")
Back to Journals » Drug Design, Development and Therapy » Volume 14
RGFP966 Suppresses Tumor Growth and Migration Through Inhibition of EGFR Expression in Hepatocellular Carcinoma Cells in vitro
Authors Yu X , Yang F, Jiang H, Fan L
Received 16 October 2019
Accepted for publication 21 December 2019
Published 10 January 2020 Volume 2020:14 Pages 121—128
DOI https://doi.org/10.2147/DDDT.S234871
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Xinying Yu, 1 Fan Yang, 2 Hong Jiang, 3 Ling Fan 1
1Second Pediatric Intensive Care Unit, Shengjing Hospital of China Medical University, Shenyang City, Liaoning Province, People’s Republic of China; 2Third Neonatal Ward, Shengjing Hospital of China Medical University, Shenyang City, Liaoning Province, People’s Republic of China; 3Second Neonatal Ward, Shengjing Hospital of China Medical University, Shenyang City, Liaoning Province, People’s Republic of China
Correspondence: Xinying Yu; Ling Fan
Shengjing Hospital of China Medical University, Shenyang City, Liaoning Province 110004, People’s Republic of China
Tel/Fax +86 24-31939077
Email [email protected]; [email protected]
Purpose: Histone deacetylase 3 (HDAC3) has been suggested to play a role in hepatocellular carcinoma (HCC). In the present report, we aimed to identify the effects of RGFP966, a specific HDAC3 inhibitor, on the cell proliferation and migration of HCC cell lines.
Methods: Human HCC cell lines, which were identified using short tandem repeat (STR) DNA profiling analysis, were used in this report. Cell proliferation assay was used to identify the growth viability of cells. Wound healing and transwell assay were used to identify the migration ability of cells. Further, a human phospho-receptor tyrosine kinases array kit was used to screen out RGFP966 effects on key receptor tyrosine kinases. Then, the mRNA expression was quantified by real-time PCR, and protein expression was identified by Western blot immunoassay.
Results: We found that RGFP966 inhibited both proliferation and migration of HCC cells. Further, RGFP966 represses the expression and phosphorylation levels of epidermal growth factor receptor (EGFR) in HCC cells. Moreover, HDAC3 is involved in the inhibition of EGFR by RGFP966. Overall, we elucidated an inhibitive function of RGFP966 in HCC progression.
Conclusion: RGFP966 inhibits EGFR signaling pathway and suppresses proliferation and migration of HCC cells.
Keywords: RGFP966, hepatocellular carcinoma, HDAC3, EGFR
Introduction
Globally, liver cancer is the fourth leading cause of cancer death in 2018.1 Hepatocellular carcinoma (HCC), which is accounting for 75–85% of all liver cancers, is the major histological type of primary liver cancer.1 Due to the advanced stage of the disease, many HCC patients are not eligible for curative surgery at the time of diagnosis. For a number of years, sorafenib was the only first-line medical therapy for patients with HCC.2 Even though encouraging studies have emerged, such as the kinase inhibitors lenvatinib,3 regorafenib,4 and cabozantinib,5 there is an unmet need for the pharmacological therapy for HCC.
Histone deacetylases (HDACs) play major roles in diverse biological functions, and HDAC inhibitors show clinical promise for the treatment of cancers, including HCC.6–8 Based on sequence similarity, HDAC family are divided into four subfamilies: classes I, IIa, IIb, III, and IV. HDAC3 was the third one to be identified of HDACs and belongs to class I HDACs.9 RGFP966, a selective histone deacetylase 3 (HDAC3) inhibitor, has been proved to be effective in inhibiting different types of tumors. RGFP966 inhibits growth of both PTEN-deficient and SPOP-mutated prostate cancer cells in culture, patient-derived organoids and xenografts in mice,10 and also holds promise for the treatment of castration-resistant prostate cancer.11 RGFP966 decreased cell growth in T cell lymphoma cell lines due to increased apoptosis that was associated with DNA damage and impaired S phase progression.12 RGFP966 markedly increased the protein level of AKAP12 and suppresses the progression and migration of colorectal cancer.13 But the effects of RGFP966 on HCC remains to be further evaluated.
Although hdac3 is shown to be essential for the maintenance of chromatin structure and genome stability and hdac3-Null livers develop hepatocellular carcinoma in mouse, HDAC3 protein expression was downregulated only in a small number of human liver cancers.14 Copy number gain of HDAC3 occurs in HCC and overexpressed HDAC3 is associated with increased tumor growth and a poor prognosis in HCC patients.8,15 Further, HDAC3 is an important regulator of STAT3-dependent cell proliferation in and liver cancer cells proliferation.15 Moreover, HDAC3 plays a critical role in regulating self-renewal of liver cancer stem cells and liver regeneration.15,16 In addition, HDAC3 may be served as a candidate biomarker for predicting the recurrence of hepatitis B virus-associated HCC following liver transplantation and a potential therapeutic target.17 Consequently, HDAC3 suppression by RGFP966 may be a potential medical-therapeutic approach for HCC.
In this study, we found that RGFP966 inhibited both proliferation and migration of HCC cells. Further, we screened out that RGFP966 represses the expression and phosphorylation levels of EGFR in HCC cells. Moreover, HDAC3 is involved in the inhibition of EGFR by RGFP966. Overall, our results suggested that RGFP966 inhibits EGFR signaling pathway leading to suppression of proliferation and migration of HCC cells.
Materials and Methods
Chemicals
RGFP966 was obtained from MedChemExpress (MCE, HY-13909), and was dissolved in DMSO.
Cell Lines and Cell Culture
Human HCC cell lines were obtained from Kunming cell bank of Chinese Academy of Sciences and were identified using short tandem repeat (STR) DNA profiling analysis. Huh7 cells were cultured in Dulbecco’s modified Eagle medium (Gibco), while PLC/PRF5 and HepG2 cells were cultured in Minimum Essential Media (Gibco), and supplemented with 10% (v/v) fetal bovine serum (Gibco). Cells were incubated at 37°C in a humidified incubator containing 5% CO2.
Cell Proliferation Assay
Cells were seeded into 96-well plates at a density of 3x103 cells/well and cultured overnight. Then, cells were treated with different concentrations of RGFP966 for indicated time. Finally, growth viability of cells was assayed using the one-solution cell proliferation assay (MTS) assay (Promega).
Wound Healing Assay
Cells were seeded into 12-well tissue culture dishes. After confluent cell monolayers were formed, a linear wound was generated with a sterile plastic pipette tip. Then, cells were washed with phosphate buffer saline (PBS) and cultured in 1% FBS with or without RGFP966 treatment. Representative images of the scratched region were photographed. After incubation for 40 hrs, image from the same area was photographed.
Transwell Assay
Transwell chamber system (Corning) with 8-μm pore filter inserts were used. 2.5 x104 or 5x104 cells were planted into upper chamber per well in serum-free medium, and medium supplemented with 10% FBS was placed in lower chamber. RGFP966 or vehicle was added into both upper chamber and lower chamber. After incubation for 40 hrs, cells were fixed with 95% ethanol and the invaded cells were stained by crystal violet.
Human Phospho-Receptor Tyrosine Kinases Array
The proteome profiler human phospho-receptor tyrosine kinases (RTK) array kit was obtained from R&D Systems (ARY001B). Whole-cell lysates from Huh7 cells with or without RGFP966 treatment were collected, and a total of 300 μg fresh protein was for determination of the relative phosphorylation of 49 human RTKs.
Cell Transfection
HA-tagged HDAC3 expression plasmid was obtained from Sino Biological. Transfections were performed according to the manufacturer’s instructions of jetPRIME reagents (Polyplus-transfection).
Western Blot
Cell lysates were prepared by extraction with RIPA lysate. Protein concentration was determined with BCA protein assay kit (Pierce). Approximately 20 μg of total protein was loaded. Antibodies against Phospho-EGFR (Tyr845) were obtained from Cell Signaling Technology. Antibodies against EGFR, HA tag, GAPDH and histone H3 were obtained from Proteintech. Antibodies against total histone H3 acetylation (H3ac) and specific acetylation on histone H3 lysine of (H3K9ac) were obtained from Abcam. Goat anti-Mouse/Rabbit IgG (HRP) were obtained from Invitrogen.
RNA Extraction, cDNA Synthesis and Real-Time PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific). cDNA synthesis was carried out with PrimeScript RT reagent Kit with gDNA Eraser (TaKaRa), and then analyzed by SYBR Green technology in Roche LightCycler96 system according to the manufacturer’s instructions. Relative quantification of EGFR expression was normalization with the endogenous GAPDH control. Primer sequences were EGFR-Sense 5ʹ-CCCACTCATGCTCTACAACCC-3ʹ; EGFR-anti-Sense 5ʹ-TCGCACTTCTTACACTTGCGG-3ʹ; GAPDH-Sense 5ʹ-GGAGCGAGATCCCTCCAAAAT-3ʹ; GAPDH-anti-Sense 5ʹ- GGCTGTTGTCATACTTCTCATGG-3ʹ.
Statistical Analysis
Statistical differences were determined by using the Student’s t test.
Results
RGFP966 Suppresses the Proliferation of HCC Cells
Given that HDAC3 play major roles in HCC development and RGFP966 is a specific inhibitor of HDAC3, we sought to investigate the effects of RGFP966 on HCC. We first measure cell proliferation by MTS method. In PLC/PRF/5, Huh7 and HepG2 cells, RGFP966 inhibited proliferation of in a dose-dependent manner, with maximum suppression observed at 25μM (Figure 1A–C). Then, growth curves were measured under RGFP966 treatment with an effective concentration. The results showed that cell proliferation was slightly inhibited in Huh7 cells or HepG2 cells, while proliferation of PLC/PRF/5 cells was more significantly inhibited (Figure 1D and E). These data suggested that RGFP966 has an anti-proliferative effect on HCC cells.
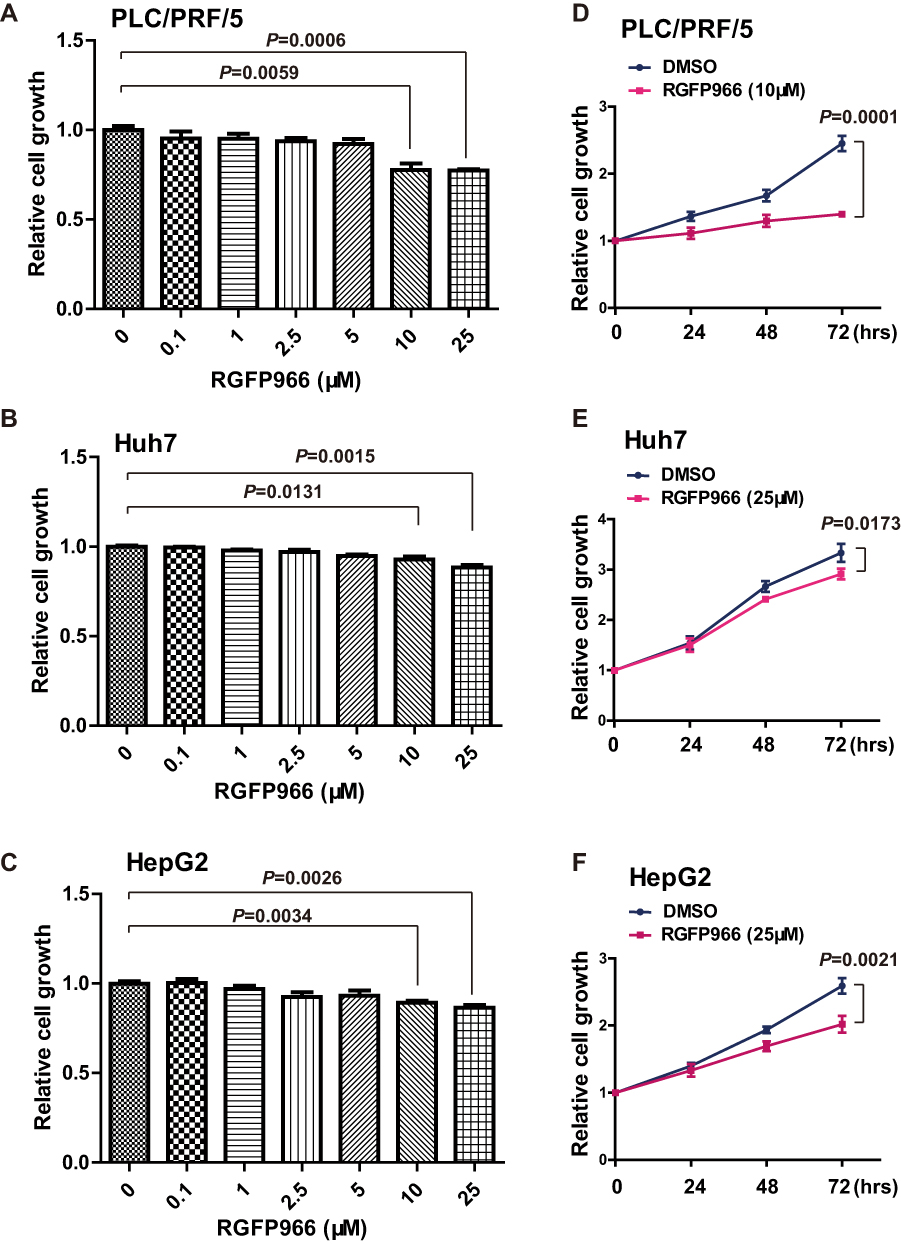 |
Figure 1 Growth repression induced by RGFP966 in HCC cells. (A–C), PLC/PRL/5, Huh7 and HepG2 cells were treated with indicated doses of RGFP966, or vehicle. 48 hrs later, relative cell numbers were determined using MTS assay by absorbance at 492 nm. Data are represented as mean ± SD from three independent experiments. P value refers to two-sided t test. (D–F), PLC/PRL/5, Huh7 and HepG2 cells were treated with RGFP966 (10 or 25μM) or vehicle. Relative cell numbers were determined at indicated times using MTS assay by absorbance at 492 nm and normalized by 0 hr group. Data are represented as mean ± SD from three independent experiments. P value refers to two-sided t test. |
RGFP966 Suppresses the Cell Migration Ability of HCC Cells
Next, we evaluated whether HCC cell migration is regulated by RGFP966. We carried out our analyses in Huh7 and PLC/PRF/5 cells, which showed a higher ability of migration. Transwell assay showed that cell migration was also suppressed by RGFP966 at 10 μM (Figure 2A and B). And wound healing assay showed that cell movement and cell migration were repressed by RGFP966 treatment (Figure 2C and D). These results show that RGFP966 suppresses the cell migration ability of HCC cells.
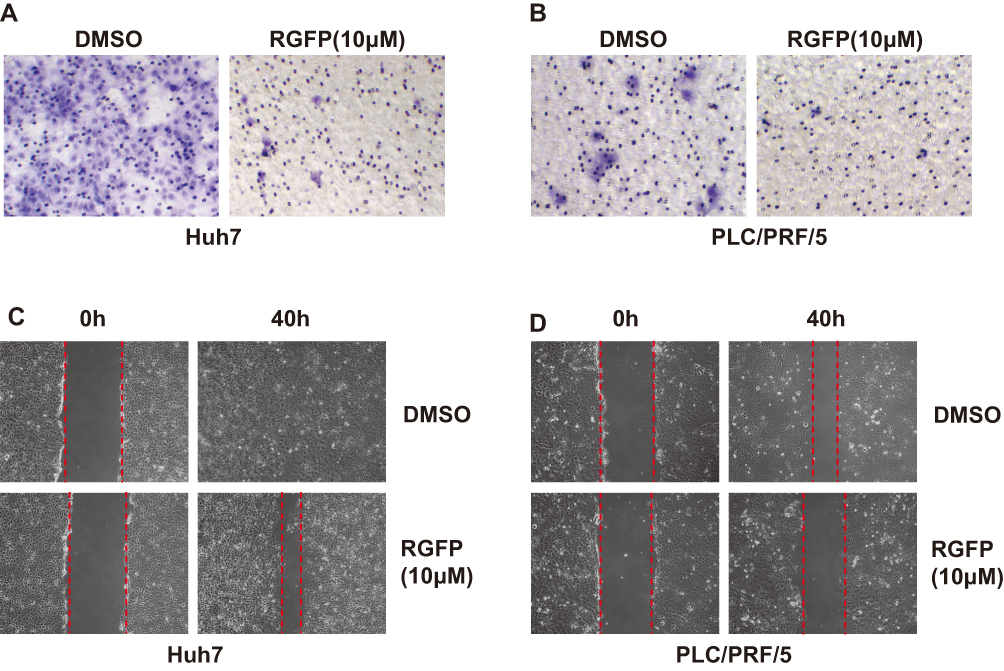 |
Figure 2 RGFP966 suppresses cell migration of HCC cells. (A) and (B), 5x104 Huh7 and PLC/PRL/5 cells were plated into transwell chamber with treatment of RGFP966 (RGFP,10μM) or vehicle. After 40 hrs, the invaded cells were stained, and representative images were photographed. (C) and (D), After a linear wound was generated, Huh7 and PLC/PRL/5 cells were treated with RGFP966 (RGFP, 10μM) or vehicle. After 40 hrs, representative images were photographed. |
RGFP966 Represses the Expression and Phosphorylation Levels of EGFR in HCC Cells
Activation of RTK pathways has been shown in several human cancers including HCC.18 In order to better understand the mechanisms of RGFP966 action in HCC cells, we wondered whether RGFP966 has effects on RTK pathways. We then evaluated the phosphorylation status of RTKs after RGFP966 treatment with Proteome Profiler Human Phospho-RTK Array Kit. Among the examined 49 RTKs, the phosphorylation levels of epidermal growth factor receptor (EGFR) were significantly repressed in Huh7 cells exposed to RGFP966 (Figure 3A). In addition, Western Blot showed RGFP966 treatment not only inhibited the phosphorylation level of EGFR Tyr845, which is phosphorylated in most hepatocellular carcinomas,19 but also induced reduction of EGFR total protein amount in a dose-dependent manner in Huh7 cells (Figure 3B). Similar results after RGFP966 treatment were obtained in PLC/PRF/5 cells (Figure 3C). These data suggested that both the phosphorylation level and total protein amount of EGFR are markedly downregulated upon RGFP966 treatment in HCC cells.
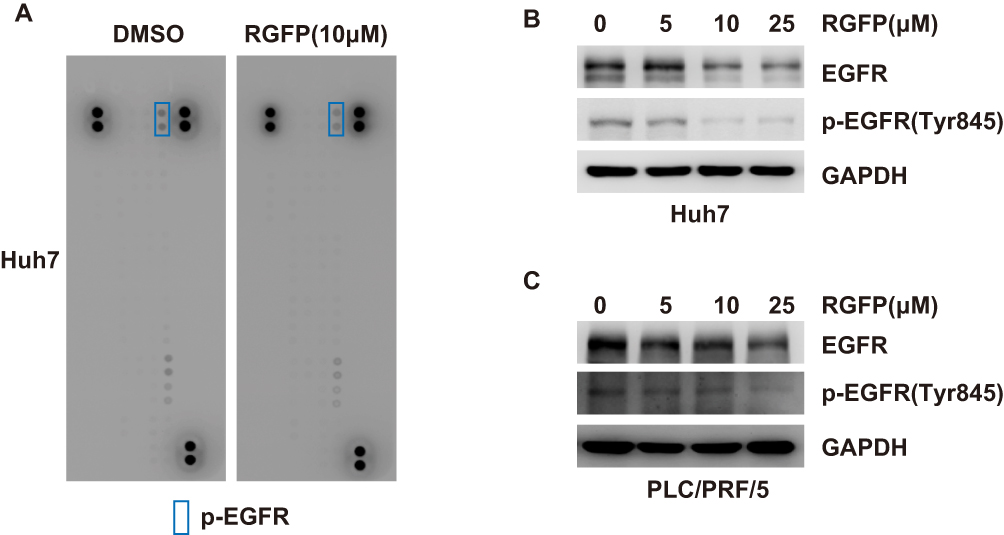 |
Figure 3 RGFP966 downregulates the expression and phosphorylation levels of EGFR in HCC cells. (A), Huh7 cells were treated with or without RGFP966 (RGFP, 10μM). And Human Phospho-RTK array was used to detect the effect of RGFP966 on relative phosphorylation of 49 different RTKs. Representative images were shown. (B) and (C) after treatment with indicated concentrations of RGFP966 (RGFP) for 48 hrs, proteins from Huh7 (B) and PLC/PRL/5 (C) cells were harvested, and Western Blot analysis was performed with the indicated antibodies. GAPDH was used as internal control. |
HDAC3 Enhances the Expression of EGFR and Is Involved in the Inhibition of EGFR by RGFP966
We therefore examined whether RGFP966 can modulate mRNA expression of EGFR in HCC cells, real-time PCR showed that RGFP966 repressed the mRNA expression of EGFR in Huh7 cells in a dose-dependent manner (Figure 4A). Having established that RGFP966 inhibits EGFR expression, we further wonder whether the inhibition is associated with HDAC3. Ectopic expression of HDAC3 can promote mRNA, total protein, and the phosphorylation of levels of EGFR (Figure 4B and C). Migration of Huh7 cells was promoted, too (Figure 4D). Moreover, global histone H3 acetylation (H3ac), and specific acetylation on residue lysine 9 of H3 (H3K9ac) under RGFP966 treatment were increased, suggesting the efficacy of HDAC3 inhibition by RGFP966 (Figure 4E). In addition, ectopic expression of HDAC3 reversed the RGFP966-induced downregulation of EGFR (Figure 4F). These data suggested that HDAC3 is involved in RGFP-induced inhibition of EGFR expression.
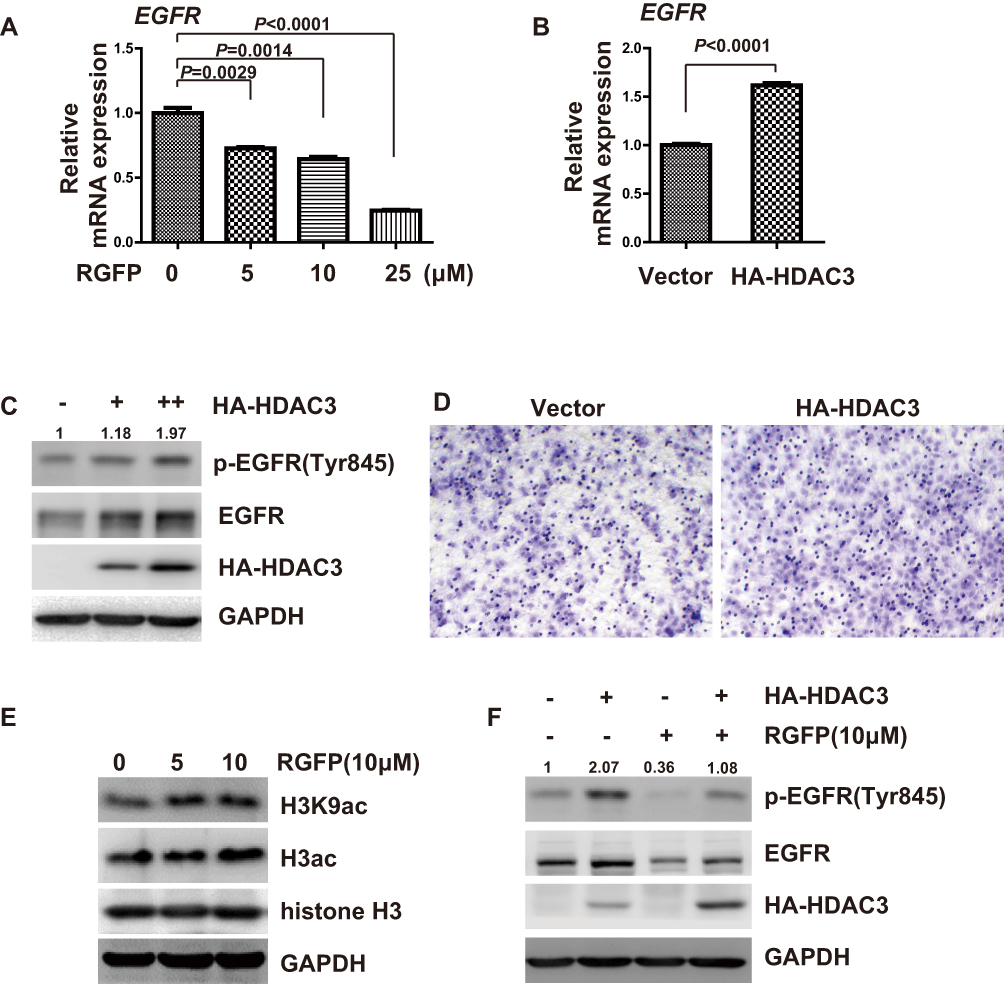 |
Figure 4 Inhibition of EGFR by RGFP966 is associated with HDAC3. (A), Huh7 cells were treated with indicated doses of RGFP966 (RGFP), or vehicle. After 40 hrs, cells were harvested, and total RNA was extracted. Relative expression of EGFR mRNA was determined by real-time PCR. GAPDH was used as internal control. Data are represented as mean ± SD from three independent experiments. P value refers to two-sided t test. (B), Huh7 cells were separately transfected with empty vector and expression vector for HA-tagged HDAC3. After 40 hrs, cells were harvested, and total RNA was extracted. Relative expression of EGFR mRNA was determined by real-time PCR. GAPDH was used as internal control. Data are represented as mean ± SD from three independent experiments. P value refers to two-sided t test. (C), Huh7 cells were separately transfected with empty vector and different doses of HA-tagged HDAC3 expression plasmids. After 48 hrs, cells were harvested, and Western Blot analysis was performed with the indicated antibodies. GAPDH was used as internal control. (D), Huh7 cells were separately transfected with empty vector and expression vector for HA-tagged HDAC3. After 24 hrs, 2.5 x104 cells were plated into a transwell chamber. After 40 hrs, the invaded cells were stained, and representative images were photographed. (E), Huh7 cells were treated with indicated doses of RGFP966 (RGFP), or vehicle. After 48 hrs, cells were harvested and Western Blot analysis was performed with the indicated antibodies. GAPDH was used as internal control. (F), Huh7 cells were separately transfected with empty vector and expression plasmids for HA-HDAC3, and then were treated with RGFP966 (RGFP, 10μM) and vehicle for 48hrs. Then, cells were harvested, and Western Blot analysis was performed with the indicated antibodies. GAPDH was used as internal control. |
Discussion
In the present report, we showed that RGFP966 has the strong potential against HCC cells, as evidenced by decreased proliferation and migration. Our finding suggests that HDAC3 inhibition by RGFP966 may offer a new approach for HCC chemotherapy.
In previous studies, pharmacological inhibition of HDACs showed clinical promise for the treatment of HCC. Panobinostat is a non-selective HDAC inhibitor. Treatment with panobinostat combined with sorafenib demonstrated the highest preclinical efficacy in HCC models, providing the rationale for clinical studies with this novel combination.8 Droxinostat, a type of histone deacetylase inhibitor, suppresses HDAC3 expression and induces histone acetylation and HCC cell death through activation of the mitochondrial apoptotic pathway and downregulation of FLIP, supporting its potential application in the treatment of HCC.7 A recent study reported that HDAC3 participates in hepatitis C virus (HCV) replication, and RGFP966 may be a potential treatment for diseases associated with HCV infection such as HCC, although no change in cell viability following treatment of 10μM RGFP966 in HCV-infected Huh7 cells.20 In accordance with this report, we found that RGFP966 mildly inhibits cell proliferation of Huh7 and HepG2 cells at a higher concentration, whereas remarkable inhibitory effect was shown in PLC/PRF/5 cells following treatment of 10μM RGFP966 (Figure 1).
EGFR, a receptor tyrosine kinase, is overexpressed in a significant proportion of hepatocellular carcinomas and can be phosphorylated at different tyrosine sites, leading to subsequent activation of different pathways.19 Notably, a recent work suggested that activation of EGFR signal in HCC cells caused the cells to exhibit EMT phenotypic changes and KIAA1199 promotes sorafenib tolerance and the metastasis of hepatocellular carcinoma by activating the EGF/EGFR-dependent epithelial-mesenchymal transition program.21 EGFR activation may also contribute to sorafenib resistance in HCC.22 In our study, RGFP966 inhibited EGFR expression and phosphorylation, which could induce the repression of cell growth and cell migration (Figures 1 and 2), but other underlying mechanisms may be involved in the processes.
The relationship between EGFR and HDAC3 seems dependent on cellular context. HDAC inhibition decreases the transcription and expression of EGFR in colorectal cancer cells.23 However, in another study, the down-regulation of HDAC3 did not affect the expression of EGFR, but HDAC3-CAGE axis regulates the activation of EGFR.24 In kidney cells, HDAC3 inhibition reduces the EGFR expression level and attenuates cellular proliferation.25 HDAC3 functions as a locus-specific corepressor to repress genes through histone deacetylation.26 In liver, HDAC3 regulates gene transcription by catalyzing the deacetylation of core histones and is involved in various biological processes.27 In our study, we found that HDAC3 upregulates EGFR transcription and RGFP966 can repress the mRNA expression of EGFR in HCC cells (Figure 4A and B) further proving that HDAC3 is directly targeted by RGFP966, but the mechanisms underlying involvement of HDAC3 in EGFR transcription of HCC cells need further investigation.
Conclusions
Overall, we elucidated an inhibitive function of RGFP966 in HCC progression. Our results suggested that RGFP966 inhibits EGFR signaling pathway by repressing EGFR expression and phosphorylation, which could lead to suppression of proliferation and migration of HCC cells. In conclusion, RGFP966 may inhibit HCC cell growth and migration through suppressing the HDAC3-EGFR signaling, and suggests a potential role in the treatment of HCC.
Abbreviations
HDAC3, histone deacetylase 3; HCC, hepatocellular carcinoma; EGFR, epidermal growth factor receptor; HCV, hepatitis C virus; RTK, phospho-receptor tyrosine kinases; STR, short tandem repeat.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi:10.3322/caac.21492
2. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16(7):411–428. doi:10.1038/s41575-019-0145-7
3. Kudo M. Lenvatinib in advanced hepatocellular carcinoma. Liver Cancer. 2017;6(4):253–263. doi:10.1159/000479573
4. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, Phase 3 trial. Lancet. 2017;389(10064):56–66. doi:10.1016/S0140-6736(16)32453-9
5. Abou-Alfa GK, Meyer T, Cheng AL, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. 2018;379(1):54–63. doi:10.1056/NEJMoa1717002
6. Lu YS, Kashida Y, Kulp SK, et al. Efficacy of a novel histone deacetylase inhibitor in murine models of hepatocellular carcinoma. Hepatology. 2007;46(4):1119–1130. doi:10.1002/hep.21804
7. Liu J, Li G, Wang X, et al. Droxinostat, a histone deacetylase inhibitor, induces apoptosis in hepatocellular carcinoma cell lines via activation of the mitochondrial pathway and downregulation of FLIP. Transl Oncol. 2016;9(1):70–78. doi:10.1016/j.tranon.2016.01.004
8. Lachenmayer A, Toffanin S, Cabellos L, et al. Combination therapy for hepatocellular carcinoma: additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J Hepatol. 2012;56(6):1343–1350. doi:10.1016/j.jhep.2012.01.009
9. Yang WM, Yao YL, Sun JM, Davie JR, Seto E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J Biol Chem. 1997;272(44):28001–28007. doi:10.1074/jbc.272.44.28001
10. Yan Y, An J, Yang Y, et al. Dual inhibition of AKT-mTOR and AR signaling by targeting HDAC3 in PTEN- or SPOP-mutated prostate cancer. EMBO Mol Med. 2018;10(4):e8478. doi:10.15252/emmm.201708478
11. McLeod AB, Stice JP, Wardell SE, Alley HM, Chang CY, McDonnell DP. Validation of histone deacetylase 3 as a therapeutic target in castration-resistant prostate cancer. Prostate. 2018;78(4):266–277. doi:10.1002/pros.23467
12. Wells CE, Bhaskara S, Stengel KR, et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS One. 2013;8(7):e68915. doi:10.1371/journal.pone.0068915
13. He P, Li K, Li SB, et al. Upregulation of AKAP12 with HDAC3 depletion suppresses the progression and migration of colorectal cancer. Int J Oncol. 2018;52(4):1305–1316. doi:10.3892/ijo.2018.4284
14. Bhaskara S, Knutson SK, Jiang G, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18(5):436–447. doi:10.1016/j.ccr.2010.10.022
15. Lu XF, Cao XY, Zhu YJ, et al. Histone deacetylase 3 promotes liver regeneration and liver cancer cells proliferation through signal transducer and activator of transcription 3 signaling pathway. Cell Death Dis. 2018;9(3):398. doi:10.1038/s41419-018-0428-x
16. Liu C, Liu L, Shan J, et al. Histone deacetylase 3 participates in self-renewal of liver cancer stem cells through histone modification. Cancer Lett. 2013;339(1):60–69. doi:10.1016/j.canlet.2013.07.022
17. Wu LM, Yang Z, Zhou L, et al. Identification of histone deacetylase 3 as a biomarker for tumor recurrence following liver transplantation in HBV-associated hepatocellular carcinoma. PLoS One. 2010;5(12):e14460. doi:10.1371/journal.pone.0014460
18. Logue JS, Morrison DK. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012;26(7):641–650. doi:10.1101/gad.186965.112
19. Kannangai R, Sahin F, Torbenson MS. EGFR is phosphorylated at Ty845 in hepatocellular carcinoma. Mod Pathol. 2006;19(11):1456–1461. doi:10.1038/modpathol.3800665
20. Zhou Y, Wang Q, Yang Q, et al. Histone deacetylase 3 inhibitor suppresses hepatitis C virus replication by regulating Apo-A1 and LEAP-1 expression. Virol Sin. 2018;33(5):418–428. doi:10.1007/s12250-018-0057-7
21. Xu Y, Xu H, Li M, et al. KIAA1199 promotes sorafenib tolerance and the metastasis of hepatocellular carcinoma by activating the EGF/EGFR-dependent epithelial-mesenchymal transition program. Cancer Lett. 2019;454:78–89. doi:10.1016/j.canlet.2019.03.049
22. Ezzoukhry Z, Louandre C, Trecherel E, et al. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. int J Cancer. 2012;131(12):2961–2969. doi:10.1002/ijc.27604
23. Chou CW, Wu MS, Huang WC, Chen CC. HDAC inhibition decreases the expression of EGFR in colorectal cancer cells. PLoS One. 2011;6(3):e18087. doi:10.1371/journal.pone.0018087
24. Kim H, Kim Y, Goh H, Jeoung D. Histone deacetylase-3/CAGE axis targets EGFR signaling and regulates the response to anti-cancer drugs. Mol Cells. 2016;39(3):229–241. doi: 10.14348/molcells.2016.2244
25. Gilbert RE, Huang Q, Thai K, et al. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int. 2011;79(12):1312–1321. doi:10.1038/ki.2011.39
26. Karagianni P, Wong J. HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene. 2007;26(37):5439–5449. doi:10.1038/sj.onc.1210612
27. Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J. 2008;27(7):1017–1028. doi:10.1038/emboj.2008.51
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.