant is rare in children and seen more often in adults. Advances in the research of the embryonal and alveolar variants have improved our understanding of certain genes and biologic pathways that are involved in RMS, but much less is known for the other variants. Multimodality therapy that includes surgery and chemotherapy with or without radiation therapy is the mainstay of treatment for RMS. Improvements in the risk stratification of the pediatric patients based on presurgical (primary tumor site, tumor size, regional lymph node involvement, presence of metastasis) and postsurgical parameters (completeness of resection or presence of residual disease or metastasis) has allowed for the treatment assignment of patients in different studies and therapeutic trials, leading to increases in 5-year survival from 25%–70% over the past 40 years. However, for adult patients, in great part due to rarity of the disease and the lack of consensus on optimal treatment, clinical outcome is still poor. Many factors have been implicated for the differing outcomes between pediatric RMS versus adult RMS, such as the lack of standardized treatment protocols for adult RMS patients and the increased prevalence of advanced presentations. Now that there are increased numbers of survivors, we can appreciate the sequelae from therapy in these patients, such as bone growth abnormalities, endocrinopathies, and infertility. Improvements in risk stratification have led to clinical trials using lower doses of chemotherapy or radiation therapy with the intention of decreasing the incidence of side effects without compromising survival outcome.
Keywords: rhabdomyosarcoma, soft-tissue sarcoma, late effects
Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children and adolescents, accounting for approximately 5% of all pediatric cancers and about one-half of all soft tissue sarcomas. It is the third most common extracranial solid tumor in children after neuroblastoma and Wilms tumor.1 Among the population age group between 0–19 years, an estimated 350 new cases of RMS are diagnosed each year in the United States.2 There is a slight male predominance, and approximately two-thirds of the cases are diagnosed in children younger than 6 years of age.1,3 The two most common histologic variants encountered in children and adolescents are the embryonal (ERMS) and alveolar (ARMS) subtypes, while the botryoid and spindle cell variants are also encountered. The ERMS mainly occurs in the head and neck and genitourinary regions, and ERMS demonstrates a bimodal age of distribution, with a larger peak between 0–5 years and a smaller peak in adolescence. More than one-half of ERMS cases occur before the age of 5 years.4 Conversely, ARMS is more likely to occur in adolescents.
RMS is infrequent in adults; soft tissue sarcomas represent <1% of all adult solid tumor malignancies, and RMS accounts for only 3% of all soft tissue sarcomas in adults.5 In this patient population, RMS can occur as a primary malignancy, but it can also be a component of a heterogeneous malignancy, such as nongerm cell or teratomatous malignant tumor.6 The histologic distribution of RMS in adults also differs from young children. The pleomorphic subtype and RMS not otherwise specified (RMS NOS) are more frequently encountered in adult patients.7
RMS can arise in a variety of anatomic sites throughout the body. The most common primary tumor sites include the head and neck region (35%), followed by the genitourinary and extremity primaries.8 However, the epidemiology of primary tumor presentation is dependent upon the histologic variant and age. For example, adolescent patients are more likely to have the ARMS compared to younger patients. Also, adult RMS patients tend to have an increased likelihood of primary tumors occurring at unfavorable anatomic sites.7 Given some of the clinical and histologic differences between young children and older patients with RMS, the purpose of this review is to provide an overview of the known biology of RMS, diagnosis, clinical management, and late effects from therapy, while highlighting certain aspects that are relevant to the adolescent and young adult population.
Associated genetic syndromes
Most cases of RMS are currently considered sporadic in origin, but there have been associations of RMS with a number of genetic syndromes. In one study, approximately 30% of children with RMS had a congenital anomaly (most commonly affecting the gastrointestinal system, central nervous system, and genitourinary system), which hints at other potential genetic syndromes that have yet to be described.9 Familial syndromes associated with RMS include: Li–Fraumeni syndrome; neurofibromatosis type 1; Rubinstein–Taybi syndrome; Beckwith–Wiedemann syndrome (overgrowth syndrome with macrosomia, organomegaly, hemihypertrophy, and omphalocele); Costello syndrome (mental retardation, neonatal feeding problems, coarse facies, low birth weight, hirsutism, loose soft skin); Noonan syndrome; hereditary retinoblastoma; and Gorlin basal cell carcinoma nevus syndrome.10–14
RMS is the most frequently observed pediatric cancer in classic Li–Fraumeni cancer syndrome families. Li–Fraumeni syndrome has been linked to germline mutations (de novo or inherited) of the tumor suppressor gene for p53. Family members of afflicted children have an increased frequency of other types of cancers, such as breast cancer, acute leukemia, soft tissue and bone sarcomas, adrenocortical carcinoma, and brain tumors.3
RMS pathology and tumor biology
RMS is recognized as one of the small, round blue cell tumors of childhood, which includes lymphoma, neuroblastoma, Ewing’s sarcoma, and primitive neuroectodermal tumors. The cell of origin for RMS remains a subject of debate. It has been suggested that committed muscle stem cells and also multipotent mesenchymal stem cells can give rise to RMS.15,16
As previously noted, the two major histologic subtypes of RMS in children are ERMS and ARMS. In adults, RMS NOS and pleomorphic RMS are more common. Although they share a commonality of being mesenchymal-derived sarcomas, each subtype likely represents a distinct entity with its own unique biology for formation and possibly with different cells of origin. For example, the expression of the PAX7 gene in ERMS and in the myogenic satellite cells points to these latter cells as possible cells of origin of this subtype of RMS. Given that the upregulation of PAX7 expression is not seen in cases of ARMS (that have PAX3-FKHR or PAX7-FKHR translocation), this supports the belief that the two histologic subtypes may originate from different cells.17
The development of experimental animal models has significantly contributed to our current understanding of the molecular factors that play a role in RMS genesis. Studies derived from these models have shown chemical and physical (heavy metals, ionizing radiation, polycyclic aromatic hydrocarbons), but also biological (viral proteins, p53 pathway alterations, rat sarcoma [RAS] or hepatocyte growth factor alterations) triggers of RMS.18 These studies have established an association of RMS with other diseases, like muscular dystrophy. This was mainly observed in aged dystrophic mice, suggesting that cumulative deoxyribonucleic acid damage – over time, together with paracrine signals – could direct the muscle cells and their progenitors toward tumor formation.19 Other animal models have also detected “druggable” kinases that are overexpressed in both human and mouse.20 Taken together, the use of animal models has not only helped us to understand RMS biology, but it also has provided an avenue to find new targets for specific histologic subtypes of RMS.
ERMS represents approximately 70% of all childhood RMS. Under light microscopy, ERMS tumor resembles immature skeletal muscle. Almost all ERMS tumors demonstrate loss of heterozygosity (LOH) in region 11p15.5; and chromosomal fragment transfer studies have demonstrated that this region represses tumor cell growth, suggesting the presence of tumor suppressor genes.14 This LOH region includes genes for IGF2, H19, and CDKN1C, all subject to parental imprinting. In ERMS, the LOH results in loss of the maternal IGF2 allele and duplication of the paternal allele, resulting in the overexpression of IGF2.21
Most ARMS tumors have a microscopic appearance that resembles lung alveoli, but a solid variant has also been identified, so that a certain percentage of alveolar pattern is not required for the diagnosis of ARMS.22 Specific chromosomal translocations are detected in 70%–80% of ARMS.23 Translocation t(2;13)(q35;q14) occurs in 60% of ARMS, and translocation t(1;13)(p36;q14) occurs in approximately 20% of ARMS. These translocations result in the expression of chimeric transcription factors PAX3-FKHR (PAX3-FOXO1) or PAX7-FKHR (PAX7-FOXO1), respectively. PAX-FKHR is a pleiotropic fusion protein that: stimulates proliferation; induces angiogenesis; inhibits apoptosis; activates the myogenic program; and inhibits simultaneously terminal differentiation.4 Approximately 20% of ARMS are translocation negative, and – interestingly – fusion-negative ARMS have allelic imbalance and LOH patterns that are indistinguishable from conventional ERMS cases. The clinical behavior and molecular characteristics of ARMS without a fusion gene are indistinguishable from ERMS cases and significantly different from fusion-positive ARMS cases.24,25 Thus, fusion gene status may play a role as a factor in risk stratification in RMS, irrespective of histology.
Other molecular aberrations found in both ARMS and ERMS involve tumor suppressor genes, such as p53 and Rb1. RMS cell lines showed a high frequency of mutations in p53 (up to 60%), but a lower frequency was observed in tumors (up to 30%).14,26–28 Mutations in CDKN2A have been found in all RMS cell lines but only 25% of RMS primary tumors.29 MYCN expression has been found in almost all RMS primary tumors, with higher levels in ARMS.30 Dysregulation of RAS genes has been associated with ERMS.31 Elevated expression of MET has also been associated with more advanced disease.32,33 Recent studies have also demonstrated that elevated messenger RNA levels, and protein expression of ALK have been associated with ARMS and worse clinical outcome.34,35 Even though the histological and genetic characteristics of ARMS and ERMS are different, these two RMS subtypes have a similar phenotype of defective differentiation, which occurs through multiple signaling pathways but involves – in many cases – the myogenic transcription factor MyoD.36
Pleomorphic RMS is more common in adults compared to children. It typically arises in the deep soft tissues of the extremities of patients older than 45 years. It is an aggressive neoplasm that is probably closer, both biologically and clinically, to adult, high-grade soft-tissue sarcomas than to pediatric RMS.37 Unfortunately, there is very little published data regarding the known biologic pathways involved in pleomorphic RMS when compared to the other major subtypes. One animal study was able to demonstrate the development of the pleomorphic RMS tumors in p53-deficient mice when KRAS was overexpressed.38 Another study suggested that ERMS and the undifferentiated pleomorphic sarcoma form a continuum, with mutant p53, Ptch1, or Rb1 in satellite cells, giving rise to undifferentiated sarcoma and ERMS originating from myoblasts that express satellite cells markers.39
Diagnosis
Patients with RMS can present with an asymptomatic mass or with signs and symptoms that are associated with the primary tumor site and are related to mass effect or complications that are secondary to the tumor. For example, patients with parameningeal tumors may initially present with cranial nerve palsies, while patients with intra-abdominal tumors may present with bowel obstruction. Pain is not a consistent presenting symptom and usually arises due to mass effect and compression upon adjacent neural structures. For active adolescents and young adults, swelling in certain areas, such as the extremities and the paratesticular site, is often mistaken as a sequelae of perceived musculoskeletal injury, and because there is often no associated pain, the area of question may not be brought to medical attention for some time.
Imaging studies should include computed tomography (CT) scan or magnetic resonance imaging of the primary tumor to determine the size and possible involvement of vital organ structures; these are parameters to be considered when surgical resection is planned. For tumors arising from the head, imaging studies should include full views of the neck to evaluate the cervical lymph node chains. Open biopsy of the suspected mass is often used to confirm the diagnosis of RMS. Biopsy incision should be oriented in a direction that allows for later wide tumor excision, respecting tissue planes. Core needle biopsy may be performed for small lesions in areas that will be treated with chemotherapy/radiation or for metastatic disease.40
In the majority of pediatric cancer centers, a metastatic evaluation includes bone marrow biopsy and technetium methylene diphosphonate bone scintigraphy (99mTc MDP bone scan) for the detection of marrow involvement and bone metastases, respectively. A CT scan of the chest is performed to evaluate for the presence of lung metastasis or thoracic lymph node involvement. A lumbar puncture for cerebrospinal fluid collection should be performed for patients with parameningeal tumors, especially those exhibiting intracranial extension. However, the metastatic evaluation for pediatric RMS patients has been under scrutiny and may need to be tailored in the future to a patient’s presenting characteristics. For example, in one study, a bone marrow aspirate biopsy and bone scan were found to be of low yield in at least one-third of patients with RMS (specifically, patients with ERMS and T1 disease because they were at particularly low risk of metastatic disease to bone and bone marrow).41
There has been significant interest in evaluating the utility of 18–fluorodeoxyglucose positron emission tomography (FDG PET) scans in evaluating disease extent. A small number of studies have compared the use of FDG PET CT and conventional imaging (CT, magnetic resonance imaging, and bone scan) in staging pediatric RMS.42,43 The PET CT was found to be better in identifying nodal, bone, and bone marrow involvement (Figure 1), while conventional imaging was better for the detection of pulmonary nodules. The authors of one study even recommended that PET CT could be used for RMS staging in lieu of 99mTc bone scan.43 In adults with sarcoma, PET has been used to monitor patients with sarcoma to evaluate tumor grade, staging, and treatment monitoring, as well as in prediction of survival.44
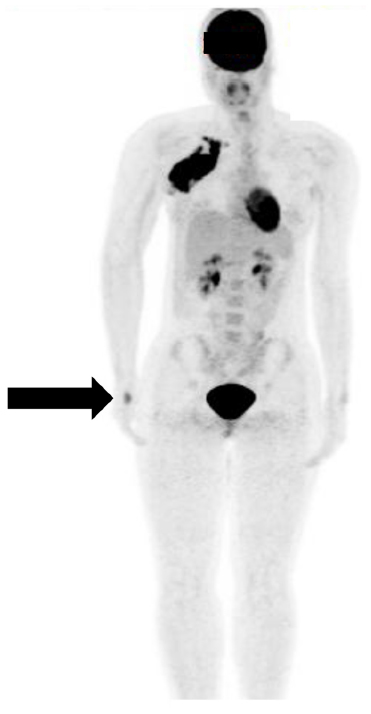 | Figure 1 FDG PET scan of an adolescent female who presented with right axillary mass. |
Other special anatomic sites merit special consideration when evaluating the extent of the RMS disease involvement. For patients with paratesticular tumors and treated according to the Children’s Oncology Group (COG) treatment protocols, an ipsilateral retroperitoneal lymph biopsy surgery is recommended for all patients ≥10 years of age. The presence of lymph node involvement would influence the treatment plan by introducing radiation therapy (RT). However, this practice is not shared by other international collaborative groups due to concern about the risk of complications from the surgery, such as nerve damage or bleeding. For patients with extremity tumors, a sentinel node biopsy is recommended.
Staging
In 1972, the Intergroup Rhabdomyosarcoma Study Group (IRSG) initially developed a grouping system for risk stratification of RMS patients based on the postoperative extent of disease (Table 1). A pretreatment clinical staging system (based on clinical examination and imaging studies) was subsequently developed to account for pretreatment patient characteristics (Table 2). This staging system takes into consideration the size and site of the primary tumor as well as the degree of invasion of adjacent structures, the presence or absence of nodal involvement, and presence of absence of distant spread (metastasis). The IRSG has now become the Soft Tissue Sarcoma Committee of COG, and current COG treatment protocols utilize a combination of the group and stage criteria for risk stratification of patients and treatment assignment. While other international collaborative groups have their own risk stratification systems, we will mainly highlight the COG system for this review.
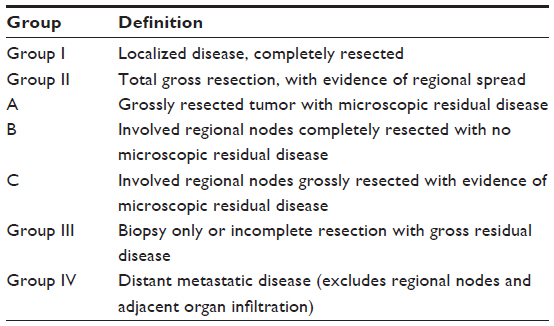 | Table 1 COG clinical group classification of RMS |
 | Table 2 Pretreatment TNM staging system for RMS |
Unlike pediatric RMS, which employs a more complex risk stratification system, only a TNM Classification of Malignant Tumors (TNM system) is used for adult patients to assign treatment.
Prognostic factors
Given that most RMS clinical trials have been performed mainly in children, the characterization of prognostic factors has been described mainly for this patient population. Favorable prognostic factors in RMS are: undetectable distant metastases at diagnosis; favorable anatomic sites (orbit, nonparameningeal head/neck, and genitourinary nonbladder/prostate regions); grossly complete surgical removal of the localized tumor at the time of diagnosis; ERMS/botryoid histology; tumor size ≤5 cm; and age older than 1 but younger than 10 years at diagnosis.45–47 The presence of regional lymph node disease alters the prognosis for patients with ARMS with outcomes similar to distant metastatic disease, thus suggesting the need for more aggressive therapy for patients with ARMS and regional lymph node disease.46,48
With regards to molecular markers, the presence of PAX3/FOXO1 fusion has been proposed by one study as a molecular prognostic marker of poor outcome in patients with nonmetastatic ARMS.49 However, the German cooperative group did not find the same conclusions.50 Comparison between the two studies is somewhat complicated since the two groups used different treatment protocols. As previously described, other genes, such as MET, MYCN, and ALK, have been proposed as potential molecular prognostic markers based on retrospective pathologic studies, but they need to be validated as part of biologic correlates for future prospective clinical trials.
Other studies evaluating various clinical parameters, such as tumor diameter versus tumor volume at diagnosis, response to initial chemotherapy, and weight loss during therapy, were not of significant prognostic value.51–53
The outcome of adolescents and adults with RMS appears to be worse than that of children. A recent series demonstrated a 5-year overall survival rate of 27% in adults versus 61% in children.7 Adult RMS has a significant incidence of metastatic recurrence.54 Tumors in adults were more likely to be at an unfavorable site and to have histologies that are unusual during childhood, particularly the pleomorphic and RMS NOS subtype. The most significant difference in outcome was in patients with localized disease: 5-year survival estimates were 82% for children and 47% for adults.7 Another study found that adolescent patients tended to have an increased likelihood for an unfavorable primary tumor site, metastatic disease, and lymph node involvement.55
Many factors have been implicated in the different outcomes from pediatric versus adult RMS. One theory is that most pediatric RMS patients are referred to specialized centers, benefiting from more accurate diagnosis and evaluation that leads to improved risk stratification and optimal treatment. The proportion of adult patients with RMS of unknown histologic subtype has been reported to be up to three times higher than that reported in children.56 Other factors include advanced disease presentation in adults, absence of standardized protocols for adult patients, and low inclusion in trials for adolescents and young adults.55,57
Treatment
Currently, multimodality treatment that includes chemotherapy, surgery with or without RT, has become the standard of care for RMS. For children and adolescents with RMS, the multidisciplinary treatment of the disease according to collaborative group clinical trials has been performed in the United States and in Europe. The cure rates for RMS in children have increased from 25% in the 1970s to 70% in the 1990s.58–61 Patients at low risk have an estimated 3-year failure-free survival (FFS) rate of 88%. Patients at an intermediate risk have a 3-year FFS rate of 55%–76%, while patients at high risk have a 3-year FFS rate of <30%. Given the excellent outcome for low-risk pediatric patients, successive generations of clinical trials have attempted to decrease the intensity of therapy to decrease the incidence of treatment-related late effects. Survival rates for adolescents and adults with RMS, on the other hand, are significantly worse compared to younger children (range 21%–56%), and more study is needed to improve these outcomes.55,57
Chemotherapy
For COG treatment protocols, the three-drug combination of vincristine, actinomycin D, and cyclophosphamide (VAC) has been the backbone of RMS therapy. For certain groups with low-risk disease, there has been interest in treating with only vincristine and actinomycin D to avoid large cumulative alkylator exposure that has been linked to secondary malignancies and sterility, but the results of a recent study found that the local control rates using actinomycin D were slightly inferior to historical published local control rates using VAC.62 Thus, the most recent COG clinical trial for low-risk RMS patients (ARST 0331)63 is attempting to determine if the addition of four doses of cyclophosphamide would be sufficient to improve the local control rate.
Patients with intermediate risk disease are treated with VAC. Successive IRSG and COG clinical trials have attempted to improve the outcome by examining different strategies, such as incorporating different agents (etoposide, ifosfamide, cisplatin, irinotecan, topotecan, and doxorubicin) or intensifying cyclophosphamide during induction, but these strategies have not significantly improved clinical outcome.64–66 The greatest need for improvement is in patients with high-risk disease. Recent COG studies have employed the use of a more intensive chemotherapy backbone using vincristine, doxorubicin, and cyclophosphamide that is alternated with ifosfamide and etoposide. Since the survival outlook for high-risk patients has been historically so poor, it appears that a plateau of efficacy has been reached with traditional cytotoxic chemotherapy. Thus, there has been significant interest in the development of targeted and other novel therapies.
European cooperative group studies have typically categorized patients in four risk groups (low-risk, standard risk, high-risk, and very high risk).57 European cooperative group studies have also differed from COG studies in that the European clinical trials have been more treatment response-based.57 This approach has been used with the goal of decreasing the number of patients exposed to RT based on good clinical response to initial chemotherapy. Thus, the European cooperative group studies tend to incorporate alkylator and anthracycline chemotherapeutic agents more readily than the COG studies.
Due to the rarity of the disease in adults, there is a lack of standardized treatment, and patients are treated with a variety of chemotherapy regimens. One retrospective study of adult RMS patients demonstrated that those patients treated according to current pediatric guidelines had similar response rates and outcome to pediatric RMS patients, suggesting that adults may be treated with similar treatment protocols as children with RMS.5 Even though assessment of the use of pediatric treatment regim ens in adult patients is of paramount importance, certain barriers – such as low patient numbers and the lack of centralization of care for adults with RMS – will create obstacles for this evaluation.
It is important to consider differences in pharmacology and pharmacodynamics between the adult and pediatric population when adapting pediatric treatment regimens to adult patients with RMS.57 In one single-institutional study, adolescent and adult patients <50 years of age were more likely to receive ifosfamide-based chemotherapy than their older counterparts, possibly related to improved tolerance to this chemotherapeutic agent.56 Another study noted that adolescents tended to experience less hematologic toxicity but more peripheral neurotoxicity when compared to younger pediatric patients.67
Surgery
The surgical management of patients with RMS is site-specific, and attempts for complete tumor resection should be made. Complete tumor resection is generally not advised if there is a significant risk for functional or cosmetic morbidity (for example, in head and neck RMS). Complete compartmental resection is not required for tumors arising within a muscle, as long as a minimum of 0.5 cm of margins is obtained.40 Tumor debulking in general should not be performed, with the possible exception of tumors localized in the retroperitoneum that are causing organ compromise, such as bowel obstruction.
If an open biopsy of the mass was initially performed for diagnostic purposes or if the patient still has gross residual tumor or microscopically uncertain margins, then pretreatment re-excision of the tumor is recommended. This consists of a wide re-excision of the previous operative site with adequate margins of normal tissue prior to adjuvant therapy and usually performed in trunk and extremity lesions.
Second-look operations (SLO) may play a role in improving the local control of tumors that are initially unresectable but show a pronounced response to induction chemotherapy and/or RT. Additionally, the patients with group III disease (postoperative gross residual disease) who have a good response to induction chemotherapy can have a complete resection by means of SLO and then receive reduced radiotherapy dose.68,69 One COG study evaluated whether the lack of tumor viability at SLO was a reliable positive prognostic factor.70 The study noted a benefit in FFS but not for overall survival.
RT
RT is an important component of multimodality therapy for pediatric RMS. Induction chemotherapy, followed by concurrent chemoradiation, is the current standard of care for patients with unresected disease, those with micro- or macroscopic residual disease after surgery for patients with lymph node involvement, and for patients with alveolar histology.69,71 RT is usually initiated between 6–12 weeks after the start of chemotherapy, omitting the concomitant administration of a radiosensitizing agent, such as an anthracycline or actinomycin D. The dose of RT is dependent upon several variables usually related to the extent of the disease, such as regional lymph node involvement. Current COG studies have been evaluating the delivery of 36 Gray (Gy) to certain groups of low-risk patients (eg, favorable site and with microscopic residual margins following initial tumor resection). Typical peak doses are 50.4 Gy for patients with a macroscopic residual tumor.
Reduction of RT volumes has been used if the tumor size decreases after induction chemotherapy. A recent study evaluating the cone-down boost according to tumor response to induction chemotherapy in pediatric patients found that for most of the patients a cone-down boost allowed for significant reductions in high-dose RT treatment volume while maintaining excellent tumor control. However in the subset of patients with parameningeal RMS and intracranial tumor extension, early RT initiation and wider margin RT to cover adjacent areas at high risk for meningeal extension may be more important for adequate disease control.71
In the case of parameningeal RMS, there are data to suggest that earlier use of RT may improve local control rates.65 Other RT techniques, such as intensity modulated RT, brachytherapy, and proton beam therapy are being used in different centers to treat RMS with the aim to reduce locoregional side effects and to achieve a higher tumor treatment dose without an increase of toxic side effects, especially at anatomically sensitive areas, such as the orbit, bladder/prostate, and parameningeal area.72–76
Role of transplant in RMS
The utility of bone marrow transplant for high-risk or relapsed RMS has been of much interest since this approach has been used in other solid tumors, such as neuroblastoma and germ cell tumor. For high-dose chemotherapy followed by autologous stem cell rescue, recent studies performed mainly in Europe have not demonstrated any significant benefit of this treatment approach compared to conventional treatment strategies.77–79 Other transplant strategies have mainly been reported as small case series or single case reports and will need further investigation as a clinical trial to validate their feasibility.80,81
Late effects
The late effects of treatment for RMS survivors are increasingly being recognized and represent an important area of study. The type of late effect encountered is largely dependent upon a variety of factors, including anatomic site of tumor involvement, types of chemotherapy received, exposure to RT, and extent of surgical resection. The presentation of late effect may occur even many years after completion of therapy.82 Due to the broad nature of this topic, only a few relevant areas will be reviewed.
Alkylating agents like cyclophosphamide and ifosfamide have been linked to secondary malignancies and have shown a dose-dependent effect on testicular function and fertility.83–85 In females, age and dose-dependent effects of alkylating agents and RT on infertility and premature ovarian failure have been reported.86–88 Given the risks of infertility, concerted efforts must be made to inform adolescent and young adult RMS patients about these risks and the opportunity for options such as sperm banking. For female patients, ovarian preservation may be more difficult depending upon the time available before starting treatment and the availability of specialists. Other chemotherapy-related toxicities include increased peripheral nervous system toxicity in adolescent patients and increased risk of cardiomyopathy or other cardiac dysfunction in patients treated with anthracycline chemotherapy.67,89,90 For adolescents and young adults, these effects may lead to increased risk of physical inactivity or decreased exercise tolerance. Thus, appropriate surveillance for these toxicities is needed so that early physical therapy or exercise plans can be instituted.
RT-related late effects are varied and depend on the treatment field and dose received. In the head and neck region, facial growth retardation, xerostomia, dental abnormalities, visual and hearing deficits, and neuroendocrine dysfunction can occur.91,92 Jaw dysfunction, due to radiation-induced fibrosis of the temporomandibular joint, has also been reported.93 For the adolescent and young adult patient, facial asymmetry, growth deficiency, and jaw dysfunction can cause significant cosmetic morbidity and can dramatically impact quality of life. Early intervention with an experienced occupational therapy and speech pathology team can help ameliorate some of the risks for trismus and jaw dysfunction.
In the pelvic region, gonadal function can be affected; there can also be musculoskeletal growth delay or bowel obstruction.76 For extremity tumors treated with radiation or surgery, problems with growth or atrophy, fibrosis, peripheral nerve damage, and range of motion can be observed.94,95 Again, these effects may impact physical activity for adolescent and young adult patients.
For a more comprehensive review of treatment-related late effects, the COG has published a comprehensive, empiric literature review-based set of guidelines that are available online.96
Conclusion
While there have been significant improvements made in the treatment of children with RMS, further work is needed to improve the clinical outcome for adolescent and young adult RMS patients – both in the understanding of the biology of the histologic subtypes found in adults and in standardizing the treatment for adult RMS patients. Increasing enrollment of adolescent and young adult patients into cooperative group studies would also represent an improvement. Current COG clinical trials allow patients up to 50 years of age to be enrolled. Finally, treatment-related late effects can have a significant impact on the lives of adolescent and young adult survivors of RMS. Thus, long-term surveillance is required so that early intervention can be applied.
Disclosure
The authors report no conflicts of interest in this work.
References
Dasgupta R, Rodeberg DA. Update on rhabdomyosarcoma. Semin Pediatr Surg. 2012;21(1):68–78. | |
Linet MS, Ries LA, Smith MA, Tarone RE, Devesa SS. Cancer surveillance series: recent trends in childhood cancer incidence and mortality in the United States. J Natl Cancer Inst. 1999;91(12):1051–1058. | |
Paulino AC, Okcu MF. Rhabdomyosarcoma. Curr Probl Cancer. 2008;32(1):7–34. | |
De Giovanni C, Landuzzi L, Nicoletti G, Lollini PL, Nanni P. Molecular and cellular biology of rhabdomyosarcoma. Future Oncol. 2009;5(9):1449–1475. | |
Ferrari A, Dileo P, Casanova M, et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer. 2003;98(3):571–580. | |
Woodruff JM, Perino G. Non-germ-cell or teratomatous malignant tumors showing additional rhabdomyoblastic differentiation, with emphasis on the malignant Triton tumor. Semin Diagn Pathol. 1994;11(1):69–81. | |
Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. J Clin Oncol. 2009;27(20):3391–3397. | |
Pappo AS, Shapiro DN, Crist WM, Maurer HM. Biology and therapy of pediatric rhabdomyosarcoma. J Clin Oncol. 1995;13(8):2123–2139. | |
Ruymann FB, Maddux HR, Ragab A, et al. Congenital anomalies associated with rhabdomyosarcoma: an autopsy study of 115 cases. A report from the Intergroup Rhabdomyosarcoma Study Committee (representing the Children’s Cancer Study Group, the Pediatric Oncology Group, the United Kingdom Children’s Cancer Study Group, and the Pediatric Intergroup Statistical Center). Med Pediatr Oncol. 1988;16(1):33–39. | |
Hennekam RC. Costello syndrome: an overview. Am J Med Genet C Semin Med Genet. 2003;117C(1):42–48. | |
Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet. 1995;56(1):112–115. | |
Sung L, Anderson JR, Arndt C, Raney RB, Meyer WH, Pappo AS. Neurofibromatosis in children with Rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma study IV. J Pediatr. 2004; 144(5):666–668. | |
Ferrari A, Bisogno G, Macaluso A, et al. Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer. 2007;109(7):1406–1412. | |
Xia SJ, Pressey JG, Barr FG. Molecular pathogenesis of rhabdomyosarcoma. Cancer Biol Ther. 2002;1(2):97–104. | |
Merlino G, Khanna C. Fishing for the origins of cancer. Genes Dev. 2007;21(11):1275–1279. | |
Charytonowicz E, Cordon-Cardo C, Matushansky I, Ziman M. Alveolar rhabdomyosarcoma: is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009;279(2):126–136. | |
Tiffin N, Williams RD, Shipley J, Pritchard-Jones K. PAX7 expression in embryonal rhabdomyosarcoma suggests an origin in muscle satellite cells. Br J Cancer. 2003;89(2):327–332. | |
Zanola A, Rossi S, Faggi F, Monti E, Fanzani A. Rhabdomyosarcomas: an overview on the experimental animal models. J Cell Mol Med. 2012;16(7):1377–1391. | |
Fanzani A, Monti E, Donato R, Sorci G. Muscular dystrophies share pathogenetic mechanisms with muscle sarcomas. Trends Mol Med. 2013;19(9):546–554. | |
Nishijo K, Chen QR, Zhang L, et al. Credentialing a preclinical mouse model of alveolar rhabdomyosarcoma. Cancer Res. 2009;69(7):2902–2911. | |
Visser M, Sijmons C, Bras J, et al. Allelotype of pediatric rhabdomyosarcoma. Oncogene. 1997;15(11):1309–1314. | |
Tsokos M, Webber BL, Parham DM, et al. Rhabdomyosarcoma. A new classification scheme related to prognosis. Arch Pathol Lab Med. 1992;116(8):847–855. | |
Fanzani A, Zanola A, Faggi F, et al. Implications for the mammalian sialidases in the physiopathology of skeletal muscle. Skelet Muscle. 2012;2(1):23. | |
Skapek SX, Anderson J, Barr FG, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children’s oncology group report. Pediatr Blood Cancer. 2013;60(9):1411–1417. | |
Williamson D, Missiaglia E, de Reyniès A, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. 2010;28(13):2151–2158. | |
Keleti J, Quezado MM, Abaza MM, Raffeld M, Tsokos M. The MDM2 oncoprotein is overexpressed in rhabdomyosarcoma cell lines and stabilizes wild-type p53 protein. Am J Pathol. 1996;149(1):143–151. | |
Taylor AC, Shu L, Danks MK, et al. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Med Pediatr Oncol. 2000;35(2):96–103. | |
Takahashi Y, Oda Y, Kawaguchi K, et al. Altered expression and molecular abnormalities of cell-cycle-regulatory proteins in rhabdomyosarcoma. Mod Pathol. 2004;17(6):660–669. | |
Iolascon A, Faienza MF, Coppola B, et al. Analysis of cyclin-dependent kinase inhibitor genes (CDKN2A, CDKN2B, and CDKN2C) in childhood rhabdomyosarcoma. Genes Chromosomes Cancer. 1996;15(4):217–222. | |
Williamson D, Lu YJ, Gordon T, et al. Relationship between MYCN copy number and expression in rhabdomyosarcomas and correlation with adverse prognosis in the alveolar subtype. J Clin Oncol. 2005;23(4):880–888. | |
Langenau DM, Keefe MD, Storer NY, et al. Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes Dev. 2007;21(11):1382–1395. | |
Becker J, Doherr MG, Bruckmaier RM, Bodmer M, Zanolari P, Steiner A. Acute and chronic pain in calves after different methods of rubber-ring castration. Vet J. 2012;194(3):380–385. | |
Hilbe M, Kaufmann Ch, Zlinszky K, Zanolari P, Ehrensperger F. New World camelids and Bovine Virus Diarrhea Virus (BVDV) infection in Switzerland. Schweiz Arch Tierheilkd. 2012;154(4):155–158. | |
Bonvini P, Zin A, Alaggio R, Pawel B, Bisogno G, Rosolen A. High ALK mRNA expression has a negative prognostic significance in rhabdomyosarcoma. Br J Cancer. 2013;109(12):3084–3091. | |
van Gaal JC, Flucke UE, Roeffen MH, et al. Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: clinical and prognostic implications. J Clin Oncol. 2012;30(3):308–315. | |
Keller C, Guttridge DC. Mechanisms of impaired differentiation in rhabdomyosarcoma. FEBS J. 2013;280(17):4323–4334. | |
Maki RG. Pediatric sarcomas occurring in adults. J Surg Oncol. 2008;97(4):360–368. | |
Rubin BP, Nishijo K, Chen HI, et al. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell. 2011;19(2):177–191. | |
Tsumura H, Yoshida T, Saito H, Imanaka-Yoshida K, Suzuki N. Cooperation of oncogenic K-ras and p53 deficiency in pleomorphic rhabdomyosarcoma development in adult mice. Oncogene. 2006;25(59):7673–7679. | |
Leaphart C, Rodeberg D. Pediatric surgical oncology: management of rhabdomyosarcoma. Surg Oncol. 2007;16(3):173–185. | |
Weiss AR, Lyden ER, Anderson JR, et al. Histologic and clinical characteristics can guide staging evaluations for children and adolescents with rhabdomyosarcoma: a report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. J Clin Oncol. 2013;31(26):3226–3232. | |
Völker T, Denecke T, Steffen I, et al. Positron emission tomography for staging of pediatric sarcoma patients: results of a prospective multicenter trial. J Clin Oncol. 2007;25(34):5435–5441. | |
Federico SM, Spunt SL, Krasin MJ, et al. Comparison of PET-CT and conventional imaging in staging pediatric rhabdomyosarcoma. Pediatr Blood Cancer. 2013;60(7):1128–1134. | |
Benz MR, Tchekmedyian N, Eilber FC, Federman N, Czernin J, Tap WD. Utilization of positron emission tomography in the management of patients with sarcoma. Curr Opin Oncol. 2009;21(4):345–351. | |
Joshi D, Anderson JR, Paidas C, Breneman J, Parham DM, Crist W; Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Age is an independent prognostic factor in rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Pediatr Blood Cancer. 2004;42(1):64–73. | |
Meza JL, Anderson J, Pappo AS, Meyer WH; Children’s Oncology Group. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children’s Oncology Group. J Clin Oncol. 2006;24(24):3844–3851. | |
Dantonello TM, Int-Veen C, Winkler P, et al. Initial patient characteristics can predict pattern and risk of relapse in localized rhabdomyosarcoma. J Clin Oncol. 2008;26(3):406–413. | |
Rodeberg DA, Garcia-Henriquez N, Lyden ER, et al. Prognostic significance and tumor biology of regional lymph node disease in patients with rhabdomyosarcoma: a report from the Children’s Oncology Group. J Clin Oncol. 2011;29(10):1304–1311. | |
Missiaglia E, Williamson D, Chisholm J, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol. 2012;30(14):1670–1677. | |
Stegmaier S, Poremba C, Schaefer KL, et al. Prognostic value of PAX-FKHR fusion status in alveolar rhabdomyosarcoma: a report from the cooperative soft tissue sarcoma study group (CWS). Pediatr Blood Cancer. 2011;57(3):406–414. | |
Ferrari A, Miceli R, Meazza C, et al. Comparison of the prognostic value of assessing tumor diameter versus tumor volume at diagnosis or in response to initial chemotherapy in rhabdomyosarcoma. J Clin Oncol. 2010;28(8):1322–1328. | |
Burke M, Anderson JR, Kao SC, et al; Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Assessment of response to induction therapy and its influence on 5-year failure-free survival in group III rhabdomyosarcoma: the Intergroup Rhabdomyosarcoma Study-IV experience – a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J Clin Oncol. 2007;25(31):4909–4913. | |
Burke ME, Lyden ER, Meza JL, et al; Children’s Oncology Group Soft Tissue Sarcoma Committee. Does body mass index at diagnosis or weight change during therapy predict toxicity or survival in intermediate risk rhabdomyosarcoma? A report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. Pediatr Blood Cancer. 2013;60(5):748–753. | |
Little DJ, Ballo MT, Zagars GK, et al. Adult rhabdomyosarcoma: outcome following multimodality treatment. Cancer. 2002;95(2):377–388. | |
Bisogno G, Compostella A, Ferrari A, et al. Rhabdomyosarcoma in adolescents: a report from the AIEOP Soft Tissue Sarcoma Committee. Cancer. 2012;118(3):821–827. | |
Dumont SN, Araujo DM, Munsell MF, et al. Management and outcome of 239 adolescent and adult rhabdomyosarcoma patients. Cancer Med. 2013;2(4):553–563. | |
Van Gaal JC, De Bont ES, Kaal SE, Versleijen-Jonkers Y, van der Graaf WT. Building the bridge between rhabdomyosarcoma in children, adolescents and young adults: the road ahead. Crit Rev Oncol Hematol. 2012;82(3):259–279. | |
Maurer HM, Beltangady M, Gehan EA, et al. The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer. 1988;61(2):209–220. | |
Maurer HM, Gehan EA, Beltangady M, et al. The Intergroup Rhabdomyosarcoma Study-II. Cancer. 1993;71(5):1904–1922. | |
Crist W, Gehan EA, Ragab AH, et al. The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol. 1995;13(3):610–630. | |
Baker KS, Anderson JR, Link MP, et al. Benefit of intensified therapy for patients with local or regional embryonal rhabdomyosarcoma: results from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol. 2000;18(12):2427–2434. | |
Raney RB, Walterhouse DO, Meza JL, et al. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J Clin Oncol. 2011;29(10):1312–1318. | |
Walterhouse D, Pappo A, Meza J, et al. Shorter duration therapy that includes vincristine (V), dactinomycin (A), and lower doses of cyclophosphamide (C) with or without radiation therapy for patients with newly diagnosed low-risk embryonal rhabdomyosarcoma (ERMS): A report from the Children’s Oncology Group (COG). J Clin Oncol. 2011;29:15(suppl): Abstract 9516. | |
Arndt CA, Hawkins DS, Meyer WH, Sencer SF, Neglia JP, Anderson JR. Comparison of results of a pilot study of alternating vincristine/doxorubicin/cyclophosphamide and etoposide/ifosfamide with IRS-IV in intermediate risk rhabdomyosarcoma: a report from the Children’s Oncology Group. Pediatr Blood Cancer. 2008;50(1):33–36. | |
Arndt CA, Stoner JA, Hawkins DS, et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: children’s oncology group study D9803. J Clin Oncol. 2009;27(31):5182–5188. | |
Spunt SL, Smith LM, Ruymann FB, et al. Cyclophosphamide dose intensification during induction therapy for intermediate-risk pediatric rhabdomyosarcoma is feasible but does not improve outcome: a report from the soft tissue sarcoma committee of the children’s oncology group. Clin Cancer Res. 2004;10(18 Pt 1):6072–6079. | |
Gupta AA, Anderson JR, Pappo AS, et al. Patterns of chemotherapy-induced toxicities in younger children and adolescents with rhabdomyosarcoma: a report from the Children’s Oncology Group Soft Tissue Sarcoma Committee. Cancer. 2012;118(4):1130–1137. | |
Breneman JC, Wiener ES. Issues in the local control of rhabdomyosarcoma. Med Pediatr Oncol. 2000;35(2):104–109. | |
Cecchetto G, Carretto E, Bisogno G, et al. Complete second look operation and radiotherapy in locally advanced non-alveolar rhabdomyosarcoma in children: A report from the AIEOP soft tissue sarcoma committee. Pediatr Blood Cancer. 2008;51(5):593–597. | |
Raney B, Stoner J, Anderson J, et al; Soft-Tissue Sarcoma Committee of the Children’s Oncology Group. Impact of tumor viability at second-look procedures performed before completing treatment on the Intergroup Rhabdomyosarcoma Study Group protocol IRS-IV, 1991–1997: a report from the children’s oncology group. J Pediatr Surg. 2010;45(11):2160–2168. | |
Eaton BR, McDonald MW, Kim S, et al. Radiation therapy target volume reduction in pediatric rhabdomyosarcoma: implications for patterns of disease recurrence and overall survival. Cancer. 2013;119(8):1578–1585. | |
Blank LE, Koedooder K, van der Grient HN, et al. Brachytherapy as part of the multidisciplinary treatment of childhood rhabdomyosarcomas of the orbit. Int J Radiat Oncol Biol Phys. 2010;77(5):1463–1469. | |
Childs SK, Kozak KR, Friedmann AM, et al. Proton radiotherapy for parameningeal rhabdomyosarcoma: clinical outcomes and late effects. Int J Radiat Oncol Biol Phys. 2012;82(2):635–642. | |
Cotter SE, Herrup DA, Friedmann A, et al. Proton radiotherapy for pediatric bladder/prostate rhabdomyosarcoma: clinical outcomes and dosimetry compared to intensity-modulated radiation therapy. Int J Radiat Oncol Biol Phys. 2011;81(5):1367–1373. | |
Magné N, Haie-Meder C. Brachytherapy for genital-tract rhabdomyosarcomas in girls: technical aspects, reports, and perspectives. Lancet Oncol. 2007;8(8):725–729. | |
Curtis AE, Okcu MF, Chintagumpala M, Teh BS, Paulino AC. Local control after intensity-modulated radiotherapy for head-and-neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys. 2009;73(1):173–177. | |
McDowell HP, Foot AB, Ellershaw C, Machin D, Giraud C, Bergeron C. Outcomes in paediatric metastatic rhabdomyosarcoma: results of The International Society of Paediatric Oncology (SIOP) study MMT-98. Eur J Cancer. 2010;46(9):1588–1595. | |
Klingebiel T, Boos J, Beske F, et al. Treatment of children with metastatic soft tissue sarcoma with oral maintenance compared to high dose chemotherapy: report of the HD CWS-96 trial. Pediatr Blood Cancer. 2008;50(4):739–745. | |
Bisogno G, Ferrari A, Prete A, et al. Sequential high-dose chemotherapy for children with metastatic rhabdomyosarcoma. Eur J Cancer. 2009;45(17):3035–3041. | |
Donker AE, Hoogerbrugge PM, Mavinkurve-Groothuis AM, et al. Metastatic rhabdomyosarcoma cured after chemotherapy and allogeneic SCT. Bone Marrow Transplant. 2009;43(2):179–180. | |
Pérez-Martínez A, Leung W, Muñoz E, et al. KIR-HLA receptor-ligand mismatch associated with a graft-versus-tumor effect in haploidentical stem cell transplantation for pediatric metastatic solid tumors. Pediatr Blood Cancer. 2009;53(1):120–124. | |
Punyko JA, Mertens AC, Gurney JG, et al. Long-term medical effects of childhood and adolescent rhabdomyosarcoma: a report from the childhood cancer survivor study. Pediatr Blood Cancer. 2005;44(7):643–653. | |
Kenney LB, Laufer MR, Grant FD, Grier H, Diller L. High risk of infertility and long term gonadal damage in males treated with high dose cyclophosphamide for sarcoma during childhood. Cancer. 2001;91(3):613–621. | |
Heyn R, Haeberlen V, Newton WA, et al. Second malignant neoplasms in children treated for rhabdomyosarcoma. Intergroup Rhabdomyosarcoma Study Committee. J Clin Oncol. 1993;11(2):262–270. | |
Green DM, Kawashima T, Stovall M, et al. Fertility of male survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Clin Oncol. 2010;28(2):332–339. | |
Chemaitilly W, Mertens AC, Mitby P, et al. Acute ovarian failure in the childhood cancer survivor study. J Clin Endocrinol Metab. 2006;91(5):1723–1728. | |
Spunt SL, Sweeney TA, Hudson MM, Billups CA, Krasin MJ, Hester AL. Late effects of pelvic rhabdomyosarcoma and its treatment in female survivors. J Clin Oncol. 2005;23(28):7143–7151. | |
Sudour H, Chastagner P, Claude L, et al. Fertility and pregnancy outcome after abdominal irradiation that included or excluded the pelvis in childhood tumor survivors. Int J Radiat Oncol Biol Phys. 2010;76(3):867–873. | |
van Dalen EC, van der Pal HJ, Kok WE, Caron HN, Kremer LC. Clinical heart failure in a cohort of children treated with anthracyclines: a long-term follow-up study. Eur J Cancer. 2006;42(18):3191–3198. | |
Oeffinger KC, Mertens AC, Sklar CA, et al; Childhood Cancer Survivor Study. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med. 2006;355(15):1572–1582. | |
Raney RB, Asmar L, Vassilopoulou-Sellin R, et al. Late complications of therapy in 213 children with localized, nonorbital soft-tissue sarcoma of the head and neck: A descriptive report from the Intergroup Rhabdomyosarcoma Studies (IRS)-II and – III. IRS Group of the Children’s Cancer Group and the Pediatric Oncology Group. Med Pediatr Oncol. 1999;33(4):362–371. | |
Paulino AC, Simon JH, Zhen W, Wen BC. Long-term effects in children treated with radiotherapy for head and neck rhabdomyosarcoma. Int J Radiat Oncol Biol Phys. 2000;48(5):1489–1495. | |
Krasin MJ, Wiese KM, Spunt SL, et al. Jaw dysfunction related to pterygoid and masseter muscle dosimetry after radiation therapy in children and young adults with head-and-neck sarcomas. Int J Radiat Oncol Biol Phys. 2012;82(1):355–360. | |
Paulino AC. Late effects of radiotherapy for pediatric extremity sarcomas. Int J Radiat Oncol Biol Phys. 2004;60(1):265–274. | |
Raney B Jr, Heyn R, Hays DM, et al. Sequelae of treatment in 109 patients followed for 5 to 15 years after diagnosis of sarcoma of the bladder and prostate. A report from the Intergroup Rhabdomyosarcoma Study Committee. Cancer. 1993;71(7):2387–2394. | |
Long-Term Follow-Up Guidelines for Survivors of Childhood, Adolescent, and Young Adult Cancers [webpage on the Internet]. Monrovia: Children’s Oncology Group; 2014. Available from: http://www.survivorshipguidelines.org/. Accessed January 3, 2014. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.