")
Back to Journals » Drug Design, Development and Therapy » Volume 17
Review of Mavacamten for Obstructive Hypertrophic Cardiomyopathy and Future Directions
Authors Dong T, Alencherry B , Ospina S, Desai MY
Received 2 September 2022
Accepted for publication 16 March 2023
Published 8 April 2023 Volume 2023:17 Pages 1097—1106
DOI https://doi.org/10.2147/DDDT.S368590
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Tin Wui Wong
Tiffany Dong,* Ben Alencherry,* Susan Ospina, Milind Y Desai
Section of Cardiovascular Imaging, Heart, Vascular and Thoracic Institute, Cleveland Clinic, Cleveland, OH, USA
*These authors contributed equally to this work
Correspondence: Milind Y Desai, Department of Cardiovascular Medicine, Miller Family Heart and Vascular Institute, Cleveland Clinic Main Campus J1-5, 9500 Euclid Avenue, Cleveland, OH, 44195, USA, Tel +1 216 445 5250, Fax +1 216 445 6155, Email [email protected]
Abstract: Hypertrophic cardiomyopathy (HCM) is a condition with abnormal hypertrophy of the left ventricle in the absence of common causes. The most common form involves the basal septum and can lead to obstruction of the left ventricular outflow tract. Patients can experience exertional symptoms such as chest pain, dyspnea and syncope. Traditional treatment has included beta blockers and nondihydropyridine calcium channel blockers with second-line therapy being disopyramide. Recently, mavacamten, a cardiac myosin inhibitor, has demonstrated improvement in quantitative measures of obstruction and symptom relief to such a degree that patients were able to defer invasive management of the disease. This review focuses on the pharmacology of mavacamten, its clinical trial data and guidance on how to incorporate this drug into clinical practice. Furthermore, it discusses emerging therapies currently being investigated for HCM.
Keywords: hypertrophic cardiomyopathy, mavacamten, septal reduction therapy, EXPLORER HCM, VALOR HCM
Introduction
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant genetic condition affecting the sarcomere. Phenotypically, the disease manifests in hypertrophy in the absence of other causes. The prevalence is estimated >1:500 in individuals.1 HCM can be subdivided into obstructive (oHCM) and nonobstructive (nHCM), with the former defined as a resting left ventricular outflow (LVOT) gradient >30mmHg. Symptoms in oHCM include dyspnea, exertional intolerance, chest pain, syncope or sudden cardiac death.
Treatment options for oHCM can be divided into medication and invasive therapies. Traditional pharmacotherapy utilized for oHCM includes nonvasodilating beta blockers (BBs), nondihydropyridine calcium channel blockers (CCBs) and disopyramide. Interventions include alcohol septal ablation and surgical septal myectomy. This review will focus on mavacamten, a novel cardiac myosin inhibitor (CMI) which was recently approved by the Food and Drug Administration (FDA) for patients with oHCM.2 Specifically, a description of mavacamten pharmacology, clinical trial evidence, application into clinical practice for oHCM and future areas of study will be discussed.
Pathophysiology and Diagnostic Evaluation of Hypertrophic Cardiomyopathy
There is a strong genetic component to HCM, which affects myocardial sarcomeres.3 With more than 2000 mutations identified, the most common genes identified are beta myosin heavy chain 7 (MYH7), which codes for the major myosin isoform, and myosin-binding protein (MYBPC3). Gene identification is useful particularly for screening family members if the proband has a positive screen. However, there remains inconsistent genotype–phenotype correlations; some patients are gene positive but do not meet clinical criteria for HCM and others meet clinical criteria without identifiable genes. Furthermore, sporadic mutations do occur in patients with no family history of HCM.4
Given the inconsistencies in genetic testing, HCM remains largely a clinical diagnosis requiring identification of left ventricular hypertrophy by echocardiography or cardiac magnetic resonance imaging (CMR). Typically, left ventricle (LV) wall thickness >15mm or >13mm with pertinent family history should raise suspicion for HCM. Hypertrophy occurs most commonly in the basal septum but can vary in different morphologies of HCM. HCM mimickers such as hypertension, aortic stenosis, amyloidosis, muscular dystrophies, Fabry’s disease and lysosomal disorders should be excluded prior to diagnosing HCM.
The identification of an intracavitary gradient separates oHCM from nHCM. A peak gradient >30mmHg at rest is consistent with obstruction. If the peak gradient is <50mmHg, provocation with amyl nitrate, Valsalva maneuver or exercise echocardiography may reveal a concealed higher gradient.5,6 Other features of obstruction include systolic anterior motion (SAM) of the mitral valve that yields a posteriorly directed mitral regurgitant jet. CMR can supplement echocardiography in equivocal cases, provide further anatomical definition of hypertrophy and subvalvular apparatus anomalies, and quantify the degree of fibrosis by late gadolinium enhancement.
Management of HCM
Medical management of HCM has included avoiding hypovolemia, decreasing contractility, and managing tachycardia. For patients with oHCM, first-line therapy includes nonvasodilating BBs and nondihydropyridine CCBs. Current guidelines from the American Heart Association and American College of Cardiology recommend titrating these medications to either effectiveness or maximally tolerated dose.5 Unfortunately, use of these agents is often limited by side effects. If symptoms persist, then the addition of disopyramide or evaluation of intervention at an experienced center should be pursued. Disopyramide is a class IA antiarrhythmic that has negative inotropic effects. Patients started on disopyramide should continue their BB or CCB because this antiarrhythmic can increase conduction through the atrioventricular node; as such, patients with non-rate controlled atrial fibrillation may not be candidates for disopyramide. In addition, it has a strong side effect profile, including anticholinergic effects, QTc prolongation, and alterations of the cytochrome P450 system, necessitating time-intensive clinician oversight.7
Indications for septal reduction therapy (SRT) include patients who are severely symptomatic due to LVOT obstruction despite maximally tolerated medical therapy, those with recurrent exertional syncope due to elevated LVOT gradient >50mmHg or those with severe dyspnea.5,6 Options for SRT include surgical myectomy and alcohol septal ablation. Surgical myectomy depends on surgical candidacy and if the patient has other indications for surgery such as intrinsic mitral valve disease, subvalvular anomalies, or coronary artery disease. Alcohol septal ablation is reserved for patients who are not surgical candidates. Both procedures carry their own risk with success dependent on operator experience. A retrospective review of patients who underwent SRT across centers in the United States from 2003 to 2011 found significant differences in outcomes stratified by hospital volumes.8 For the 6386 patients who underwent myectomy, the incidence of hospital death was 15% compared to 3.8% (p < 0.001) in the lowest versus the highest volume tertiles. In addition, there was a higher incidence of permanent pacemaker implantation (10% vs 8.9%, p < 0.01) and bleeding (3.3% vs 1.7%, p < 0.01). For the 4862 patients who underwent alcohol septal ablation, findings were similar with a mortality rate of 2.3% in the lowest volume tertile compared to 0.6% in the highest tertile (p = 0.02) and higher rates of post-procedure renal failure (6.2% vs 2.4%, p < 0.001). Thus, current guidelines recommend patients being considered for invasive management of HCM be referred to dedicated HCM centers.5 For patients with nHCM, treatment is sparse; BBs and nondihydropyridine CCBs can be utilized for exertional angina and dyspnea, but have minimal efficacy. If symptoms persist, then diuretics may be trialed for heart failure symptoms. Surgical apical myectomy can be considered for carefully selected patients with apical HCM experiencing symptoms refractory to medical therapy per guidelines.5,6
Biochemistry and Pharmacokinetics of Mavacamten
As a cardiac myosin inhibitor, mavacamten (Figure 1) directly targets the hypercontractility that plays a central role in the pathophysiology of HCM. Normally, ATP is hydrolyzed to ADP once bound to myosin through ATPase. This reaction generates energy stored in the myosin head. When phosphate dissociates from myosin, myosin binds to actin. These bridges are released and shortening occurs as filaments slide past each other, creating myocardial contraction. In HCM, there is an upregulation of cardiac contractility with only 15–20% of myosin heads in an inactive state compared 40–50% in the inactive state normally.9
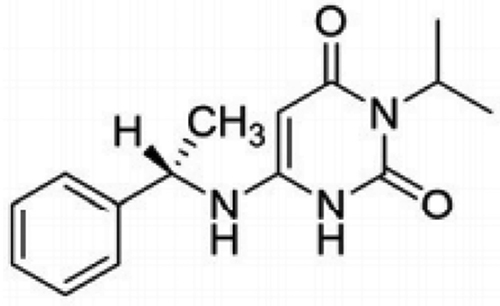 |
Figure 1 Molecular structure of mavacamten, 6-[[(1S)-1-phenylethyl]amino]-3-propan-2-yl-1H-pyrimidine-2,4-dione. Notes: Reprinted from Grillo MP, Erve JCL, Dick R, et al. In vitro and in vivo pharmacokinetic characterization of mavacamten, a first-in-class small molecule allosteric modulator of beta cardiac myosin. Xenobiotica; the fate of foreign compounds in biological systems. 2019;49(6):718–733, publisher Taylor & Francis Ltd, http://www.tandfonline.com reprinted by permission of the publisher.10 |
Mavacamten selectively inhibits beta-cardiac myosin ATPase through allosteric binding, decreasing the amount of myosin-actin bridges. In addition, this first-in-class drug targets the rate limiting step by preventing phosphate release.10 Furthermore, the drug slows the rates of myosin binding to actin in both the ADP-bound and ADP-released state.11 Combined, these mechanisms decrease the force generated by sarcomeres and reduce cardiac contractility.
Its pharmacokinetic profile includes excellent oral bioavailability of >85% and rapid absorption, with time to maximum concentration of 1 hour.2 The drug has a high distribution volume and long elimination phase. The mean half-life elimination is about 8 days in normal CYP2C19 metabolizers.12 Administration in Child-Pugh C hepatic impairment is unknown, while Child-Pugh Class A and B have increased drug exposure, but no dose adjustment is currently recommended.
Regarding pharmacodynamics, there is a dose-dependent manner in which left ventricular EF is decreased.13 This drop in EF noted in the Phase 2 trial was reversible approximately a month after receiving the drug. Currently, the drug carries a boxed warning for risk of heart failure and recommends against starting mavacamten in patients with left ventricular ejection fraction (EF) <55%. Cessation of the drug is recommended if EF <50%, heart failure symptoms develop or worsening clinical status.
Mavacamten Trials
Preclinical animal studies with mice heterozygous for the human mutation in myosin heavy chain have shown that mavacamten decreases ATPase activity, myocardial tension and fractional shortening in a dose-dependent manner. Furthermore, mice on mavacamten showed decreased contractility and profibrotic gene expression.14 Similar findings have been replicated in a study comparing wild-type mice to those with knockout of the cardiac myosin-binding protein-C. Exposure to mavacamten in both groups leads to a dose-dependent decrease in myocardial force that was most noticeable at low calcium activation.15 In Maine coon or mixed-breed founder cats that had oHCM, mavacamten was also seen in a dose-dependent manner to decrease contractility, LVOT gradients and SAM.16
Since these animal studies, there have been multiple human studies published demonstrating the efficacy of mavacamten. These include PIONEER-HCM, EXPLORER-HCM and VALOR-HCM. Table 1 summarizes these trials.
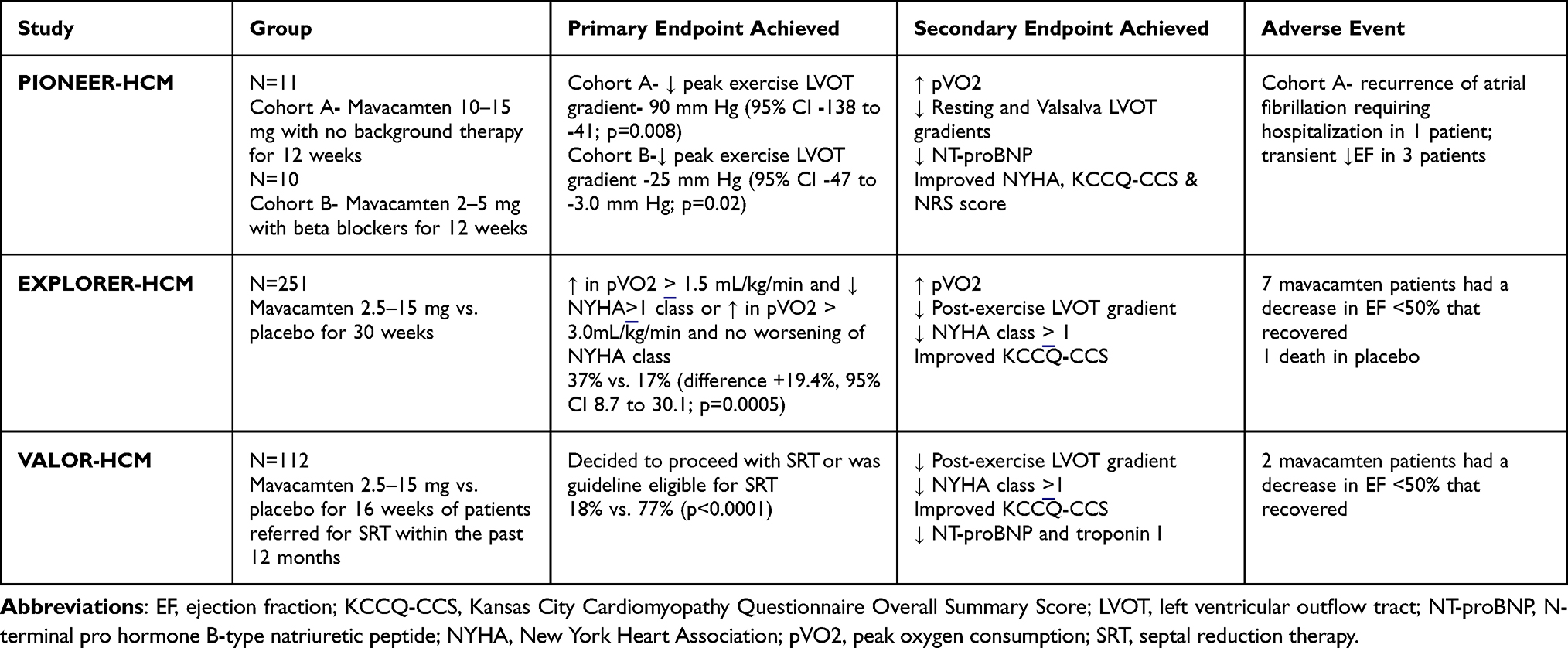 |
Table 1 Summary of Hypertrophic Cardiomyopathy Trials. |
PIONEER-HCM (Pilot Study Evaluating MYK‐461 in Subjects with Symptomatic Hypertrophic Cardiomyopathy and Left Ventricular Outflow Tract Obstruction) was a phase 2 open-label study whose primary endpoint was reduction in post-exercise LVOT gradient in obstructive HCM.13 The investigators divided the 21 patients into cohort A, whose group stopped all other HCM medications at least 14 days prior to initiation, and cohort B, whose group was maintained on their current HCM regimen. The mavacamten starting dosage was either 10 or 15mg per day for cohort A depending on weight, while cohort B was started on 2mg per day dosing. These doses could be titrated up at week 4. Exclusion criteria included those with exertional syncope within the past 6 months, sustained ventricular tachycardia, obstructive coronary artery disease, atrial fibrillation upon screening, persistent atrial fibrillation or atrial fibrillation with a resting heart rate of 100 beats per minute or greater in the past year. Key patient characteristics included 43% female with age range of 22–70 years, 57% having NYHA II while 43% had NYHA III symptoms. Twenty of the 21 patients completed the 12 weeks with one patient in cohort A stopping the drug due to recurrence of atrial fibrillation requiring hospitalization.17
PIONEER-HCM met its primary endpoint with a decrease in post-exercise peak LVOT gradient after 12 weeks. Cohort A LVOT gradient decreased from a mean of 103+50 mmHg to 19+13mmHg, while cohort B post-exercise gradient decreased from 86+43mmHg to 64+26mmHg. Cohort A also had a greater reduction in resting EF with a mean change of −15% [95% CI −23% to −6%]. The effect may have been greater in cohort A due to higher drug concentration with concentrates of 350–695ng/mL. Clinically, the results carried over with improvement in dyspnea scores. In terms of safety, the drug was well tolerated with 80% reporting only mild symptoms including fatigue, nausea and dyspnea. Aside from the one patient in cohort A who stopped mavacamten due to atrial fibrillation, three other patients experienced atrial fibrillation, which may have been related.17
EXPLORER-HCM (Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy) was a Phase 3 trial comparing oHCM patients on mavacamten versus placebo and examined if there was a change in peak oxygen consumption (pVO2) with improvement of at least one NYHA class or >3.0mL/kg/min pVO2 increase alone.18 The study involved 251 patients with the 49% in the mavacamten group. There was 1:1 randomization to either the study group with initiation of mavacamten 5mg daily or placebo for 30 weeks. Mavacamten was titrated up to 15mg daily during this period. Exclusion criteria were similar to PIONEER-HCM. Patients were maintained on standard therapy except for those on dual therapy with beta blockers and calcium channel blockers or monotherapy with disopyramide.19
The study met its primary endpoint with an improvement in pVO2 >1.5 mL/kg/minute with at least one NYHA class improvement or pVO2 by 3.0 mL/kg/minute without worsening of baseline NYHA class at 30 weeks in 37% of the mavacamten group compared to 17% in the placebo arm (difference +19.4%, 95% CI 8.7 to 30.1; p = 0.0005). In addition, secondary outcomes that were significant included post-exercise LVOT gradient, improvement in pVO2 and improved Kansas City Cardiomyopathy Questionnaire (KCCQ) scores with mavacamten. The number needed to treat (NNT) was also quite promising with NNT of 2, 3 and 4 for reduction of LVOT gradient <50mmHg, improvement in NYHA > 1 class grade and improvement to NYHA class 1, respectively.20
Regarding adverse events, safety and tolerability were similar in both groups. There were seven patients in the mavacamten group and three patients in the placebo group whose EF decreased to <50%. The seven patients in the study group recovered their EF after appropriate washout. One patient from the placebo arm passed away. Unlike in PIONEER-HCM, there was no difference in the incidence of atrial fibrillation.19
The long-term extension of this study (MAVA-LTE) is an ongoing five-year study with 231 patients from EXPLORER HCM. The majority (85%) of patients were on 5 or 10mg and on average the patients were 60 years old with 39% being female. Preliminary data at a median of 62 weeks show persistent change in resting LVOT gradient of −32.8mmHg, decrease in Valsalva LVOT gradient −46.4mmHg and decrease in N-terminal pro-B-type natriuretic peptide of 488ng/L while reduction in LVEF was −9%. There was also a notable decrease in NYHA class with 68% dropping at least one NYHA class and the percentage of patients in NYHA Class III decreasing from 29% to 4.9%.21
The most significant study thus far on patient outcomes is VALOR-HCM (Mavacamten in Adults with Symptomatic Obstructive HCM Who Are Eligible for Septal Reduction Therapy).22 The primary endpoint of this study examined if oHCM patients who were recommended for SRT were still eligible after taking mavacamten for 16 weeks. A total of 112 HCM patients with an LVOT gradient >50mmHg at rest or provocation were randomized in a 1:1 fashion based on the type of SRT recommended. Those randomized to the mavacamten group were started on a dose of 5mg that was uptitrated every 4 weeks after an echocardiogram. Patients remained on their baseline medical therapy. Aside from LVOT gradient >50mmHg and having a baseline EF >60%, other inclusion criteria were severe dyspnea or chest pain despite maximally tolerated medical therapy, NYHA class III or IV or NYHA class II with exertional syncope or presyncope.
Regarding the primary endpoint, patients were determined to qualify for SRT if those who had a baseline NYHA class II remained class II at 16 weeks with an LVOT gradient >50mmHg or if there was a decision to proceed with SRT. Conversely, patients who had a baseline NYHA class III or IV and improved to NYHA class II without exertional syncope or syncope during the trial did not qualify for SRT. Secondary endpoint included improvement in at least ONE NYHA class, resting LVOT gradient, Valsalva LVOT gradient and the KCCQ-23 score after 16 weeks.
Group characteristics included mean age of 60+12 years with 51% males. Patients in VALOR-HCM had a baseline resting LVOT gradient of 49mmHg and a post-exercise gradient of 84mmHg with 93% experiencing NYHA III or IV symptoms. Baseline medical therapy encompassed 46% on beta blockers, 15% on calcium channel blockers, 32% on combination therapy and 20% on disopyramide.
The primary endpoint was reached with 17.9% in the mavacamten group and 76.8% in the placebo group proceeding with SRT (p < 0.0001). As two patients in the mavacamten group and two patients in the placebo group decided to proceed with SRT, the primary endpoint was driven by patients who remained guideline eligible for SRT but decided against SRT. Secondary endpoints were also met including decrease in post-exercise peak LVOT gradient −37.2mmHg (95% CI −48.1 to −26.2mmHg), reduction by at least 1 NYHA functional class in 41.1% (95% CI 24.5–57.7%) and improvement in KCCQ-23 CSS by 9.4 points (95% CI 4.9–14.0).22
Overall, the drug was well tolerated with 7.1% of the study group experiencing nausea compared to 1.8% of participants in the control group. Rates of arrhythmias were low with 9.1% in the placebo group having nonsustained ventricular tachycardia compared no patients in the mavacamten group. None of the participants had chronic heart failure, sudden cardiac death (SCD) or syncope. Two patients in the mavacamten group did have a decrease in EF <50% requiring drug cessation for 4 weeks. However, both were able to resume the drug and were enrolled in the long-term extension study with systolic function recovery.
Notable study limitations were the lack of diversity in the trial, patient decision bias and small sample size for outcomes and/or safety. Because the trial duration of 16 weeks was determined by the study’s investigators to be an ethical amount of time for deferral of SRT, patients who were motivated to receive mavacamten could have declined SRT at the end to ensure they received the drug, as both the control and study group had the option to initiate or continue mavacamten, respectively. Lastly, the small sample size may limit favorable outcomes as well as adverse events. Longer duration studies currently underway will also help delineate further safety and/or tolerability issues. The long-term extension study of VALOR HCM will run for 96 weeks with participants blinded to the dosage of mavacamten followed by an 8 weeks post-treatment visit.
Application into Clinical Practice
The most recent guidelines from the American Heart Association and American College of Cardiology do not include a drug from the CMI class in the treatment cascade.23 As depicted in Figure 2, we foresee the application of mavacamten into practice depending on several clinical factors, including efficacy of guideline medical therapy, patient candidacy for SRT, and mavacamten tolerance. The Food and Drug Administration’s approval of mavacamten to treat patients with obstructive HCM and progressive symptoms will surely change this algorithmic approach for oHCM patients.2 At the time of this review, mavacamten is prescribed only through the Risk Evaluation and Mitigation Strategy (REMS) program, to ensure safe and monitored administration of the drug.2
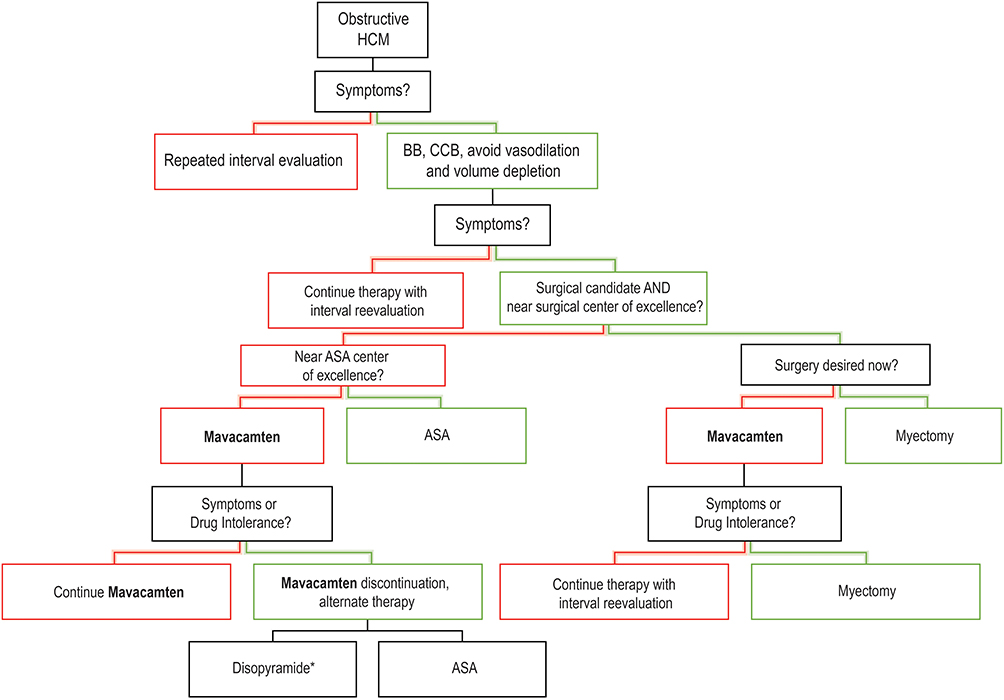 |
Figure 2 Authors proposed treatment cascade of obstructive HCM. Abbreviations: BB, beta blocker; CCB, calcium; red color branches, no; green color branches, yes. Notes: Treatment cascade adapted from 2020 AHA/ACC Guidelines for the Diagnosis and Treatment of Patients with Hypertrophic Cardiomyopathy. Data from Ommen et al23 *Minimal quality evidence and unfavorable systemic side effect profile make this option the least recommended. |
Despite the low incidence of LV systolic dysfunction in the clinical trials of mavacamten, the potential ramifications of this outcome with therapy are significant. The REMS program is focused on three aspects: the provider, the pharmacist, and the patient. Providers must be REMS certified to prescribe mavacamten; this responsibility includes proper counseling of patients on the risks, expected symptoms, and medication interactions with mavacamten use. Pharmacy, the second aspect of the program, has the role of reviewing patient medication regimens and reinforcing drug interactions and avoidances. Lastly, the patient must commit to regular screening echocardiograms during their mavacamten therapy. Specifically, the initiation phase includes a limited echocardiogram within 21–28 days of their first dose for LVEF and left ventricular outflow tract gradient monitoring. After this, there are two additional echocardiograms four weeks apart. The maintenance phase follows and includes a limited echocardiogram every 12 weeks. If the LVEF decreases below 50%, treatment must be interrupted and serial echocardiograms in 4-week intervals performed until the LVEF increases to greater than 50%. Mavacamten can be restarted once this threshold is met, at a lower dosage.2,24
Among the PIONEER-HCM, EXPLORER-HCM, and VALOR-HCM trials, the majority of patients were on background therapy that included BBs or nondihydropyridine CCBs.17,19,22 Due to this, mavacamten will likely not be a substitute for the first-line therapy, especially as newer studies have shown the benefits of B-blockers in oHCM.25 The trial participants were included only if on maximally tolerated medical therapy with breakthrough symptoms; therefore, mavacamten will likely be indicated only for those with symptoms after a trial of first-line therapy.
In its nascent stages, mavacamten requires similar follow-up, as dictated by the clinical trial methods.17,19,22 In comparing side effects, while mavacamten is a similar cytochrome P450 enzyme inducer, it does not affect the anti-cholinergic system nor alter the QTc interval. As trials studying mavacamten have focused on short-term monitoring, long-term side effects are unknown and need further study, including the influence on lactation, pregnancy, LVEF, and other drug–drug interactions. Considering the side effect profile and the mounting evidence for the CMI drug class, we support mavacamten as second-line therapy for obstructive HCM based on recent clinical trial results. Upcoming long-term extension studies of randomized trials will help inform the risk profile of chronic mavacamten therapy, especially with regard to probability of LVEF reduction.26–28
For the patients with symptom limitation on first-line medical therapy and access to surgical and procedural centers of excellence, consideration for SRT is the next viable option.23 Aside from this, we suggest several roles for the initiation of mavacamten. First, for those with access to SRT and centers of excellence, if the patient should not want an invasive therapy, mavacamten may play a role in symptom reduction as the next best therapeutic option. Next, the patient who would like to delay SRT will certainly derive benefit from the drug. The VALOR-HCM trial helped address this scenario. As described previously, it randomized patients to a 16-week period of mavacamten or placebo if they met criteria for SRT with an ejection fraction >60%. While the trial duration was short, it demonstrated a significant reduction in LVOT gradient, improvement in symptoms as assessed by the Kansas City Cardiomyopathy Questionnaire, and reduction in serum NT-proBNP.22 Having another pharmacologic option prior to SRT is important as not all SRT is effective in total reduction of symptoms, most notably among young patients with severe hypertrophy who undergo surgical myectomy.29 Lastly, for those patients who are being diagnosed with more frequency with oHCM and progressive symptoms who do not have access to centers of excellence for SRT, mavacamten should be foundational in their therapy after the initial BB or CCB options. Figure 3 displays the predicted uses for mavacamten.
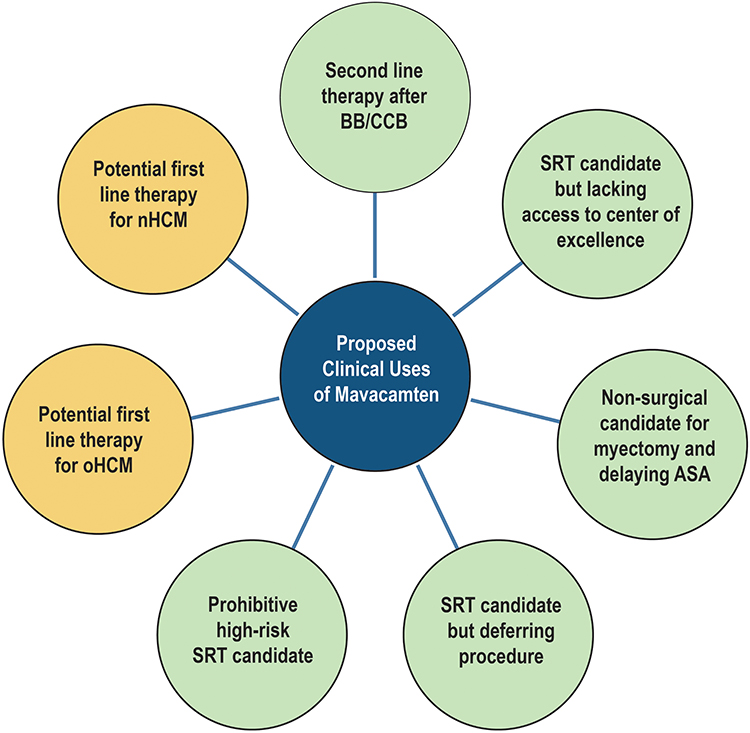 |
Figure 3 Proposed strategies for mavacamten use. Color legend: Green, contemporary; Yellow, proposed. Abbreviations: BB, beta blocker; CCB, calcium channel blocker; oHCM, obstructive hypertrophic cardiomyopathy; ASA, alcohol septal ablation; SRT, septal reduction therapy. |
Two practical considerations for the clinical use of mavacamten are the intensive monitoring required for drug administration and the projected economic cost of its use. According to the FDA release, mavacamten administration requires frequent echocardiograms during both the 12-week initiation phase and subsequent maintenance period. While we feel the monitoring intensity is appropriate, it will pose challenges for pragmatic use.2,30 In a recent study by the Institute for Clinical and Economic Review, an economic model projected that mavacamten would produce more quality-adjusted life-years compared to first-line therapy but at a much higher cost.31 These models will change if more clinical trials show increased benefit of the drug and prices are reduced but remain a topic of consideration given the need for cost-saving in the current healthcare environment.
There were several populations excluded from VALOR-HCM, EXPLORER-HCM, and PIONEER-HCM. While the comprehensive list is noted in each trial appendix, notable common characteristics that have not been studied are patients with atrial fibrillation at time of drug initiation, second degree AV-block, Child-Pugh class C liver disease or chronic kidney disease manifesting as a glomerular filtration rate ≤ 30 mL/min/1.73 m2.17,19,22 Whether mavacamten can be safely administered in these populations is currently unclear.
Other Populations for Mavacamten
Apart from oHCM, two other groups that are under investigation are nHCM and heart failure with preserved ejection fraction (HFpEF). Both pathologies represent populations with minimal proven therapies, igniting more interest in developing targeted treatments. MAVERICK-HCM is a Phase II study designed to examine dosing and effects of mavacamten in nHCM. The authors studied a small population of 59 patients and met the primary safety objective. The drug was generally well tolerated, with the majority of adverse events described as mild including palpitations, dizziness, and fatigue. Although exploratory in nature, NT-proBNP demonstrated a favorable response to drug therapy, suggestive of reduction in myocardial wall stress.32 This small phase II study is hypothesis-generating and will need to be evaluated in larger, randomized controlled trials.
Patients with HFpEF represent a heterogeneous population, which makes trial enrollment difficult to assess specific subgroups.33 Given mavacamten’s positive lusitropic effect, the drug is being studied in the HFpEF population in the EMBARK-HF trial. The eligibility criteria include obesity, clinical heart failure, invasive hemodynamic evidence of elevated filling pressures, elevated natriuretic peptides, and echocardiographic findings suggesting HFpEF. Results will help inform the utility of myosin inhibitors for those with similar characteristics of the study population and HFpEF.34
Future Role for Drugs and Gene Therapies
A limitation of mavacamten is its longer half-life of around 7 days which may make drug monitoring and side effects challenging. An alternative second-generation myosin inhibitor, aficamten, is under study currently in the REDWOOD HCM trial, which completed its phase 2 clinical trial.28 The trial included oHCM patients and divided them into three groups with a placebo group, aficamten alone or dose adjusted aficamten with background therapy for 10 weeks. The study met its primary endpoint of safety and its secondary endpoints of reduction in LVOT gradient both at rest and with Valsalva after 10 weeks. Larger scale results from the Phase III SEQUOIA-HCM clinical trial are pending as of this manuscript publication.
As described above, hypertrophic cardiomyopathy has been associated with over 450 genetic mutations, with most directed at the sarcomere and myofilaments of cardiac muscle. The most common are MYH7 and MYBPC3, and there are several variations of mutations among these genes; yet still, in a genetic study only 34% of patients with phenotypic HCM were genotype positive for a known culprit mutation.35 Cardiac myosin inhibitors may keep symptoms at bay and reduce obstruction, but finding a treatment for the genetic underpinning of HCM remains elusive. Given new technologies such as CRISPR and other genetic modification techniques, a recent study examining endogenous, germline-specific DNA repair mechanisms corrected mutations that are thought to cause HCM. The future of gene editing may lead to a greater emphasis on determining pathogenic gene mutations and the targeted therapies that would follow, representing a cure as opposed to symptom relief and management of obstruction.
Conclusion
The discovery of mavacamten has demonstrated great potential in treating oHCM. Mavacamten improves both quantitative and qualitative measures including peak LVOT gradient at rest and post-exercise, cardiac biomarker profile, and patient perception of symptoms. Perhaps most strikingly, drug initiation delayed the need for SRT in VALOR-HCM. Although long-term studies are currently ongoing, the side effect profile appears to be tolerable with only transient decreases in EF. Given the promising findings, clinical incorporation of mavacamten for oHCM should be strongly considered for those with refractory symptoms despite maximal medical therapy, those with limited access to centers of excellence or those who prefer to defer SRT.
Furthermore, the use of mavacamten may expand beyond oHCM with promising phase 2 studies in HCM patients without obstructive physiology and in the HFpEF population. In addition, another myosin inhibitor, aficamten, is being studied for oHCM and results of a phase 3 clinical trial are pending. While gene editing remains elusive, myosin inhibitors have proven to be an effective drug for treating oHCM and should be incorporated into clinical practice with appropriate monitoring. With the advent of mavacamten, a first-in-class drug, the medical management of oHCM has changed drastically with many more promising developments on the horizon.
Disclosure
Tiffany Dong and Ben Alencherry are co-first authors for this study. Dr Milind Y Desai is a consultant for Bristol Myers Squibb and Medtronic. The authors report no other conflicts of interest in this work.
References
1. Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2015;65(12):1249–1254. doi:10.1016/j.jacc.2015.01.019
2. Administration FD. Approved drug proucts: CAMZYOS (mavacamten) capsules for oral use; 2022.
3. Maron BJ. Clinical course and management of hypertrophic cardiomyopathy. N Engl J Med. 2018;379(7):655–668. doi:10.1056/NEJMra1710575
4. Ingles J, Burns C, Bagnall RD, et al. Nonfamilial hypertrophic cardiomyopathy: prevalence, natural history, and clinical implications. Circ Cardiovasc Genet. 2017;10:2. doi:10.1161/CIRCGENETICS.116.001620
5. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy. Circulation. 2020;142(25):e558–e631.
6. Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–2779.
7. Verlinden NJ, Coons JC. Disopyramide for hypertrophic cardiomyopathy: a pragmatic reappraisal of an old drug. Pharmacotherapy. 2015;35(12):1164–1172. doi:10.1002/phar.1664
8. Kim LK, Swaminathan RV, Looser P, et al. Hospital volume outcomes after septal myectomy and alcohol septal ablation for treatment of obstructive hypertrophic cardiomyopathy: US nationwide inpatient database, 2003-2011. JAMA Cardiol. 2016;1(3):324–332. doi:10.1001/jamacardio.2016.0252
9. Edelberg JM, Sehnert AJ, Mealiffe ME, Del Rio CL, McDowell R. The impact of mavacamten on the pathophysiology of hypertrophic cardiomyopathy: a narrative review. Am J Cardiovasc Drugs. 2022;22:497–510. doi:10.1007/s40256-022-00532-x
10. Grillo MP, Erve JCL, Dick R, et al. In vitro and in vivo pharmacokinetic characterization of mavacamten, a first-in-class small molecule allosteric modulator of beta cardiac myosin. Xenobiotica. 2019;49(6):718–733. doi:10.1080/00498254.2018.1495856
11. Kawas RF, Anderson RL, Ingle SRB, Song Y, Sran AS, Rodriguez HM. A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J Biol Chem. 2017;292(40):16571–16577. doi:10.1074/jbc.M117.776815
12. Tower-Rader A, Ramchand J, Nissen SE, Desai MY. Mavacamten: a novel small molecule modulator of β-cardiac myosin for treatment of hypertrophic cardiomyopathy. Expert Opin Investig Drugs. 2020;29(11):1171–1178. doi:10.1080/13543784.2020.1821361
13. Heitner SB, Jacoby D, Lester SJ, et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy. Ann Intern Med. 2019;170(11):741–748. doi:10.7326/M18-3016
14. Green EM, Wakimoto H, Anderson RL, et al. A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science. 2016;351(6273):617–621. doi:10.1126/science.aad3456
15. Mamidi R, Li J, Doh CY, Verma S, Stelzer JE. Impact of the myosin modulator mavacamten on force generation and cross-bridge behavior in a murine model of hypercontractility. J Am Heart Assoc. 2018;7(17):e009627. doi:10.1161/JAHA.118.009627
16. Stern JA, Markova S, Ueda Y, et al. A small molecule inhibitor of sarcomere contractility acutely relieves left ventricular outflow tract obstruction in feline hypertrophic cardiomyopathy. PLoS One. 2016;11(12):e0168407. doi:10.1371/journal.pone.0168407
17. Heitner SB, Jacoby D, Lester SJ, et al. Mavacamten treatment for obstructive hypertrophic cardiomyopathy a clinical trial. Ann Intern Med. 2019;170:741–748.
18. Spertus JA, Fine JT, Elliott P, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): health status analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2021;397(10293):2467–2475. doi:10.1016/S0140-6736(21)00763-7
19. Olivotto I, Oreziak A, Barriales-Villa R, et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396:759–769. doi:10.1016/S0140-6736(20)31792-X
20. Semigran M, Wang Y, Xie J, Xu Y, Sutton MB, Desai NR. Abstract 10969: Estimated number needed to treat with mavacamten vs placebo to improve functional capacity and left ventricular outflow tract obstruction in patients with symptomatic obstructive hypertrophic cardiomyopathy. Circulation. 2021;144(Suppl_1):A10969–A10969. doi:10.1161/circ.144.suppl_1.10969
21. Rader F, Choudhury L, Saberi S, et al. Long-term safety of mavacamten in patients with obstructive hypertrophic cardiomyopathy: interim results of the mava-long term extension (LTE) study. J Am Coll Cardiol. 2021;77(18_Supplement_1):532. doi:10.1016/S0735-1097(21)01891-X
22. Milind DY, Owens A, Geske Jeffrey B, et al. Myosin inhibition in patients with obstructive hypertrophic cardiomyopathy referred for septal reduction therapy. J Am Coll Cardiol. 2022;80(2):95–108. doi:10.1016/j.jacc.2022.04.048
23. Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American heart association joint committee on clinical practice guidelines. J Am Coll Cardiol. 2020;76(25):e159–e240. doi:10.1016/j.jacc.2020.08.045
24. FDA approves new drug to improve heart function in adults with rare heart condition [press release]. FDA.gov; 2022.
25. Dybro AM, Rasmussen TB, Nielsen RR, Andersen MJ, Jensen MK, Poulsen SH. Randomized trial of metoprolol in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2021;78:2505–2517. doi:10.1016/j.jacc.2021.07.065
26. Bristol-Myers Squibb. Extension Study of Mavacamten (MYK-461) in Adults With Symptomatic Obstructive Hypertrophic Cardiomyopathy Previously Enrolled in PIONEER 2018 https://ClinicalTrials.gov/show/NCT03496168.
27. Bristol-Myers Squibb. A Long-Term Safety Extension Study of Mavacamten in Adults Who Have Completed MAVERICK-HCM or EXPLORER-HCM 2018 https://ClinicalTrials.gov/show/NCT03723655
28. Cytokinetics REDWOOD-HCM: Randomized Evaluation of Dosing With CK-3773274 in HCM 2020 https://ClinicalTrials.gov/show/NCT04219826
29. Wells S, Rowin EJ, Boll G, et al. Clinical profile of nonresponders to surgical myectomy with obstructive hypertrophic cardiomyopathy. Am J Med. 2018;131:e235–e239. doi:10.1016/j.amjmed.2017.12.031
30. Dalo JD, Weisman ND, White CM. Mavacamten, a first-in-class cardiac myosin inhibitor for obstructive hypertrophic cardiomyopathy. Ann Pharmacother. 2022;2022:10600280221117812.
31. Beinfeld M. Mavacamten for hypertrophic cardiomyopathy: effectiveness and value; 2022.
32. Ho CY, Mealiffe ME, Bach RG, et al. Evaluation of mavacamten in symptomatic patients with nonobstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2020;75:2649–2660. doi:10.1016/j.jacc.2020.03.064
33. Patel RB, Shah SJ. Inclusion criteria for heart failure with preserved ejection fraction clinical trials: making the case for precision diagnosis and greater inclusivity. J Card Fail. 2022;28:732–735. doi:10.1016/j.cardfail.2022.03.345
34. Bristol-Myers Squibb A study of mavacamten in participants with HFpEF and elevation of NT-proBNP with or without elevation of cTnT (EMBARK-HFpEF) 2021 https://ClinicalTrials.gov/show/NCT04766892
35. Bos JM, Will ML, Gersh BJ, Kruisselbrink TM, Ommen SR, Ackerman MJ. Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2014;89:727–737. doi:10.1016/j.mayocp.2014.01.025
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.