")
Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
RF1 Gene Mutation in Familial Hemophagocytic Lymphohistiocytosis 2: A Family Report and Literature Review
Authors Shi Y, Qiao Z, Bi X, Zhang C, Fu J, Jia Y, Yang G
Received 9 July 2021
Accepted for publication 24 November 2021
Published 16 December 2021 Volume 2021:14 Pages 1637—1645
DOI https://doi.org/10.2147/PGPM.S326921
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Yuan Shi,* Zhidong Qiao,* Xiaoduo Bi, Chenxin Zhang, Junxian Fu, Yuexin Jia, Guanglu Yang
Department of Pediatric, The Affiliated Hospital of Inner Mongolia Medical University, Hohhot, 010000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guanglu Yang
Department of Pediatric, The Affiliated Hospital of Inner Mongolia Medical University, No. 1 Tongdao North Road, Hohhot, Inner Mongolia Autonomous Region, Hohhot, 010000, People’s Republic of China
Tel +86 13451311622
Email [email protected]
Objective: Gene mutation analysis was performed on a family with familial hemophagocytic lymphohistiocytosis (FHL) so as to provide an accurate etiological diagnosis, leading to genetic counseling for the family members.
Methods: The clinical data of two probands (siblings) with FHL in one family were analyzed, and eight genes related to the onset of the primary hemophagocytic lymphohistiocytosis (pHLH) (PRF1, UNC13D, STX11, STXBP2, SH2D1A, BIRC4/XIAP, Rab27a, LYST) were detected and analyzed in the probands and their parents with whole exome sequencing.
Results: Proband 1 was a two-year-old male with the clinical manifestations of fever, hepatosplenomegaly, and a decreased peripheral blood cell count, sCD25: 12504pg/mL. The results of genetic testing showed that there was a c.1349C>T heterozygous missense mutation and a c.853_855del heterozygous mutation in the PRF1 in proband 1. Proband 2 was an eight-year-old female with the clinical manifestations of convulsions and disturbance of consciousness with fever. The genetic test results were the same as those of proband 1. There was a single heterozygous mutation in the parents of the probands, and both probands had compound heterozygous mutations.
Conclusion: According to the clinical manifestations, laboratory tests, and results of the family molecular genetic testing, the probands could be clinically diagnosed as FHL2. The results of gene sequencing revealed that this was an autosomal recessive family with familial hemophagocytic syndrome. A rare pathogenic mutation (c.853_855del) in the PRF1 was discovered in the two patients with HLH.
Keywords: FHL, PRF1, novel mutation
Hemophagocytic lymphohistiocytosis (HLH) is also referred to as hemophagocytic syndrome (HPS). HLH is a disease caused by the dysfunction of the immune system. The etiologies include molecular genetic abnormalities, infections, tumors, and autoimmune diseases. The main clinical manifestations are repeated fever, a decreased blood cell count to varying degrees, hepatosplenomegaly, and hepatic function abnormalities,1 together with manifestations of the central nervous system in many cases.2 Studies have found that children with HLH have cytotoxic cell dysfunction, meaning that antigens cannot be cleared in the normal manner.3 In turn, the monocytes and macrophages are constantly stimulated by antigens and are abnormally activated to produce a large number of cytokines, causing tissue damage. HLH progresses quickly with potentially dangerous consequences. If it is not treated in time, or treated ineffectively, survival duration is short and the prognosis is poor.4 HLH is traditionally divided into two subtypes, according to whether there are genetic defects: primary (hereditary) HLH and secondary (acquired) HLH. Hereditary HLH comprises the familial HLH (FHL) types 1–5 and HLH caused by primary immunodeficiency syndrome, and mainly occurs in infants or during early childhood. Acquired HLH is secondary to infections, rheumatic diseases and tumors.5
FHL2 is the most common familial form of HLH with immune deficiency. FHL2 patients have mutations in genes affecting the expression of perforin which contribute to excessive activation and hyperplasia of CD8+ cytotoxic T lymphocytes (CTL) and natural killer (NK) cells. Perforin, a cytolytic protein, is a major factor responsible for the cytolytic activities of CTL and NK cells. Perforin, a pore-forming protein, creates pores in the target cytomembrane, generating apoptotic death through granzymes. PRF1 mutations cause decreased or absent expression of perforin protein, resulting in cytotoxic immunodeficiency. The loss of cytotoxic immune function contribute to an uncontrolled and persistent immune stimulus response in FHL2 patients, accompanied by high levels of cytokine release. If the immune system and excessive cytotoxic T-cell and NK-cell stimulation incontrollable. They can cause systemic inflammation and multiorgan failure.6
In 2004, the International Society of Tissue Cells revised the diagnostic criteria for familial and reactive HLH.7 In the present study, gene mutation analyses were performed on one family, consisting of two children with FHL and their parents, with a view to providing an accurate etiological diagnosis, followed by etiological diagnosis and genetic counseling for the family members.
Case Report
Proband 1, a 2-year-old male, reported “fever and hepatosplenomegaly” at the time of the hospital visit. There was no abnormality in the personal history and past history of the proband. The parents were not consanguineous and had no similar medical history. We perfected the laboratory examination and found: He had decreased peripheral blood cells, bone marrow cell morphology shows: lymphocytes increase, hemophagocytic cells are more common, and rised sCD25: 12504pg/mL. The ferritin level is normal.
Proband 2 (the sibling of proband 1), 8 years old, was first diagnosed with acute disseminated encephalomyelitis (ADEM) in a hospital in Beijing after presenting with “convulsions, disturbance of consciousness and fever for two days” more than a year ago. Proband 2’s symptoms recovered after immunotherapy. Magnetic resonance imaging (MRI) of the head during the hormone reduction period indicated that there were lesions that were aggravated, and this anomaly was followed by neurological symptoms. With the re-application of hormone therapy, proband 2’s condition improved, but the intracranial lesions still showed progressive aggravation. Transient cytopenia, hepatosplenomegaly, and decreased NKC activity were observed during the course of the disease with normal hepatic function, levels of triacylglyceride and coagulation function in multiple detections. Eventually, after disease analysis and genetic examinations, proband 2 was given the diagnosis of “familial haemophagocytosis syndrome (FHPS) with the onset of the central nervous system”. And the proband 2’s bone marrow cell morphology shows: lymphocytes increase, hemophagocytic cells are more common.
The proband 1 met the following five, out of a total of eight, clinical diagnostic criteria of HLH-2004: fever, splenomegaly, cytopenia, hemophagocytosis and sCD25:12504pg/mL. While the proband 2 also met five: fever, splenomegaly, cytopenia, hemophagocytosis and decreased NKC activity. Since the two probands were strongly suspected of being HLH, genetic testing of 12 subjects in three generations of the family was carried out, and the blood was drawn and sent to the Beijing Kangxu Medical Laboratory. The proband 1 did full exon detection, using the method of next-generation sequencing, while the proband 2 did site verification, using sanger sequencing. The genotypes of these two sites can be determined by sanger sequencing. The results are shown in Figure 1. There was a heterozygous missense mutation in exon 3 c.1349C>T (nucleotide No. 1349 in the coding region was changed from C to T), which was detected in the probands, their father, grandfather and father’s sister, and c.853_855del (nucleotide No. 853_855 in the coding region) hybrid combined frameshift mutation, which was detected in the probands, their mother, grandfather, and mother’s brother. The probands’s genes sequencing results in Figure 2. The c.1349C>T heterozygous missense mutation caused amino acid N450 to change from Thr to Met; while the c.853_855del heterozygous frameshift mutation caused the deletion of amino acid N285. The protein damage prediction results were analyzed by SIFT, Polyphen2 and Mutation Taster. The c.1349C>T heterozygous missense mutation is c. 1349c > t heterozygous missense mutation is harmful and pathogenic. While c.853_855del heterozygous frameshift mutation is suspected to be pathogenic. The above-mentioned mutations might have caused the protein function to be affected, which in turn would lead to the occurrence of HLH. The mutation site c.1349C>T (p.Thr450Met) has been included in the dbSNP database with the ID of rsl8965890, and literature reports related to HLH disease can be retrieved.8 While the pathogenicity of the mutation c.853_855del has not been reported in the Chinese literature, nor has it been included in the dbSNP database. Both the maternal and fraternal grandfathers, the maternal grandmother, the parents, the second aunt, and an uncle had single heterozygous mutations. None of the above-mentioned mutations were polymorphic changes, which have an extremely low frequency of occurrence in the population. The compound heterozygous variants found in the PRF1 gene of the probands were inherited from their parents, and the above variants might be pathogenic variants that caused the onset of the disease in the probands.
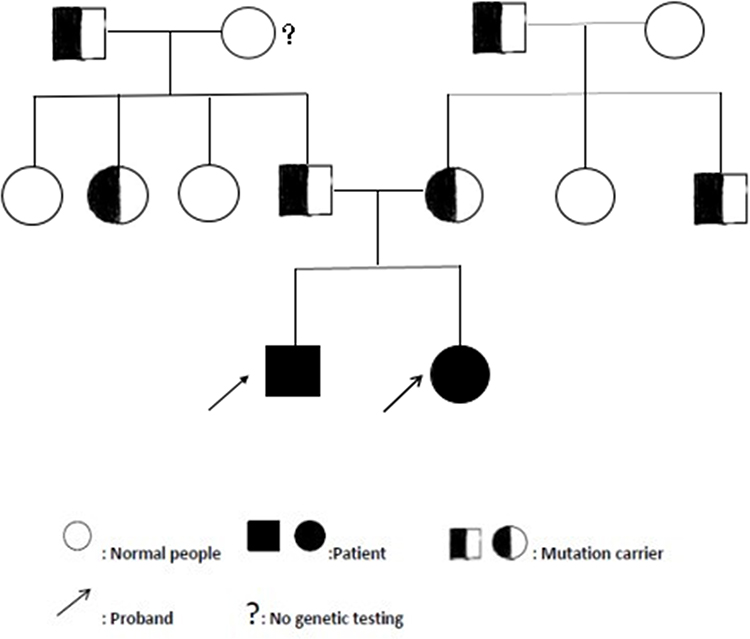 |
Figure 1 HLH pedigree map. |
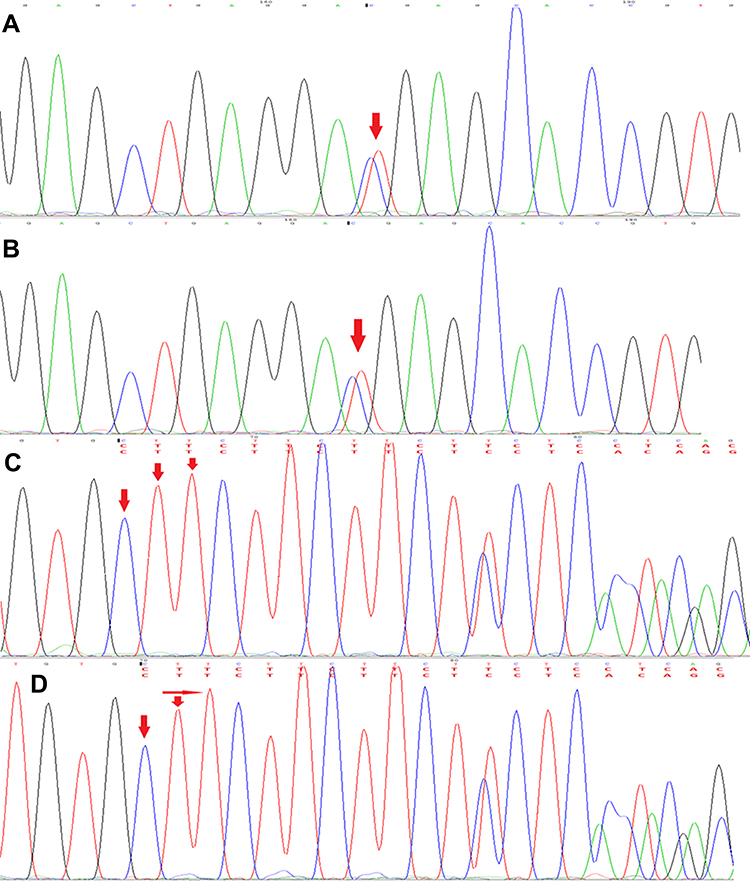 |
Figure 2 The probands’s genes sequencing results (A). The sequencing peak of c.1349C>T of Proband 1. (B) The sequencing peak of c.1349C>T of Proband 2. (C) The sequencing peak of c.853_855del of Proband 1. (D) The sequencing peak of c.853_855del of Proband 2. |
The proband 1 was Proband 1 received bone marrow transplantation successfully in January 2020. And proband 2 is scheduled to receive bone marrow transplantation this year.
Informed written consent was obtained from patients and their guardians for the publication of the case details and any accompanying images.
Literature Review
The CNKI and Wanfang databases were searched using the keywords of “familial hemophagocytic syndrome”, “PRF1” and “novel mutation”, and the PubMed database was searched using the keywords of “FHL”, “PRF1” and “novel mutation” from January 1st, 2009 to June 30th, 2020.
From a comprehensive database of gene mutations related to the disease, the four genes for FHL, namely, PRF1, UNC13D, STXBP2, and STX11, were investigated. There were 433 different mutations of these genes to be found in the literature review, of which UNC13D had the maximum amount of mutations (189), followed by PRF1 (157), STXBP2 (66), and then STX11 (21).9
Thus, it was shown that mutations in the PRF1 accounted for the main proportion of the occurrences of FHL. Genotypic characteristics and immune indicators were evaluated in a study concerning 311 cases of HPS in China. It was reported that in the 128 cases where genetic testing was complete, the different types of genetic variation could be clarified. The mutation rate of the UNC13D gene was said to be the highest (29%), and then LYST (21%), PRF1 (17%) and STXBP2 (10%). The most common of the main pathogenic genes in 39 cases of primary HPS was PRF1 (16/39, 41%), follow by UNC13D (10/39, 26%) and BIRC4 (6/39, 15%). Also, there were differences in the genes related to primary HPS in children and adolescents, in that PRF1 was more prominent in children and adolescents while in grown-up UNC13D was the most common, followed by PRF1.10
There was a report in X of a new fatal case of congenital pyrimidine metabolic abnormality and rapid progression of multiple organ failure. It was found that the patient had two single gene defects at the same time, including the c.853_855del. At present, this gene defect has not been reported in China. The mixed phenotype of the two coexisting single gene defects might have been the main reason for the rapid deterioration of the condition in the patient, leading to multiple organ failure and early death.11
Discussion
HLH is a group of clinical syndromes in which lymphocytes and macrophages are excessively activated and then proliferate due to multiple etiologies. A large number of inflammatory cytokines are released causing a “cytokine storm”, resulting in multiple organ dysfunction. Current studies believe that the main pathogenesis of the disease is an immune regulation disorder or immune imbalance, causing the excessive secretion of lymphokines and monocyte factors by T lymphocytes and mononuclear macrophages. In addition, a cascade of inflammatory mediators will lead to corresponding clinical symptoms. The highly activated macrophages, which have strong cell activation, proliferation, and hemophagocytic abilities, together with the increased levels of inflammation, can inhibit blood cell production and reduce the amount of blood cells.1
Classify FHL as five subtypes, types 1 to 5, all of which are autosomal recessive diseases. Except for FHL1, in which the gene defect is unknown, four different pathogenic genes have been identified: PFR1 (FHL2), UNC13D (FHL3), STX11 (FHL4), and STXBP2 (FHL5).12 The incidence of gene defects in FHL, of which FHL2 and FHL3 are the most common, is largely correlated with race.13 The PRF1 is located at 10q21-22 and contains three exons. The coding regions are the 2nd and 3rd exons. The perforin encoded in PRF1 contains 555 amino acids, including three transmembrane regions, the EGF-like region and three functional regions in the C2 region. The perforin can produce cytotoxic particles in the cytoplasm of target cells, which can lead to apoptosis. The NK cells and CTL cells mainly kill the target cells through the perforin/granzyme pathway. When this gene is mutated, the expression of perforin is down-regulated, and so the stability is reduced. When cytotoxic cells make contact with the target cell, the damaged perforin cannot smoothly form a channel on the membrane of the target cell. This will prevent the perforin from delivering the granzyme into the target cell to induce the apoptosis and osmotic dissolution, and, eventually, lead to impaired function in the killing of the target cells.
The PRF1 mutation is the most frequency mutation, accounting for 20–40% of all FHL cases. The highest rate of PRF1 mutation was reported in African-American families (>50%), and then the population of Turkey (43%), Japan (42%), and India (41%).14 According NCBI clinvar database (https://www.ncbi.nlm.nih.gov/clinvar), a total of 173 PFR1 mutations have been reported now, among which 28 mutations were definitely pathogenic, all occurring in the second and third exons of the coding region. Some special mutation sites are more common in subjects from specific races and regions.14 The nonsense mutations (c.1122G>A) of PRF1 mutations are more common in the Turkish population, the frameshift mutations (c.1090_1091delCT and c.50delT) are more common in Japanese and African-American populations, while the missense mutations, p.Ala91Val and p.Trp129Ser, have appeared in the Italian and Indian populations, respectively. The missense mutations (c.445G>A) are common in Spanish patients while in Japanese patients with HLH, the most common mutation is c.1090_1091delCT (accounting for approximately 62.5% of the PRF1 mutations), followed by c.207delC (about 37.5% of PRF1 mutations).15,16
In addition, mutations and variants of the PRF1 have also been reported in other diseases, including perforin deficiency syndrome, multiple sclerosis, type 1 diabetes, non-Hodgkin’s lymphoma, and leukemia. So far, more than 115 pathogenic gene variants have been reported for PRF1, which mainly is the missense and nonsense mutations. Generally, the severity of the disease depends on the residual activity of perforin (Table 1).17
 |
 |
 |
Table 1 Mutation of PRF1 Gene in Database |
According to data from European and American HLH registration centers, FHL3 accounts for about 30–50% of FHL, second only to FHL2. However, data from Japan, South Korea, and other countries in Asia show that FHL3 is the most common and can account for up to 89% of FHL in this part of the world.18 According to the statistics from the Histiocytosis Research Group of the Chinese Academy of Pediatrics in 2016, there were 323 cases of HLH in 12 hospitals in China. Among these cases, FHL accounted for 27.9%, of which 41.7% were FHL3, exceeding FHL2, which accounted for 29.2%.19 These data indicate that the mutation type in FHL has a geographical location and race difference. FHL3 is an autosomal recessive inherited disease. So far, 112 mutations have been discovered. The UNC13D gene mutation causes the abnormal function of the Munc13-4 protein, leading to severe damage to the killing function of the NK cells. The mutation sites are all over the gene, and nonsense/missense mutations predominate (53.6%).20
Conclusion
In the present case, both the two probands in the family were confirmed with a diagnosis of familial HPS. A pathogenic mutation in the PRF1 was found in the probands, and there have been related literature reports concerning the heterozygous mutation of c.1349C>T. The pathogenicity of the c.853_855del heterozygous mutation has not been reported in China. However, it might also cause protein dysfunction, and this might be the cause of HLH in the probands. Meanwhile, detailed etiological diagnosis and genetic counseling has also been provided for the families of the patients.
Abbreviations
FHL, familial hemophagocytic lymphohistiocytosis; pHLH, primary hemophagocytic lymphohistiocytosis; HLH, Hemophagocytic lymphohistiocytosis; HPS, hemophagocytic syndrome; MRI, Magnetic resonance imaging; FHPS, familial haemophagocytosis syndrome.
Ethics Approval and Consent to Participate
This study was conducted with approval from the Ethics Committee of The Affiliated Hospital of Inner Mongolia Medical University. This study was conducted in accordance with the declaration of Helsinki. Informed written consent was obtained from patients and the children’s guardians for the publication of the case details and any accompanying images.
Funding
Natural Science Foundation of Inner Mongolia Autonomous Region, Project Number: 2015MS08103; Major Project of Inner Mongolia Medical University, Project Number: YKD2012KJBW005; Youth Science and Technology Talents Program-Class A, Project Number: NJYT-17-A19; Major Scientific Research Project of Affiliated Hospital of Inner Mongolia Medical University, Project number: NYFYZD022; Inner Mongolia Autonomous Region Neurological Disease Clinical Medicine Research Center, project number: MYYXT201903.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Shi LH, Tian L, Huang S, et al. Clinical features of phagocytic syndrome in children and the significance of genetic testing. Chin J Pract Pediatr. 2019;34(6):449–452.
2. Chen X, Wang F, Zhang Y, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet. 2018;94(2):200–212. doi:10.1111/cge.13363
3. Jiang ZF, Shen KL, Shen Y, et al. Chu Fu tang practical pediatrics. 8th ed; 2018. ISBN 978-7-117-19978-0/R-19979.
4. Rousselin A, Alavi Z, Le Moigne E, et al. Hemophagocytic syndrome in pregnancy: case report, diagnosis, treatment, and prognosis. Clin Case Rep. 2017;5(11):1756–1764. doi:10.1002/ccr3.1172
5. Luo ZB, Tang YM. Research progress of primary phagocytic syndrome. Chin J Pediatr. 2014;52(4):267–270.
6. Sevim H, Kocaefe YÇ, Onur MA, Uçkan-çetinkaya D, Gürpınar ÖA. Bone marrow derived mesenchymal stem cells ameliorate inflammatory response in an in vitro model of familial hemophagocytic lymphohistiocytosis 2. Stem Cell Res Ther. 2018;9(1):198. doi:10.1186/s13287-018-0941-y
7. Mian A, Kumari K, Kaushal S, et al. Fatal familial hemophagocytic lymphohistiocytosis with perforin gene (PRF1) mutation and EBV-associated T-cell lymphoproliferative disorder of the thyroid. Autops Case Rep. 2019;9(3):e2019101. doi:10.4322/acr.2019.101
8. Ding Q, Guo X, Li Q. [Analysis of PRF1gene variant in a child with late-onset familial hemophagocytic lymphohistiocytosis type 2 and severe central nervous system disease]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2019;36(6):592–594. Chinese. doi:10.3760/cma.j.issn.1003-9406.2019.06.015
9. Viñas-Giménez L, Padilla N, Batlle-Masó L, et al. FHLdb: a comprehensive database on the molecular basis of familial hemophagocytic lymphohistiocytosis. Front Immunol. 2020;31(11):107. doi:10.3389/fimmu.2020.00107
10. Zhang J, Sun Y, Shi X, et al. Genotype characteristics and immunological indicator evaluation of 311 hemophagocytic lymphohistiocytosis cases in China. Orphanet J Rare Dis. 2020;15(1):112. doi:10.1186/s13023-020-01390-z
11. Pérez-Torras S, Mata-Ventosa A, Drögemöller B, et al. Deficiency of perforin and hCNT1, a novel inborn error of pyrimidine metabolism, associated with a rapidly developing lethal phenotype due to multi-organ failure. Biochim Biophys Acta Mol Basis Dis. 2019;1865(6):1182–1191. doi:10.1016/j.bbadis.2019.01.013
12. Fager Ferrari M, Leinoe E, Rossing M, et al. Germline heterozygous variants in genes associated with familial hemophagocytic lymphohistiocytosis as a cause of increased bleeding. Platelets. 2018;29(1):56–64. doi:10.1080/09537104.2017.1293808
13. Benson LA, Li H, Henderson LA, et al. Pediatric CNS-isolated hemophagocytic lymphohistiocytosis. Neurol Neuroimmunol Neuroinflamm. 2019;6(3):e560. doi:10.1212/NXI.0000000000000560
14. Mhatre S, Madkaikar M, Desai M, Ghosh K. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis (FHL) patients in India. Blood Cells Mol Dis. 2015;54(3):250–257. doi:10.1016/j.bcmd.2014.11.023
15. Sun SW, Guo X, Zhu YP, et al. Clinical phenotype and gene mutation analysis of a familial family with phagocytic syndrome. Chin J Med Gene. 2014;31(05):570–573. doi:10.3760/cma.j.issn.1003-9406.2014.01.006
16. Sheth J, Patel A, Shah R, Bhavsar R, Trivedi S, Sheth F. Rare cause of Hemophagocytic Lymphohistiocytosis due to mutation in PRF1 and SH2D1A genes in two children - A case report with a review. BMC Pediatr. 2019;19(1):73. doi:10.1186/s12887-019-1444-4
17. Bordbar MR, Modarresi F, Farazi Fard MA, Dastsooz H, Shakib Azad N, Faghihi MA. A case report of novel mutation in PRF1 gene, which causes familial autosomal recessive hemophagocytic lymphohistiocytosis. BMC Med Genet. 2017;18(1):49. doi:10.1186/s12881-017-0404-9
18. Huang JB, Wang J, Jiang L, et al. Late fetal onset of familial phagocytic syndrome - A case report. Chin J Exp Hematol. 2017;25(6):1825–1828. doi:10.7534/j.issn.1009-2137.2017.06.045
19. Xu XJ, Wang HS, Ju XL, et al.; Histiocytosis Study Group of the Chinese Pediatric Society. Clinical presentation and outcome of pediatric patients with hemophagocytic lymphohistiocytosis in China: a retrospective multicenter study. Pediatr Blood Cancer. 2017;64(4):e26264. doi:10.1002/pbc.26264
20. Kamoun F, Hsairi M, Grandin V, Ben Ameur S, De Saint Basile G, Hachicha M. Familial haemophagocytosis lymphohisticytosis type 3: a case report. Arch Pediatr. 2017;24(1):33–35. doi:10.1016/j.arcped.2016.10.021
21. Del Giudice E, Savoldi G, Notarangelo LD, et al. Acute inflammatory demyelinating polyradiculoneuropathy associated with perforin-deficient familial haemophagocytic lymphohistiocytosis. Acta Paediatr. 2003;92(3):398–401. doi:10.1111/j.1651-2227.2003.tb00566.x
22. Ishii E, Ueda I, Shirakawa R, et al. Genetic subtypes of familial hemophagocytic lymphohistiocytosis: correlations with clinical features and cytotoxic T lymphocyte/natural killer cell functions. Blood. 2005;105(9):3442–3448. doi:10.1182/blood-2004-08-3296
23. Clementi R, Zur Stadt U, Savoldi G. Six novel mutations in the PRF1 gene in children with haemophagocytic lymphohistiocytosis. J Med Genet. 2001;38(9):643–646. doi:10.1136/jmg.38.9.643
24. Zur Stadt U, Beutel K, Kolberg S, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006;27(1):62–68. doi:10.1002/humu.20274
25. Marcenaro S, Gallo F, Martini S, et al. Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13-4 defect and discriminates between genetic subtypes of the disease. Blood. 2006;108(7):2316–2323. doi:10.1182/blood-2006-04-015693
26. Bryceson YT, Rudd E, Zheng C, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110(6):1906–1915. doi:10.1182/blood-2007-02-074468
27. Göransdotter Ericson K, Fadeel B, Nilsson-Ardnor S, et al. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet. 2001;68(3):590–597. doi:10.1086/318796
28. Horne A, Ramme KG, Rudd E, et al. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br J Haematol. 2008;143(1):75–83. doi:10.1111/j.1365-2141.2008.07315.x
29. Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci U S A. 2009;106(24):9809–9814. doi:10.1073/pnas.0903815106
30. Nagai K, Yamamoto K, Fujiwara H, et al. Subtypes of familial hemophagocytic lymphohistiocytosis in Japan based on genetic and functional analyses of cytotoxic T lymphocytes. PLoS One. 2010;5(11):e14173.
31. Suga N, Takada H, Nomura A, et al. Perforin defects of primary haemophagocytic lymphohistiocytosis in Japan. Br J Haematol. 2002;116(2):346–349. doi:10.1046/j.1365-2141.2002.03266.x
32. Ueda I, Morimoto A, Inaba T, et al. Characteristic perforin gene mutations of haemophagocytic lymphohistiocytosis patients in Japan. Br J Haematol. 2003;121(3):503–510. doi:10.1046/j.1365-2141.2003.04298.x
33. Wang JS, Wang Z, Wu L, Chen X, Feng C. Etiology analysis of 38 cases of hemophagocytic syndrome. Chin j Exp Hematol. 2010;18(05):1316–1320.
34. Zhizhuo H, Junmei X, Yuelin S, et al. Screening the PRF1, UNC13D, STX11, SH2D1A, XIAP, and ITK gene mutations in Chinese children with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2012;58(3):410–414. doi:10.1002/pbc.23216
35. Chia J, Thia K, Brennan AJ, et al. Fatal immune dysregulation due to a gain of glycosylation mutation in lymphocyte perforin. Blood. 2012;119(7):1713–1716. doi:10.1182/blood-2011-08-374355
36. Sumegi J, Nestheide SV, Barnes MG, et al. Gene-expression signatures differ between different clinical forms of familial hemophagocytic lymphohistiocytosis. Blood. 2013;121(7):e14–24. doi:10.1182/blood-2012-05-425769
37. Seo JY, Song JS, Lee KO, et al.; Korea Histiocytosis Working Party. Founder effects in two predominant intronic mutations of UNC13D, c.118-308C>T and c.754-1G>C underlie the unusual predominance of type 3 familial hemophagocytic lymphohistiocytosis (FHL3) in Korea. Ann Hematol. 2013;92(3):357–364. doi:10.1007/s00277-012-1628-6
38. Dias C, McDonald A, Sincan M, et al. Recurrent subacute post-viral onset of ataxia associated with a PRF1 mutation. Eur J Hum Genet. 2013;21(11):1232–1239. doi:10.1038/ejhg.2013.20
39. Wada T, Sakakibara Y, Nishimura R, et al. Down-regulation of CD5 expression on activated CD8+ T cells in familial hemophagocytic lymphohistiocytosis with perforin gene mutations. Hum Immunol. 2013;74(12):1579–1585. doi:10.1016/j.humimm.2013.09.001
40. Zhang K, Chandrakasan S, Chapman H, et al. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood. 2014;124(8):1331–1334. doi:10.1182/blood-2014-05-573105
41. Mhatre S, Madkaikar M, Jijina F, Ghosh K. Unusual clinical presentations of familial hemophagocytic lymphohistiocytosis type-2. J Pediatr Hematol Oncol. 2014;36(8):e524–7. doi:10.1097/MPH.0000000000000102
42. Hussein AA, Hamadah T, Qandeel M, et al. Hematopoietic stem cell transplantation of an adolescent with neurological manifestations of homozygous missense PRF1 mutation. Pediatr Blood Cancer. 2014;61(12):2313–2315. doi:10.1002/pbc.25166
43. Abdalgani M, Filipovich AH, Choo S, et al. Accuracy of flow cytometric perforin screening for detecting patients with FHL due to PRF1 mutations. Blood. 2015;126(15):1858–1860. doi:10.1182/blood-2015-06-648659
44. Romero CA, Sánchez IP, Gutierrez-Hincapié S, et al. A novel pathogenic variant in PRF1 associated with hemophagocytic lymphohistiocytosis. J Clin Immunol. 2015;35(5):501–511. doi:10.1007/s10875-015-0169-x
45. Willenbring RC, Ikeda Y, Pease LR, Johnson AJ. Human perforin gene variation is geographically distributed. Mol Genet Genomic Med. 2018;6(1):44–55. doi:10.1002/mgg3.344
46. Willenbring RC, Johnson AJ. Finding a balance between protection and pathology: the dual role of perforin in human disease. Int J Mol Sci. 2017;18(8):1608. doi:10.3390/ijms18081608
47. Liu C, Li M, Wu X, Yao X, Zhao L. Type 2 familial hemophagocytic lymphohistiocytosis in half brothers: a case report. Medicine. 2018;97(30):e11577. doi:10.1097/MD.0000000000011577
48. Almalky MA, Saleh SHA, Baz EG, Fakhr AE. Clinico-laboratory profile and perforin gene mutations of pediatric hemophagocytic lymphohistiocytosis cases: a five-year single center study. Pan Afr Med J. 2020;27(36):354.
49. Sato H, Kawasaki N, Kawasaki M, et al. Three consecutive cases of familial hemophagocytic lymphohistiocytosis, including a case due to maternal uniparental disomy. J Pediatr Hematol Oncol. 2020;42(8):e819–e821. doi:10.1097/MPH.0000000000001681
50. Shabrish S, Kelkar M, Yadav RM, et al. The spectrum of clinical, immunological, and molecular findings in familial hemophagocytic lymphohistiocytosis: experience from India. Front Immunol. 2021;5(12):612583. doi:10.3389/fimmu.2021.612583
51. Godby RC, Kraemer RR, May J, et al. Co-occurrence of familial hemophagocytic lymphohistiocytosis Type 2 and chronic active Epstein-Barr virus in adulthood. Am J Med Sci. 2021;361(3):388–393. doi:10.1016/j.amjms.2020.10.004
52. Ahmari AA, Alsmadi O, Sheereen A, et al. Genetic and clinical characteristics of pediatric patients with familial hemophagocytic lymphohistiocytosis. Blood Res. 2021;56(2):86–101. doi:10.5045/br.2021.2020308
53. Liu XY, Nie YB, Chen XJ, Gao XH, Zhai LJ, Min FL. Adult onset type 2 familial hemophagocytic lymphohistiocytosis with PRF1 c.65delC/c.163C>T compound heterozygous mutations: a case report. World J Clin Cases. 2021;9(10):2289–2295. doi:10.12998/wjcc.v9.i10.2289
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.